
Abstract
Extra-axial chordoma is an exceedingly rare tumor, with only 28 cases reported in the literature to date. Axial and extra-axial chordoma exhibits complete morphologic and immunophenotypic (expression of brachyury) overlap. However, in consideration of the non-canonical presentation, extra-axial chordoma is under-recognized and often misdiagnosed, most often as extraskeletal myxoid chondrosarcoma or myoepithelioma. To increase our understanding of the clinicopathologic features of extra-axial chordoma, six cases have been retrieved from the files of the Istituto Ortopedico Rizzoli and of the General Hospital of Treviso. The clinicoradiologic, morphologic, and molecular features have been analyzed, and the follow-up was updated. Our series included four female and two male patients; their ages ranged from 20 to 67 years (mean 45.8 years). All patients presented with a single mass localized in four cases in the soft tissue (posterior arm, left leg, dorsal aspect of the foot, and popliteal fossa), and in two cases in the bone (radius and second metacarpal bone). Grossly, the neoplasm was lobulated, with a fleshy cut surface and a diameter ranging between 0.8 and 8 cm (mean 3.4 cm). Morphologically, all six cases showed an epithelioid cell proliferation organized in nests and cords demarcated by fibrous septa and set in an abundant extracellular myxoid matrix. Neoplastic cells featured hyperchromatic nuclei and abundant vacuolated cytoplasm. Immunohistochemically, all six cases were strongly positive for EMA, cytokeratin AE1/AE3, S100, and brachyury. INI1 nuclear expression was retained. Smooth muscle actin, calponin, p63, and GFAP were all negative. Fluorescent in situ hybridization (FISH) analysis did not reveal rearrangements involving NR4A3, FUS, and EWSR1 genes. At follow-up (mean 55 months), all patients were alive without disease after local surgical treatment. One patient underwent thigh amputation following multiple local recurrences and inguinal node metastases treated with marginal resection. In conclusion, primary extra-axial chordoma is an extremely rare neoplasm with distinct morphological and immunohistochemical features. Immunomorphology and molecular analysis allow distinction from both extraskeletal myxoid chondrosarcoma and myoepithelioma. Complete surgical resection appears to be curative.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chordoma represents a rare malignant bone tumor (annual incidence is of approximately 0.08 cases per 100,000 people) exhibiting notochordal differentiation that occurs almost exclusively in the axial skeleton, with a predilection for the vertebral bodies, the sacrococcygeal bone and the base of the skull [1]. The extra-axial location is extremely unusual, with only 28 cases (16 of soft tissue and 12 of bone) reported in the literature to date [2,3,4,5,6,7,8,9,10,11,12]. Axial and extra-axial chordoma exhibit complete morphologic and immunophenotypic overlap, featuring an epithelioid cell proliferation set in a myxoid background, exhibiting variably vacuolated eosinophilic cytoplasm (so-called physaliphorous cells), centrally located nuclei and immunohistochemical expression of brachyury [1, 13].
However, in consideration of the non-canonical presentation, extra-axial chordoma is under-recognized and often misdiagnosed, especially when large and deep-seated, mostly as extraskeletal myxoid chondrosarcoma and myoepithelioma. Brachyury is a transcription factor playing a key role in notochordal differentiation and represents a very specific marker for notochordal tumors. A strong nuclear immunohistochemical expression of brachyury was reported in the majority of neoplastic cells of two cases of soft tissue myoepithelioma [14, 15]. Of course, in consideration of the already mentioned immunomorphologic overlap, it cannot be excluded that those cases actually represented examples of extra-axial chordoma.
Myoepithelial neoplasms represent a clinicopathologic heterogeneous group of tumors, ranging from benign to highly aggressive lesions, which, based on their anatomical location as well on the presence/absence of ductal differentiation, are sometimes designated under different labels: soft tissue myoepithelioma, parachordoma, and mixed tumors [15]. EWSR1 and FUS gene rearrangements are observed in approximately 65% of soft tissue myoepithelioma. NR4A3 gene rearrangement is instead detected in almost all cases of extraskeletal myxoid chondrosarcoma, therefore serving as a powerful diagnostic confirmatory tool [15].
To increase our understanding of the clinicopathologic and molecular features of extra-axial chordoma, we herein described six cases of this exceedingly rare tumor entity.
Material and methods
Cases
The study is based on six patients with extra-axial chordoma, without history of prior axial chordoma or other skeletal notochordal tumor evaluated by radiological imaging (TC, MRI, and PET). The samples relative to four patients were obtained from the Pathology Department of the Rizzoli Institute from 1981 to 2016, and the other two cases were external consultations of the Department of Pathology of the General Hospital of Treviso. Follow-up data was available for all six patients. Institutional Review Board permission was obtained for the study (protocol numbers: 0009581 and 0011714).
Immunohistochemistry
Serial, 4-mm thick paraffin sections mounted on pre-coated slides were processed according to standardized automated procedures (Ventana Medical Systems, Tucson, AZ) and immunostained with the following antibodies: protein S-100 (rabbit polyclonal antibody, Ventana, pre-diluted), pan-cytokeratin AE1/AE3 (mouse monoclonal antibody, clone AE1/AE3/PCK26, Ventana, pre-diluted), EMA (mouse monoclonal antibody, clone E29, Ventana, pre-diluted), INI1 (mouse monoclonal antibody, clone BAF47, Cell Marque, pre-diluted), GFAP (rabbit monoclonal antibody, clone EP672Y, Cell Marque, pre-diluted), smooth muscle actin (mouse monoclonal antibody, clone MMAB, 1A4, Ventana, pre-diluted), p63 (mouse monoclonal antibody, clone 4A4, Ventana, pre-diluted), calponin (rabbit monoclonal antibody, clone EP798Y, Ventana Cell Marque, pre-diluted), and brachyury (Santa Cruz Biotechnology; Rabbit polyclonal antibody, 1:50 dilution). Pretreatment for antigen retrieval was performed at 95 °C with Tris-EDTA pH 8 for 20 min. Staining was performed with an UltraView Universal DAB Detection Kit (Ventana Medical Systems), as previously reported [16]. Appropriate positive and negative controls were included in each run, and the absence of a primary antibody was used as a negative control.
FISH
FISH was performed with the EWSR1 (22q12), the FUS (16p11), VYsis LSI Dual Color Break-apart DNA probe (Abbott Molecular, Des Plaines, IL), and the SPEC NR4A3 (9q22.33) Dual Color Break-apart probe (ZytoVision GmbH, Bremerhaven, Germany) according to the manufacturers’ protocol as we have previously published [17]. Briefly, tissue sections of 4 mm stained with H&E from each tumor were prepared, and areas of representative non-necrotic neoplasm were marked. A minimum of 100 tumor cell nuclei with intact morphology, as determined by DAPI counterstaining, were counted in the previously marked neoplastic area. A positive result was defined as the presence of a visible translocation (separation of red and green signals > 3 signal diameters) in > 10% of the cells.
Results
Clinical details are summarized in Table 1. The six patients consisted of four females and two males with an age range from 20 to 67 years (mean 46 years). All patients presented with a single localized mass. Four cases occurred in the soft tissue (posterior arm, left leg, foot, and popliteal fossa), and two cases in the bone (radius and second metacarpal bone). Clinically, in all patients, the lesions were initially asymptomatic but became painful with activity during a time variable between 6 months to 2 years.
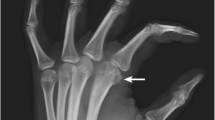
Imaging was available for four patients (two bone lesions and two in the soft tissues). Bone tumors were parosteal and cortical, respectively. Both were lytic, well-limited, and surrounded by an inflammatory reaction, with an intermediate signal on T1-weighted images, and high on T2 (Fig. 1a–c). One soft tissue lesion, eroded the cortex of the proximal tibia, was well-circumscribed, with a low signal on T1-weighted images, with more intense parts due to bleeding, and a high and heterogeneous signal on T2-weighted images. The 4th lesion arose inside the knee joint had a high signal on proton density. The bone was uninvolved (Figs. 2, 3, and 4).

Case 3. (a) On CT, the lesion is cortical, lytic, well-limited, and surrounded by a thick periosteal reaction. On magnetic resonance, the lesion has an intermediate signal on the T1-weighted images and high signal on the T2-weighted image. There is intramedullary edema (b: coronal T1 and c: axial T2-weighted images)

Case 3. On hematoxylin and eosin staining relative to the surgical specimen, neoplastic cells that infiltrate the trabecular bone, showing a lobulated growth pattern, are evident (a hematoxylin and eosin staining, ×100 magnification; b ×200 magnification). c The nuclei of the vacuolated neoplastic cells show a strong nuclear immunoreactivity for brachyury (×400 magnification)

Case 4. Large cells with clear to eosinophilic cytoplasms set in extracellular myxoid matrix are present (a hematoxylin and eosin staining, ×100 magnification; b hematoxylin and eosin staining, ×400 magnification). The nuclei of neoplastic cells are positive for brachyury antibody (×400 magnification)

Case 6. The neoplastic cells demonstrate pale eosinophilic to somewhat foamy cytoplasm with ovoid to round nuclei, immunohistochemically positive for brachyury (a hematoxylin and eosin staining, ×200 magnification; b hematoxylin and eosin staining, ×400 magnification; c brachyury expression, ×400 magnification)
All patients underwent surgical treatment. Complete resection was obtained in five cases and marginal excision in one case. Grossly, soft tissue neoplasms were well-circumscribed, composed of multilobular white or yellowish nodules, featuring a partially gelatinous cut surface and variable amounts of hemorrhage. By contrast, bone lesions were lobulated, with a fleshy cut surface. Tumor ranged in size from 0.8 to 8 cm (mean 3.4 cm).
Microscopically, at low power, both soft tissue and bone tumors exhibited a lobular pattern of growth with fibrous bands of varying thickness separating neoplastic cells. In all six cases, neoplastic cells featured an epithelioid morphology and were organized in nests and cords set in an abundant extracellular myxoid matrix. At high power, neoplastic cells featured hyperchromatic nuclei and partially vacuolated clear to eosinophilic cytoplasm, as seen in classic physaliferous cells. All but one case showed mild cytologic atypia, displaying low to moderate cellularity. Only a case had focal areas of increased cellularity in absence of severe cytological atypia and with little intervening myxoid stroma. No tumors contained foci of chondrogenic differentiation or areas of dedifferentiation.
Immunohistochemically, all six cases showed strong expression of EMA, cytokeratin AE1/AE3, S100, and brachyury. INI1 nuclear expression was retained. Smooth muscle actin, calponin, p63, and GFAP were all negative. At FISH analysis, no tumor showed NR4A3, EWSR1, and FUS gene rearrangements.
At follow-up (mean 36 months; range 4–74 months), all patients are alive without disease following local surgical treatment. No patients underwent adjuvant chemotherapy or radiotherapy after the first surgical resection. One patient treated with marginal resection underwent thigh amputation following two local recurrences developed at 6 and 9 months from diagnosis, respectively. The same patient presented inguinal lymph node metastases 22 months after the first resection. The other five patients have not developed local recurrence or distant metastases at most recent follow-up.
Discussion
Extra-axial chordomas have never been systematically studied because of their extreme rarity and the difficulty in separating them from similar-appearing lesions. In fact, based on microscopic features, their distinction from extraskeletal myxoid chondrosarcoma and myoepithelial tumor may be really challenging. Before the introduction of both immunohistochemical staining for brachyury and molecular analysis, the term “parachordoma” (the original label “chordoma periphericum” was coined by Maria Dabska in 1977) [18] became popular to describe a heterogeneous group of neoplasms that most likely includes extraskeletal myxoid chondrosarcomas, myoepithelial neoplasms/mixed tumors, and chordoma occurring in deep soft tissue in extra-axial sites [19, 20]. Nuclear expression of brachyury, a member of the T-box proteins that play a key role in early development of the spine, is currently regarded as sensitive as well as specific marker for notochordal differentiation. Interestingly, Miettinen et al. reported two out of 32 cases of myoepithelioma of the soft tissue featuring strong and diffuse brachyury expression in the majority of neoplastic cells [14]. To make diagnostic confusion, even greater loss of nuclear expression of SMARCB1/INI1 (that is seen in approximately 20% of both extraskeletal myxoid chondrosarcoma and myoepithelial tumors) has been observed in subset of pediatric axial chordomas [21, 22]. In consideration of the described immunomorphologic similarities, the integration of molecular genetic finding appears mandatory in order to support accurate diagnoses. Genetically, the vast majority of deep-seated myoepithelial tumors feature distinct molecular alterations. The most common genetic aberration is represented by EWSR1 gene rearrangement, present in approximately 50% of myoepithelial tumors [12]. Recently, a distinctive variant of cutaneous myoepithelial tumor, designated as syncytial myoepithelioma, was found to have a higher percentage (82%) of EWSR1 gene rearrangement [23]. It is interesting to observe as EWSR1-driven myoepithelial tumors most often exhibit a uniform rounded cell morphology associated with cytoplasm clearing, therefore overlapping morphologically with extra-axial chordoma [14]. Recently Huang reported FUS gene rearrangement in 6 out of 66 EWSR1-negative myoepithelial tumors, reporting the FUS-rearranged myoepithelial tumors had wide clinical presentations, morphologic patterns, and consistently lacked ductal differentiation or osteochondroid matrix formation. Regarding extraskeletal myxoid chondrosarcoma, immunohistochemical stains (variable immunopositivity for S100 and EMA) are of limited diagnostic help and morphologic overlap with myoepithelioma almost complete. In this situation, the demonstration of NR4A3 (nuclear receptor subfamily 4, group A, member 3) gene rearrangement is mandatory to support the diagnosis [24]. Our series of extra-axial chordoma exhibited the typical immunohistochemical features of notochordal neoplasms which include a strong expression of EMA, cytokeratin AE1/AE3, S100, and brachyury, associated with retained expression of SMARCB1/INI1 protein without rearrangement of the EWSR1, NR4A3, or FUS genes. In addition to myoepithelioma and extraskeletal myxoid chondrosarcoma, differential diagnosis of extra-axial chordoma includes metastatic axial chordoma that can be excluded only following careful clinical work-up.
As far as prognosis is concerned, primary extra-axial chordoma (34 cases including our 6 cases published so far) tends to exhibit an indolent clinical course. Median follow-up of the reported cases is 66 months (2 to 212 months) [2,3,4,5,6,7,8,9,10,11,12]. Seven of these 31 patients developed local recurrence after a mean of 18 months (range, 6–75 months) and were associated with lung metastases in two patients. At the last follow-up, most (n = 27) of these 32 patients were alive without evidence of diseases; three were alive with disease, one died for unknown disease, and one died of cardiac failure without evidence of neoplastic disease. Despite the limited number of cases, these data would suggest that extra-axial chordoma exhibits a less aggressive behavior compared with axial skeletal locations but of course this may be largely dependent upon more favorable anatomic location [1]. There is no evidence for a role for either radiation therapy or chemotherapy in the extra-axial chordoma, and surgical resection with wide margins is to be recommended.
In conclusion, primary extra-axial chordoma is an extremely a rare neoplasm with distinct morphological and immunohistochemical features that allow distinction from morphologic mimics such as both extraskeletal myxoid chondrosarcoma and myoepithelioma. Our data would further confirm the assumption that extra-axial chordoma is characterized by less aggressive behavior when compared to axial skeletal chordoma, making complete surgical resection most often curative.
References
Flanagan A, Yamaguchi T (2013) Notochordal tumours. In: Fletcher CD, Bridge JA, Hogendoorn PC, Mertens F (eds) In World Health Organization classification of tumours of soft tissue and bone. IARC Press, Lyon, pp 326–329
Rekhi B (2016) Primary, large extra-axial chordoma in proximal tibia: a rare case report with literature review and diagnostic implications. APMIS 124:238–242
Lauer SR, Edgar MA, Gardner JM, Sebastian A, Weiss SW (2013) Soft tissue chordomas: a clinicopathologic analysis of 11 cases. Am J Surg Pathol 37:719–726
Tirabosco R, Mangham DC, Rosenberg AE, Vujovic S, Bousdras K, Pizzolitto S, De Maglio G, den Bakker MA, Di Francesco L, Kalil RK, Athanasou NA, O'Donnell P, McCarthy EF, Flanagan AM (2008) Brachyury expression in extra-axial skeletal and soft tissue chordomas: a marker that distinguishes chordoma from mixed tumor/myoepithelioma/parachordoma in soft tissue. Am J Surg Pathol 32:572–580
Rekhi B, Sable M, Jambhekar NA (2012) Histopathological, immunohistochemical and molecular spectrum of myoepithelial tumours of soft tissues. Virchows Arch 461:687–697
Nielsen GP, Mangham DC, Grimer RJ, Rosenberg AE (2001) Chordoma periphericum: a case report. Am J Surg Pathol 25:263–267
Huang SC, Chen HW, Zhang L, Sung YS, Agaram NP, Davis M, Edelman M, Fletcher CD, Antonescu CR (2015) Novel FUS-KLF17 and EWSR1-KLF17 fusions in myoepithelial tumors. Genes Chromosomes Cancer 54(5):267–275
Suzuki H, Yamashiro K, Takeda H, Nojima T, Usui M (2011) Extra-axial soft tissue chordoma of wrist. Pathol Res Pract 207(5):327–331
Bitzer A, McCarthy EF, Morris CD (2017) Extra-axial chordoma of the hand. J Hand Surg Am 42(11):933.e1–933.e5
Koh JS, Chung JH, Lee SY, Cho KJ (2000) Parachordoma of the tibia: report of a rare case. Pathol Res Pract 196:269–273
Flucke U, Palmedo G, Blankenhorn N, Slootweg PJ, Kutzner H, Mentzel T (2011) EWSR1 gene rearrangement occurs in a subset of cutaneous myoepithelial tumors: a study of 18 cases. Mod Pathol 24(11):1444–1450
Kurzawa P, Fundowicz M, Dopierała M, Larque AB, Nielsen GP (2017) Primary extra-axial, para-articular chordoma of the knee. A case report and the review of literature. Histopathology Nov 20. doi: https://doi.org/10.1111/his.13440
Yamaguchi T, Imada H, Iida S, Szuhai K (2017) Notochordal tumors: an update on molecular pathology with therapeutic implications. Surg Pathol Clin 10(3):637–656
Miettinen M, Wang Z, Lasota J, Heery C, Schlom J, Palena C (2015) Nuclear brachyury expression is consistent in chordoma, common in germ cell tumors and small cell carcinomas, and rare in other carcinomas and sarcomas: an immunohistochemical study of 5229 cases. Am J Surg Pathol 39(10):1305–1312
Fletcher CDM, Antonescu CR, Heim S, Hornick JL (2013) Myoepithelioma/myoepithelial carcinoma/mixed tumour. In: Fletcher CD, Bridge JA, Hogendoorn PC, Mertens F (eds) In World Health Organization classification of tumours of soft tissue and Bone. IARC Press, Lyon, pp 208–209
Righi A, Gambarotti M, Longo S, Benini S, Gamberi G, Cocchi S, Vanel D, Picci P, Bertoni F, Simoni A, Franchi A, Dei Tos AP (2015) Small cell osteosarcoma: clinicopathologic, immunohistochemical, and molecular analysis of 36 cases. Am J Surg Pathol 39(5):691–699
Benini S, Cocchi S, Gamberi G, Magagnoli G, Vogel D, Ghinelli C, Righi A, Picci P, Alberghini M, Gambarotti M (2014) Diagnostic utility of molecular investigation in extraskeletal myxoid chondrosarcoma. J Mol Diagn 16(3):314–323
Dabska M (1977) Parachordoma. A new clinicopathologic entity. Cancer 490:1586–1592
Folpe AL, Agoff SN, Willis J, Weiss SW (1999) Parachordoma is immunohistochemically and cytogenetically distinct from axial chordoma and extraskeletal myxoid chondrosarcoma. Am J Surg Pathol 23:1059–1067
Fisher C (2000) Parachordoma exists—but what is it? Adv Anat Pathol 7:141–148
Mobley BC, McKenney JK, Bangs CD, Callahan K, Yeom KW, Schneppenheim R, Hayden MG, Cherry AM, Gokden M, Edwards MS, Fisher PG, Vogel H (2010) Loss of SMARCB1/INI1 expression in poorly differentiated chordomas. Acta Neuropathol 120:745–753
Tirabosco R, Jacques T, Berisha F, Flanagan AM (2012) Assessment of integrase interactor 1 (INI-1) expression in primary tumours of bone. Histopathology 61:1245–1247
Jo VY, Antonescu CR, Zhang L, Dal Cin P, Hornick JL, Fletcher CD (2013) Cutaneous syncytial myoepithelioma: clinicopathologic characterization in a series of 38 cases. Am J Surg Pathol 37(5):710–718
Finos L, Righi A, Frisoni T, Gambarotti M, Ghinelli C, Benini S, Vanel D, Picci P (2017) Primary extraskeletal myxoid chondrosarcoma of bone: report of three cases and review of the literature. Pathol Res Pract 213(5):461–466
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study was approved by local Ethics Committee (20th November 2017). All information regarding the human material used in this study was managed using anonymous numerical codes.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Righi, A., Sbaraglia, M., Gambarotti, M. et al. Extra-axial chordoma: a clinicopathologic analysis of six cases. Virchows Arch 472, 1015–1020 (2018). https://doi.org/10.1007/s00428-018-2334-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-018-2334-0