
Abstract
Chordomas are rare, slowly growing, locally aggressive bone neoplasms that arise from embryonic remnants of the notochord, showing dual epithelial-mesenchymal differentiation. The high plasticity probably is the main reason for the high variety in phenotypes of chordoma, from its high heterogeneity on a cellular level to its subtype variations depending on tissue location, with its potential to develop from an inactive quiescent form to an aggressive cancer with extreme adaptability and resistance to drugs and other treatments. Gene expression profiles of formalin-fixed, paraffin-embedded skull chordoma, spine chordoma, and normal tissue specimens were generated and compared. Using strict criteria, we identified 222 differentially expressed transcripts unique to skull base chordoma, 261 unique to spine chordoma, and 192 common to both chordoma subtypes. Further analysis of these three groups of transcripts allowed the selection of three subsets of highly differentially expressed genes as potential biomarkers, disease drivers, and therapeutic targets in both chordoma subtypes. Immunohistochemistry revealed LMX1A to be dominant in skull base chordoma, SALL3 to be unique to spine chordoma, and T to be common to both chordoma subtypes. In both chordoma subtypes, the genes with the highest expression were predominantly development-related genes, mostly transcription factors. Our findings indicate that these developmental genes play important oncogenic roles in chordoma, mainly causing high plasticity and resistance to therapy in both these cancer subtypes but also determining their differentiation status and proliferation activity, pointing to features expected of heterogeneous stem cell-like tissues with similarities to their notochord origins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chordomas are rare, slowly growing, locally aggressive bone neoplasms that arise from embryonic remnants of the notochord and show a dual epithelial-mesenchymal differentiation [1]. These tumors typically occur in the axial skeleton and have a proclivity for the spheno-occipital region of the skull base and sacral regions. They arise from the sacrum in approximately 50–60% of cases, from the skull base region (spheno-occipital/nasal) in approximately 25–35% of cases, from the cervical vertebrae in approximately 10% of cases, and from the thoracolumbar vertebrae in approximately 5% of cases. Chordoma has been considered of low metastatic potential; however, distant metastasis to lung, bone, soft tissue, lymph node, liver, and skin has been reported in up to 43% of patients [2, 3]. Typical of chordoma are physaliferous cells, which appear in clusters of large cells separated by fibrous septa into lobules and surrounded by basophilic extracellular matrix rich in mucin and glycogen. By morphology, chordomas are divided into classical (conventional), chondroid, and dedifferentiated [4]. Although chordomas have been studied histologically, very little is known about the molecular mechanisms that drive these tumors [4].
Next generation sequencing (NGS) and its integration with related technologies have enabled extensive genome analysis of groups of individuals, including the identification and quantification of their transcriptomes.
Compared with microarray technology, RNA-Seq is more accurate and provides much more information about the cancer transcriptome [5, 6].
The purpose of the present study was to identify candidate biomarkers specific and common to spine chordoma or skull base chordoma. For this, RNA-Seq of spine chordoma was performed and analyzed together with the RNA-Seq data generated in our previously published skull base chordoma. In-silico pathway analysis was run to determine such genes’ potential biological and disease functions, to help selecting potential biomarkers. Finally, to confirm our selected set of candidate biomarkers on protein level, and for validation of our RNA-Seq results, IHC analysis was carried out. Additionally, to identifying biomarkers that may also serve as therapeutic targets in spine and/or skull base chordoma, the present study helps to elucidate the phenotypic variations of these two chordoma subtypes.
Materials and methods
Tissue specimens
The study population consists of 37 patients with primary skull base chordoma and 32 patients with primary spine chordoma for whom clinical data and FFPE specimens were available. Six nonmatching FFPE specimens of normal nasal turbinate tissue were selected as a reference. Institutional review board approved this study. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Histological diagnosis was confirmed by dedicated head and neck pathologist (DB), soft tissue pathologist (VGP), and neuropathologist (GNF).
Sample preparation and RNA sequencing
Total RNA was extracted from 32 spine chordoma and enriched by ribosomal RNA depletion. A whole-transcriptome strand-specific cDNA library was successfully generated for 30 samples as described previously [4].
RNA sequencing (RNA-Seq) of library samples of 30 spine chordoma was performed on a HiSeq 2000 platform (Illumina) with 50-bp paired-end reads from each end of the RNA insert.
The resulting RNA-Seq data of the 30 spine chordoma samples were then combined with the previously generated RNA-Seq data of 21 skull base chordoma, and six normal tissue specimens [4] for RNA-Seq analysis. Across all 57 samples, the range of the adjusted read depths was 50–68 million; the median number of on-target reads per sample was 52 million.
All RNA sequencing data are available for download from the National Center for Biotechnology Information (NCBI) sequence reader archive (reference SRP 109781; NCBI, National Institutes of Health, Bethesda, MD).
RNA-Seq analysis of differentially expressed genes and isoforms
RNA-Seq analysis was performed with the RNAv9 pipeline (EA Quintiles) as described previously [4]. The analysis included gene and transcript identifier information from EA Quintiles’ database covering 34,495 gene and 88,933 isoform transcripts.
RNA-Seq data quality control analyses of the 57 tissue specimens, mainly principal component analyses of upper-quartile normalized and log-transformed gene counts for uncentered and gene-centered data, yielded statistically consistent and reliable data for a subtype comparison analysis of three normal tissue (N2, N3, N4), 14 skull base chordomas (T3, T8, T11, T17, T18, T19, T22, T26, T28, T30, T31, T32, T34, T36), and 12 spine chordoma (CT21, CT23, ST3, ST4, ST6, ST9, ST10, TM28, TT11, TT13, TT15, TT17) samples. For each chordoma subtype, this resulted 27,349 gene transcripts and 70,420 isoform transcripts, representing the whole human transcriptomes for skull base chordoma and for spine chordoma, including long noncoding RNA and excluding ribosomal, small noncoding RNA, and control transcripts (Supplemental Tables 1 and 2). Genes and transcripts were automatically identified and annotated during the alignment analysis.
These whole transcriptome tables were used to select the gene and isoform transcripts characteristic of either skull base chordoma or spine chordoma using the strict criteria of a log2 fold change > 1 plus, for genes, an FDR < 0.015 with an unadjusted p value < 5.1E-4 or, for transcript isoforms, an FDR < 0.022 with an unadjusted p value < 2.0E4 (Supplemental Table 3). Unknown annotations were manually updated using the NCBI Entrez Gene, ENSEMBL, and UCSC Genome Browser databases.
Next the tables of the gene and isoform transcripts characteristic of skull base chordoma or spine chordoma were compared to generate three groups of genes and transcripts: (1) those unique to skull base chordoma, (2) those unique to spine chordoma, and (3) those common to both chordoma subtypes (Supplemental Table 4).
These three gene groups of 222, 261, and 192 differentially expressed genes and transcripts of Supplemental Table 4 were then used to create panel lists of 30–41 genes and transcripts strongest differentially up- and downregulated, and categorized according to their biological functions and subcellular locations (Supplemental Table 5).
Heatmaps with cluster analyses
Heatmaps with cluster analyses for 14 skull base and 12 spine chordoma samples in comparison to three normal samples were generated, for the top 100 differentially expressed genes and isoforms, based on their log2 FC expression values. Similarity clusters were included, calculated as average linkage clustering based on the Euclidean distance of the samples, comparing their top 100 gene and top 100 isoform expressions. All cluster and heatmap analyses and the visualization of differentially expressed genes were conducted by EA Quintiles using the R2.15.1 software package (www.r-project.org) (Fig. 1a and b).

Heatmaps for a skull base chordomas (black to light red, increasing gene upregulation; dark green to light green, increasing gene downregulation) and b spine chordomas (dark green to black to light red, increasing gene upregulation; green to light green, increasing gene downregulation)
Sample cluster analysis and pathway network analyses
To show dissimilarity among all samples, we performed dendrogram cluster analysis as described previously [7] (Fig. 2a).

a Sample dissimilarity cluster analysis (black, normal tissues [N]; red, skull base chordomas; blue, spine chordomas). b Differentially expressed (DE) genes common to both skull base and spine chordomas were related to embryo, organism, and tissue development (red, upregulated; green, downregulated). c DE genes specific to skull base chordomas were related to embryo, organ, and nervous system development and function (red, upregulated; green, downregulated). d DE genes specific to spine chordomas were related to organ morphology and embryonic, skeletal, and muscular development (red, upregulated; green, downregulated)
Computational analysis of the gene data from Supplemental Table 4 was performed using Ingenuity Pathway Analysis (IPA; content version 33559992, release date 2017–03-28; QIAGEN Silicon Valley, Redwood, CA) to identify disease associations, pathway networks, and biomarkers among the genes unique to skull base chordoma, genes unique to spine chordoma, and genes common to both chordoma subtypes (Fig. 2b–d).
Immunohistochemistry
To validate the gene expression profiles identified by RNA-Seq analysis, we used immunohistochemistry (IHC) to assess the protein expression of certain candidate genes highly upregulated in and considered important for skull base or spine chordomas.
IHC was performed with antibodies against RAB3B (monoclonal; Lifespan Biosciences, Seattle, WA), which is expressed in normal tissue as well as skull base and spine chordomas; T (monoclonal; AbCam, Cambridge, MA) and HOXA3 (polyclonal; Lifespan Biosciences), expressed in both skull base and spine chordomas; LMX1A (polyclonal; Sigma-Aldrich, St. Louis, MO), expressed primarily in skull base chordomas; and SALL3 (polyclonal; Novus Biologicals, Littleton, CO), expressed in spine chordomas only.
All tissue specimens were assessed as described previously [4].
Results
Clinical and histologic data
The clinical characteristics of the 26 patients whose chordoma specimens were included in the subtype comparison analysis are given in Supplemental Table 6A. Ages at diagnosis ranged from 6 to 74 years for patients with skull base chordomas and from 33 to 80 years for spine chordomas. Most patients with either skull base or spine chordomas were men (n = 9 for both). Among patients with skull base chordomas, the most frequent primary tumor site was the clivus (n = 10); among those with spine chordomas, most primary tumors were equally distributed among the thoracic, lumbar, and sacral spine (n = 4 for each). The dominant morphologic subtype for both skull base chordoma patients and spine chordoma patients was conventional (n = 9 and 12, respectively). Figure 3 illustrates examples of the conventional and chondroid types of chordoma.

Morphologic chordoma subtypes. Conventional (a) and chondroid (b) chordoma specimens were stained with hematoxylin and eosin. The tumor is immunoreactive with anti-keratin cocktail (c) and with anti T, brachyury monoclonal antibodies (d)
Genome wide RNA-Seq analysis
Average linkage cluster analysis of the top 100 genes and transcripts revealed that HOXA1 and HOXA2 from the HOXA cluster, together with ZIC1, ZIC4, and FOXD1, were upregulated in skull base chordoma (Fig.1a), whereas HOXA2, HOXA3, HOXA4, HOXA5, and HOXC8 from the HOXA and HOXC clusters, together with T, were upregulated in spine chordoma (Fig.1b).
Hierarchical cluster analysis for dissimilarities among the 29 chordoma samples, based on the expression data for the top 5000 isoform transcripts, revealed no major group separations, rather a continuous change from the normal samples at the bottom to the top samples, mainly represented by skull base specimens (Fig. 2a). However, the analysis revealed two distinct cluster groups. The top cluster group was dominated by seven skull base chordoma specimens and four spine chordoma specimens, and the second cluster group was dominated by five specimens of skull base and spine chordoma each and even included one normal tissue specimen. This clustering was probably correlated with the severity and stage of each tumor tissue sample.
The panel lists the top 30–41 genes and transcripts strongest differentially up- and downregulated in the three groups: unique to skull base chordoma, unique to spine chordoma, and common to both chordoma subtypes, and categorized according to their biological functions and subcellular locations (Supplemental Table 5), present the main candidates assumed to play major roles in defining the disease subtypes of skull base and spine chordoma. Comparing these panels revealed the major differences and commonalities between the skull base and spine chordoma subtypes.
The most striking differences are easily seen at upregulated transcription-related genes. Both chordoma subtypes expressed the transcription factors T [8], LMX1A [9], FOXD1 [10], and the HOXA cluster genes HOXA1, HOXA2, and HOXA3 [11], all of which are involved in development and differentiation. Spine chordomas also expressed other transcription-related genes, including other HOXA cluster members (HOXA4, HOXA5, and HOXA10), a HOXB cluster member (HOXB6), and HOXC cluster members (HOXC6, HOXC8, and HOXC10) [11] as well as the development-related transcriptional regulators SALL3 [12], stem cell marker HOPX [13], and the cell cycle activator ZNF695 [14]. Spine chordomas also dominantly expressed the transcription factor ZNF385B, whose function is unclear; it might be involved in apoptosis [15]. In contrast, skull base chordomas expressed no other HOX cluster genes but strongly expressed the development-related transcription factors ZIC1 [16], ZIC4 [17], and LHX4 known to activate the oncogenic WNT/beta catenin/TCF4 pathway [18], and the posttranscriptional regulator NANOS, which promotes tumor cell invasion [19] and helps preserve stem cell status [20].
The second most notable differences between skull base and spine chordoma appear in the categories of upregulated signaling-related genes and proteins. In spine chordoma, the most prominent signaling gene was sonic hedgehog (SHH), known for its signaling pathway of the same name, which is involved in development and cell-to-cell differentiation [21]. Spine chordoma also had upregulation of the DDIT4L gene, indicating the inhibition of the mTOR signaling pathways [22], which are involved in stem cell differentiation, cell proliferation, and inhibition of apoptosis [23]. Other upregulated genes in spine chordoma included ARHGAP40, MRGPRX3, POPDC3, PKD1L2, and ADGRD1, which are involved in the signaling of G protein and its direct targets. In contrast, skull base chordoma had upregulation of PBK, which is involved in the oncogenic FOXM1/PBK/beta catenin pathway and in the inhibition of the p53/p21 signaling pathway, which accelerates cell cycle progression and proliferation [24]; as well as upregulation of DIO1, which is a part of the thyroid hormone T3 signaling pathways essential for growth, differentiation, and basal metabolism [25]. Skull base chordoma overexpressed only one component involved in G protein signaling, the orexin receptor HCRTR1. Other genes and transcripts differentially overexpressed between skull base and spine chordomas were mostly related to intracellular structures, remodeling, catabolism, vesicle transport, membrane components, extracellular matrix, and cell adhesion (Supplemental Tables 4 and 5). These genes and transcripts also make major contributions to the phenotype of each chordoma subtype.
The most highly downregulated genes and transcripts in each chordoma subtype mainly affect cellular functions related to development and differentiation, to membrane and cell structures, and to tumor suppression and immune defense responses (Supplemental Table 5). Interesting in this context is the high number of downregulated immune system-related genes and transcripts, noticeably the encoded follicular dendritic cell-secreted protein FDCSP, which is linked to dendritic cell immune response [26], and the innate immune response-related genes BPIFA1, BPIFB1, BPIFB2, and BPIFB3 [27], which were suppressed in both skull base and spine chordomas.
Finally, we found that 21, 19, and 8 unknown genes or transcripts were up- or downregulated in skull base chordoma only, spine chordoma only, and both chordoma subtypes, respectively. We assume that most of them belong to noncoding RNAs with unknown regulatory functions, but at least 12 of them also seem to be coded into proteins with unknown functions.
Pathway network analysis
IPA for disease- and biological function-related genes and transcripts identified 207 such genes and transcripts in skull base chordoma, of which 155 (75%) were cancer-related; 239 in spine chordoma, of which 178 (74%) were cancer-related; and 181 in both skull base and spine chordoma, of which 144 (80%) were cancer-related. The IPA results confirmed the strong contribution of development- and differentiation-related genes and, as the network analyses demonstrated, identified not only those which are common to both chordoma subtypes but also those which are specific to each disease subtype (Fig. 2b–d).
IHC for biomarker analysis
Detailed findings of the IHC analysis are given in Supplemental Table 6B.We performed IHC with antibodies against the proteins of five representative development-related genes (T, LMX1A, HOXA3, SALL3, and LHX4) and the membrane protein-encoding gene RAB3B, all of which the RNA-Seq analysis revealed to be highly upregulated in one of both chordoma subtypes. These genes’ corresponding differential gene expression values, calculated as log2 fold changes, were 14, 11, 9, 9, 7, and 8, respectively.
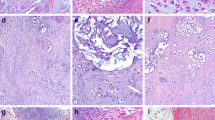
The IHC results confirmed our RNA-Seq gene results except for differences in the degree of subtype specificity. IHC indicated that LMX1A expression was higher in skull base chordoma than in spine chordoma (Fig. 4b), whereas the RNA-Seq analysis included LMX1A in the group of genes common to both chordoma subtypes. IHC showed high SALL3 expression in spine chordoma and low SALL3 expression in skull base chordoma (Fig. 4s), whereas the RNA-Seq analysis included SALL3 in the group of genes specific to spine chordoma. IHC showed equal expression of T (Fig. 3c), HOXA3 (Fig. 4a), LHX4 (Fig. 4b), and RAB3B (Fig. 4c) in skull base and spine chordomas. At the gene level, these IHC results matched those of the RNA-Seq analysis, except for LHX4, whose transcript was differentially overexpressed in the skull base chordoma-specific group, and RAB3B, whose transcript was differentially overexpressed in the spine chordoma-specific group.

IHC analyses of spine and skull base chordomas with a anti-HOXA3 polyclonal antibody, b anti-LMX1A polyclonal antibody, c anti-RAB3B monoclonal antibody, and d anti-SALL3 polyclonal antibody
Together, these results indicate that IHC with antibodies against LMX1A (to stain for skull base chordoma) and SALL3 (to stain for spine chordoma) enables us to differentiate between the chordoma subtypes. These results also clearly show that the differences between the chordoma subtypes, at least for the chosen target proteins, are not absolute, since all of the tested proteins were expressed in both chordoma subtypes but at greatly different levels.
Discussion
The findings of our previous skull base chordoma study [4] and those of the present study suggest that both spine and skull base chordomas arise from the notochord stem cells. This assertion is best supported by findings in the present study that 192 genes were equally differentially expressed in both skull base and spine chordomas (Supplemental Table 4 ) and that many genes and transcripts were dominantly, rather than exclusively, differentially expressed in spine or skull base chordomas (Supplemental Table 1). This is also supported at the protein level by the present study’s IHC results, which indicated that proteins were dominantly, rather than exclusively, differentially expressed between the subtypes. Therefore, the differences between these two chordoma subtypes are likely the result of signaling with local surrounding tissues and of certain genomic damages, which prevent the changed cells from fully differentiating and protect them against immune and damage responses (e.g., innate and dendritic cell immune responses, apoptosis, senescence). In this regard, the most obvious differences between these two chordoma subtypes are the differentially expressed upregulated developmental genes: skull base chordomas seem to have an aborted expression of HOX gene cluster pathways at the first HOX cluster (the HOXA cluster), whereas spine chordomas have differential expression of not only HOXA cluster members but also HOXB and HOXC cluster members.
Beside of their promoter regulation, gene clusters are mainly regulated by epigenetics and over enhancers [28,29,30], therefore defects affecting these regulatory mechanisms, rather than direct mutations of the developmental genes, may cause the changes in the differentiation program of skull base chordomas versus spine chordomas. Skull base chordomas seem to bypass these defects by changing the differentiation pathway to overexpressing the developmental transcription factors ZIC1 [16], ZIC4 [17], and LHX4 [18].
The main factors here responsible for these chordoma subtypes depend on supporting network pathways of transcripts, genes, and proteins. In spine chordoma, many of the components of these supporting network pathways, such as SALL3 [12], ZNF385B [15] transcription factors, and the SHH [31] signaling pathway, seem to remain active and help drive differentiation closer to a somatic state. In contrast, skull base chordoma does not seem to be able to differentiate out of stem cells states and thus can more quickly develop into more stable and increasingly aggressive cancer stem cells.
Most of the strongly downregulated, differentially expressed transcripts and genes in spine and skull base chordoma in the present study are mainly related to immune and damage response, and their roles in these chordoma subtypes seem fairly obvious.
The findings of the present study indicate that spine and skull base chordomas are caused and driven by genetic damage leading to the deregulation of development- and differentiation-related pathways, which results in notochordal stem cells differentiating into cancer stem cells rather than into surrounding somatic tissue. The high numbers of activated, upregulated development- and differentiation-related genes likely provide these cells with the high potential to adapt to very different surroundings and/or drugs and other treatments and may even enable them to hibernate in a quiescent state during unfavorable conditions.
On the basis of the results of the present study along with the results from our previous work on ACC of salivary gland [7] and results of our not yet published genomic studies of neoplastic tissues, we strongly assume that most, if not all, tumor tissues majorly depend on activated development- and differentiation-related genes and their proteins to allow them to develop into stable and aggressive cancer stem cells and to provide them with the adaptability against the patients’ own immune and defense responses and against medical drugs and other treatments. Because most developmental genes are deactivated in most human tissues (i.e., in somatic cells), future cancer research should focus on investigating the development- and differentiation-related pathways, genes, and proteins specific to each cancer type as potential therapeutic targets. Thus, simple inhibition of the highest differentially overexpressed development-related gene or protein (e.g., by stable synthetic microRNA) might lead to even more adaptable/aggressive cancer stem cells, and the strategy for intercepting a developmental pathway—for example, by inhibiting differentiation pathways at developmental checkpoints to drive cells to apoptosis or into senescence, or by influencing signaling to rescue their normal differentiation pathways—must be chosen carefully.
References
Williams BJ, Raper DM, Godbout E et al (2013) Diagnosis and treatment of chordoma. J Natl Compr Cancer Netw 11:726–731
Chambers PW, Schwinn CP (1979) Chordoma. A clinicopathologic study of metastasis. Am J Clin Pathol 72:765–776
Higinbotham NL, Phillips RF, Farr HW, Hustu HO (1967) Chordoma. Thirty-five-year study at memorial hospital. Cancer 20:1841–1850
Bell D, Raza SM, Bell AH, Fuller GN, DeMonte F (2016) Whole-transcriptome analysis of chordoma of the skull base. Virchows Arch 469:439–449
Mardis ER (2014) Sequencing the AML genome, transcriptome, and epigenome. Semin Hematol 51:250–258
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Bell D, Bell AH, Bondaruk J, Hanna EY, Weber RS (2016) In-depth characterization of the salivary adenoid cystic carcinoma transcriptome with emphasis on dominant cell type. Cancer
Faial T, Bernardo AS, Mendjan S et al (2015) Brachyury and SMAD signalling collaboratively orchestrate distinct mesoderm and endoderm gene regulatory networks in differentiating human embryonic stem cells. Development 142:2121–2135
Chao TK, Yo YT, Liao YP et al (2013) LIM-homeobox transcription factor 1, alpha (LMX1A) inhibits tumourigenesis, epithelial-mesenchymal transition and stem-like properties of epithelial ovarian cancer. Gynecol Oncol 128:475–482
Yagi K, Satou Y, Satoh N (2004) A zinc finger transcription factor, ZicL, is a direct activator of Brachyury in the notochord specification of Ciona intestinalis. Development 131:1279–1288
Seifert A, Werheid DF, Knapp SM, Tobiasch E (2015) Role of Hox genes in stem cell differentiation. World J Stem Cells 7:583–595
Heng X, Breer H, Zhang X et al (2012) Sall3 correlates with the expression of TH in mouse olfactory bulb. J Mol Neurosci 46:293–302
Lin CC, Hsu YC, Li YH et al (2017) Higher HOPX expression is associated with distinct clinical and biological features and predicts poor prognosis in de novo acute myeloid leukemia. Haematologica 102:1044–1053
Li R, Campos J, Iida J (2015) A gene regulatory program in human breast cancer. Genetics 201:1341–1348
Iijima K, Yamada H, Miharu M et al (2012) ZNF385B is characteristically expressed in germinal center B cells and involved in B-cell apoptosis. Eur J Immunol 42:3405–3415
Sankar S, Yellajoshyula D, Zhang B, Teets B, Rockweiler N, Kroll KL (2016) Gene regulatory networks in neural cell fate acquisition from genome-wide chromatin association of Geminin and Zic1. Sci Rep 6:37412
Aruga J, Nozaki Y, Hatayama M, Odaka YS, Yokota N (2010) Expression of ZIC family genes in meningiomas and other brain tumors. BMC Cancer 10:79
Cha N, Liu W, Yang N et al (2014) Oncogenicity of LHX4 in colorectal cancer through Wnt/beta-catenin/TCF4 cascade. Tumour Biol 35:10319–10324
Bonnomet A, Polette M, Strumane K et al (2008) The E-cadherin-repressed hNanos1 gene induces tumor cell invasion by upregulating MT1-MMP expression. Oncogene 27:3692–3699
Draper BW, McCallum CM, Moens CB (2007) nanos1 is required to maintain oocyte production in adult zebrafish. Dev Biol 305:589–598
Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW (2016) Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers (Basel) 8
Corradetti MN, Inoki K, Guan KL (2005) The stress-inducted proteins RTP801 and RTP801L are negative regulators of the mammalian target of rapamycin pathway. J Biol Chem 280:9769–9772
Morquette B, Morquette P, Agostinone J et al (2015) REDD2-mediated inhibition of mTOR promotes dendrite retraction induced by axonal injury. Cell Death Differ 22:612–625
Yang YF, Pan YH, Cao Y et al (2017) PDZ binding kinase, regulated by FoxM1, enhances malignant phenotype via activation of beta-catenin signaling in hepatocellular carcinoma. Oncotarget
Li Q, Qi X, Jia W (2016) 3,3′,5-triiodothyroxine inhibits apoptosis and oxidative stress by the PKM2/PKM1 ratio during oxygen-glucose deprivation/reperfusion AC16 and HCM-a cells: T3 inhibits apoptosis and oxidative stress by PKM2/PKM1 ratio. Biochem Biophys Res Commun 475:51–56
Lorenzi L, Doring C, Rausch T et al (2017) Identification of novel follicular dendritic cell sarcoma markers, FDCSP and SRGN, by whole transcriptome sequencing. Oncotarget 8:16463–16472
Liu H, Zhang X, Wu J, French SW, He Z (2016) New insights on the palate, lung, and nasal epithelium clone (PLUNC) proteins: based on molecular and functional analysis of its homolog of YH1/SPLUNC1. Exp Mol Pathol 100:363–369
Ma Z, Li M, Roy S et al (2016) Chromatin boundary elements organize genomic architecture and developmental gene regulation in Drosophila Hox clusters. World J Biol Chem 7:223–230
Rhie SK, Guo Y, Tak YG et al (2016) Identification of activated enhancers and linked transcription factors in breast, prostate, and kidney tumors by tracing enhancer networks using epigenetic traits. Epigenetics Chromatin 9:50
Moorthy SD, Davidson S, Shchuka VM et al (2017) Enhancers and super-enhancers have an equivalent regulatory role in embryonic stem cells through regulation of single or multiple genes. Genome Res 27:246–258
Mangum R, Varga E, Boue DR et al (2016) SHH desmoplastic/nodular medulloblastoma and Gorlin syndrome in the setting of Down syndrome: case report, molecular profiling, and review of the literature. Childs Nerv Syst 32:2439–2446
Funding source
This study was supported by start-up funds from MD Anderson (DB) and presented at AACR Advances in Sarcoma, Philadelphia May 2017.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Bell, A.H., DeMonte, F., Raza, S.M. et al. Transcriptome comparison identifies potential biomarkers of spine and skull base chordomas. Virchows Arch 472, 489–497 (2018). https://doi.org/10.1007/s00428-017-2224-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-017-2224-x