
Abstract
Molecular tumor profiling is now a routine part of patient care, revealing targetable genomic alterations and molecularly distinct tumor subtypes with therapeutic and prognostic implications. The widespread adoption of next-generation sequencing technologies has greatly facilitated clinical implementation of genomic data and opened the door for high-throughput multigene-targeted sequencing. Herein, we discuss the variability of cancer genetic profiling currently offered by clinical laboratories, the challenges of applying rapidly evolving medical knowledge to individual patients, and the need for more standardized population-based molecular profiling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Since the sequencing of the first reference human genome by the Human Genome Project [4, 5, 21], our ability to make connections between genomics and biology has grown exponentially. The field of medical oncology has seen a dramatic change in cancer therapy from the use of relatively nonselective cytotoxic agents to rationally designed therapies targeting specific molecular alterations (e.g., tyrosine kinase inhibitors in lung cancers with activating mutations in the epidermal growth factor receptor (EGFR) genes). Table 1 contains a partial list of current Food and Drug Administration (FDA) approved targeted cancer therapies for solid and hematologic malignancies with their molecular targets and corresponding companion diagnostics, if applicable. Some of these agents are classified as new molecular entities, which contain active moieties that have not been previously approved by the FDA, while the majority fall under new indications for existing drugs.
Notably, agents that target highly specific molecular abnormalities (e.g., the breakpoint cluster region (BCR)-Abelson murine leukemia viral oncogene homolog (ABL) gene fusion) by definition restrict the patient population that can benefit from a specific approach. In contrast, successfully modulating less specific targets (either surface receptors or targets common to many biologic processes) will likely affect a large number of patients suffering from cancer and other disorders. Checkpoint inhibitors, for example, target the programmed cell death pathway (i.e., PD-1/PD-L1). By blocking this pathway, these drugs help reactivate the body’s own immune system to fight cancer cells. In a landmark decision, the FDA recently accelerated the approval of pembrolizumab for the treatment of adult and pediatric patients with microsatellite instability-high or mismatch repair deficient solid tumors that have progressed after prior treatment and who have no satisfactory alternative treatment options [10]. As the first tissue/site-agnostic genetically matched indication of its kind, pembrolizumab’s approval by the FDA signifies the remarkable progress that has been made with the use of immunotherapy and the specific events that enable malignant disease.
Although biomarker discovery is thriving, incorporation of multi-analyte molecular assays into clinical practice continues to lag behind. Some of the challenges to clinical adoption include assay variability, inadequate reporting, and practical obstacles such as the following: lack of resources, personnel, or expertise in smaller clinical laboratories [11]. In addition, knowledge of the specific tumor markers that have the greatest value in care evolve over time, leading to inconsistencies in analysis and interpretation.
Significant efforts have been made to standardize the development and reporting of diagnostic, prognostic, and predictive tumor markers [23] since the FDA started requiring co-development of targeted therapies with companion diagnostic tests. Regulatory oversight of assay development, however, is relatively limited, especially regarding the steps required to incorporate tumor markers into clinical care and to expand testing for therapeutic drug classes rather than a single agent. An important exception to the requirement for co-development of companion diagnostics is for promising therapeutics intended to treat tumors for which no satisfactory alternative treatment exists and the benefits outweigh the risks of using a non-FDA approved diagnostic test. Most companion diagnostics are relatively simple, analyzing 1 to 2 genetic biomarkers or are based on multivariate index assays that derive a score, probability, or classification (e.g., Oncotype DX). As a result, most medium to large clinical labs design their own laboratory developed tests (LDTs) to more rapidly translate new scientific knowledge into medical practice. These tests detect genetic variations relevant to approved targeted therapies, as well as emerging biomarkers actively under investigation in clinical trials or other research studies.
Not surprisingly, there is considerable variation between laboratories in the type of LDTs that they offer. Both academic institutions and commercial reference laboratories have developed customized gene panels for targeted sequencing. These panels, however, are not static and are continually updated as new knowledge become available (Tables 2 and 3). Some panels are organ or system-specific, whereas others are pan-cancer tests. The largest pan-cancer panels typically involve 200 to 400 genes. Many academic institutions validate ready-made vendor kit solutions, such as the Ion Torrent AmpliSeq™ Cancer Hotspot Panel (Thermo Fisher Scientific, Waltham, MA), Oncomine™ Comprehensive Assay (Thermo Fisher Scientific), or Illumina TruSeq™ and TruSight™ sequencing panels (Illumina, San Diego, CA), while others use custom-designed panels with genes of their choosing. The overlap between genes covered by each panel is variable, which reflects the general lack of standardization of molecular testing and difficulty of defining a clinically relevant cancer gene set.
Although some tumors depend almost exclusively on the oncogenic activity of a protein mutation in its early stages of development, most malignant conditions accumulate multiple genetic alterations within subclonal populations that ultimately dictate tumor growth. This genetic heterogeneity of tumors along with minimal tissue requirements for testing and financial and time constraints makes it challenging to perform accurate and comprehensive genomic testing on tumor samples. In addition, rapid advances in testing technology and understanding of tumor biology can quickly render a testing method or target obsolete.
Unfortunately, there is little consensus on how physicians should use multiplex gene testing for personalized cancer care beyond the genes in the National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology (NCCN Guidelines) [7, 11, 27]. For example, testing for EGFR, KRAS, ALK, and ROS1 are the only genes clearly described in the Principles of Pathologic Review in the latest version (6.2017) of the NCCN Guidelines for non-small cell lung cancer [9]. Although targeted agents for emerging biomarkers, including HER2 mutations, BRAF V600E mutation, RET rearrangements, and MET amplification or exon 14 skipping mutation, are acknowledged, they have not been incorporated into the decision trees. Instead, the NCCN “strongly advises broader molecular profiling” and encourages enrollment of any patient with cancer in a clinical trial. The scope and type of molecular profiling, however, is not explained in detail [8, 9].
Nevertheless, great interest remains from all stakeholders in cancer care, including patients, to translate the latest research findings into clinical care. The desire for early adoption before assessment of clinical utility is performed, has led many to question the true value of molecular testing beyond what is deemed actionable at the time testing is performed, especially if multiplex testing includes both clinically useful markers and markers with undetermined clinical utility or significance. However, variants identified through multiplex testing are being used to direct patients to appropriate trials and researchers of molecular events that may influence a particular patient’s response to treatment.
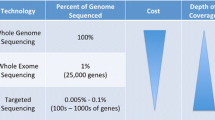
Although multiplex testing is frequently used in practice, as the number of genes implicated in cancer genetics, prognosis, and treatment increase, some are considering shifting to whole exome sequencing (WES). WES has the obvious advantage of probing a larger portion of the protein-coding genome, including all cancer-related genes present in the smaller targeted gene panels. This technique is becoming increasingly facile and cost-effective, and some groups have even demonstrated its feasibility in the clinical environment. What does this mean for the next generation of clinical oncology molecular tests? Is bigger always better? Instead of continually expanding targeted gene panels, should labs switch to whole exome (22,000 protein-coding genes) or whole genome (including non-coding regions) sequencing?
In the research setting, WES has led to the identification of novel gene fusions, cryptic splice sites, and genes not previously implicated in the tumor being tested. The large volume of data generated and the computational and analytical challenges associated with the interpretation of results have limited its widespread clinical implementation. Whole genome sequencing (WGS), which could theoretically allow better detection of non-coding and structural changes, is even further away from clinical application. WES/WGS also generates an excess of information of questionable clinical utility, highlighting our inability to interpret the functional and clinical impact of any variant that has not been well studied in the literature. Demonstrating the significance of each and every variant, however, may not be practical or even feasible. Many variants are exceedingly rare, so to find enough patients to run a clinical trial to determine whether they are significant is next to impossible given the inconsistent application of tumor profiling by clinicians, variability of assays performed, and barriers to consenting patients for public data sharing.
Although the clinical utility of large scale sequencing in patients may be low today, such information could be extremely important in the future for researchers who want to interrogate these real-world data repositories to get prognostic information. Real-world data can not only provide information on how a drug compares to the standard of care actually used in clinical practice versus the comparator testing in a clinical trial, but provide a clearer picture of realistic endpoints that clinicians can readily understand. For example, time to next treatment or change in therapy and the reason for that change, such as progression or drug toxicity, is likely to be noted in medical records and a more relevant endpoint than progression-free survival outcomes. Standardizing existing clinical datasets and integrating them with real-world data streams can speed the development of new drugs or expand labeled indications while also reducing costs of development. If leveraged correctly, information gathered from electronic health records and patient registries can allow for data to be collected on more patients and in unselected patient populations. Although randomized controlled trials are still necessary to discern small treatment effects from other factors, studies using real-world evidence can be used for therapies that demonstrate large treatment effects in relatively heterogeneous patient populations.
Efforts to standardize variant interpretation and unify independently curated (and sometimes conflicting) variant databases with health and disease-related outcomes data is ongoing [6, 14, 16, 22, 23, 29, 33]. Databases of genetic variants have the potential to speed evidence development for multiplex testing, since they are typically generated by multiple sources. Additionally, aggregated data can also provide a stronger evidence base for multiplex testing in the real-world than any single clinical trial can produce. As understanding of cancer pathogenesis progresses, the classification of each variant relative to the reference genome and population-based databases will hopefully become more refined.
Cancer, however, is multifactorial and multigenic, involving not only random genomic alterations and environmental modifiers, but epigenetic and pleiotropic phenomena as well. Intra- and intertumoral genetic heterogeneity complicates the task of identifying alterations that drive tumor progression from those that do not, and regional differences within a tumor that are further exaggerated by different selective pressures (e.g., variable access to oxygen, differences in tumor microenvironment, inconsistent exposure to targeted and non-targeted cancer therapies). These factors also need to be interpreted within the context of normal human genomic variation [13, 19, 20, 24, 30], which is an inherent part of somatic interpretation.
Since there are still many “dark” regions of the genome yet to be characterized or understood [2, 25], a reasonable approach for clinical laboratories looking to improve precision medicine efforts may be the adoption of larger pan-cancer tests with whole-gene sequencing. Memorial Sloan Kettering Cancer Center (MSKCC) and the Dana-Farber Institute/Brigham and Women’s Hospital (DFI/BWH) have created robust research infrastructures around initiatives that prioritize sequencing of all cancer patients with large custom-designed panels (approximately 300 to 500 genes) at their respective institutions [18, 34]. These initiatives have allowed every clinical patient, regardless of age, stage, or tumor type, to contribute to our understanding of cancer, particularly in genes not commonly sequenced as part of current standard of care guidelines. The size of these panels has not only facilitated novel discovery, but more importantly, provided a broad enough landscape to simultaneously assess tumor mutational burden, copy number variation, and tumor subclones. Along with public data sharing of outcomes data, more efforts like these would greatly accelerate cancer research and therapeutic development. Independence Blue Cross of Philadelphia, a private insurance provider, has also embraced the unique paradigm of comprehensive tumor profiling in the clinical setting for the purposes of discovery by offering coverage of NantHealth’s GPC Cancer™ test (Table 4) [3]. This test includes not only whole exome and transcriptome sequencing, but also comparative germline testing and quantitative proteomics by mass spectrometry. Complex multiomic data generation not only contributes to our collective understanding of tumor biology, but also provides rationale for combination therapies that target complex biologic processes.
Recent clinical sequencing reports from various groups [1, 26] point to the value of incorporating RNA sequencing (RNA-seq) with DNA sequencing to evaluate the expression of mutant alleles, to detect both known and novel gene fusions, and to confirm splice variants. RNA-seq may be especially valuable in pediatric tumors, which tend to have fewer recurrent point mutations compared with adult tumors. One case series of young patients showed that RNA-seq alone accounted for approximately 20% of actionable findings which would have been missed with WES [26]. Integrated RNA and DNA sequencing strategies may also aid in the identification of patient-specific immunogenic neoantigens expressed in the tumors which are becoming increasingly relevant for personalized cancer vaccine development [15].
Newer testing methods, such as liquid biopsies for solid tumors, have the advantage of being non-invasive and do not suffer from the same type of sample bias associated with tissue biopsies. This technology is still in its infancy and is limited to the detection of metastatic disease burden based on known molecular status of the primary tumor [17]. Nevertheless, liquid biopsy testing is slowly becoming available for patients who are not candidates for tissue biopsy and are at a high risk for resistance mutations to targeted therapy (Table 5). It is likely that this technology will not replace tissue-based testing, but 1 day may be used for minimal residual disease monitoring in a subset of patients with canonical tumor alterations.
The future of precision oncology lies in the detailed molecular characterization of a large number of patients, across ethnic, socioeconomic, and geographic subgroups, and prospective linking of multiplex testing results with individual clinical data [31]. Such concerted efforts, supported by government and private payers, will revolutionize genetic discovery, furthering our knowledge for all cancers and improving the care of patients with molecularly defined cancer in the generations to come.
Although it remains to be seen how clinical interpretation, annotation, and integration of such complex molecular data will evolve [28], broad application of standardized tumor profiling, irrespective of whether molecular testing is required to guide standard-of-care therapy, is needed to lay the groundwork for more systematic real-world studies.
Change history
01 August 2017
An erratum to this article has been published.
References
Byron SA, Van Keuren-Jensen KR, Engelthaler DM, Carpten JD, Craig DW (2016) Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat Rev Genet 17:257–271. doi:10.1038/nrg.2016.10
Chi KR (2016) The dark side of the human genome. Nature 538:275–277. doi:10.1038/538275a
CMTP (2017) New genomic technologies in clinical medicine: current evidence and decision-making http://www.cmtpnet.org/docs/resources/Genomic_Forum_Meeting_Briefing.pdf. Accessed May 9, 2017
Collins FS, Green ED, Guttmacher AE, Guyer MS, Institute UNHGR (2003) A vision for the future of genomics research. Nature 422:835–847. doi:10.1038/nature01626
Consortium IHGS (2004) Finishing the euchromatic sequence of the human genome. Nature 431:931–945. doi:10.1038/nature03001
Doig KD, Fellowes A, Bell AH, Seleznev A, Ma D, Ellul J, Li J, Doyle MA, Thompson ER, Kumar A, Lara L, Vedururu R, Reid G, Conway T, Papenfuss AT, Fox SB (2017) PathOS: a decision support system for reporting high throughput sequencing of cancers in clinical diagnostic laboratories. Genome Med 9:38. doi:10.1186/s13073-017-0427-z
Engstrom PF, Bloom MG, Demetri GD, Febbo PG, Goeckeler W, Ladanyi M, Loy B, Murphy K, Nerenberg M, Papagni P, Robson M, Sweetman RW, Tunis S, Demartino J, Larsen JK, Network NCC (2011) NCCN molecular testing white paper: effectiveness, efficiency, and reimbursement. J Natl Compr Cancer Netw 9(Suppl 6):S1–16
Ettinger DS, Wood DE, Aisner DL, Akerley W, Bauman J, Chirieac LR, D'Amico TA, DeCamp MM, Dilling TJ, Dobelbower M, Doebele RC, Govindan R, Gubens MA, Hennon M, Horn L, Komaki R, Lackner RP, Lanuti M, Leal TA, Leisch LJ, Lilenbaum R, Lin J, Loo BW, Martins R, Otterson GA, Reckamp K, Riely GJ, Schild SE, Shapiro TA, Stevenson J, Swanson SJ, Tauer K, Yang SC, Gregory K, Hughes M (2017) Non-small cell lung cancer, version 5.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 15:504–535
Ettinger DS, Wood DE, Aisner DL, Akerley W, Bauman J, Chirieac LR, D'Amico TA, DeCamp MM, Dilling TJ, Dobelbower M, Doebele RC, Govindan R, Gubens MA, Hennon M, Horn L, Komaki R, Lackner RP, Lanuti M, Leal TA, Leisch LJ, Lilenbaum R, Lin J, Loo BW, Martins R, Otterson GA, Reckamp K, Riely GJ, Schild SE, Shapiro TA, Stevenson J, Swanson SJ, Tauer K, Yang SC, Gregory K, Hughes M (2017) Non-small cell lung cancer, version 6.2017, NCCN Clinical Practice Guidelines in Oncology. https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf. Accessed May 21, 2017
FDA Hematology/Oncology (Cancer) Approvals & Safety Notifications. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm279174.htm. Accessed May 25, 2017
Febbo PG, Ladanyi M, Aldape KD, De Marzo AM, Hammond ME, Hayes DF, Iafrate AJ, Kelley RK, Marcucci G, Ogino S, Pao W, Sgroi DC, Birkeland ML (2011) NCCN Task Force report: evaluating the clinical utility of tumor markers in oncology. J Natl Compr Canc Netw 9 Suppl 5:S1–32 quiz S33
Garcia EP, Minkovsky A, Jia Y, Ducar MD, Shivdasani P, Gong X, Ligon AH, Sholl LM, Kuo FC, MacConaill LE, Lindeman NI, Dong F (2017) Validation of OncoPanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer doi:10.5858/arpa.2016-0527-OA.
Garofalo A, Sholl L, Reardon B, Taylor-Weiner A, Amin-Mansour A, Miao D, Liu D, Oliver N, MacConaill L, Ducar M, Rojas-Rudilla V, Giannakis M, Ghazani A, Gray S, Janne P, Garber J, Joffe S, Lindeman N, Wagle N, Garraway LA, Van Allen EM (2016) The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med 8:79. doi:10.1186/s13073-016-0333-9
Griffith M, Spies NC, Krysiak K, McMichael JF, Coffman AC, Danos AM, Ainscough BJ, Ramirez CA, Rieke DT, Kujan L, Barnell EK, Wagner AH, Skidmore ZL, Wollam A, Liu CJ, Jones MR, Bilski RL, Lesurf R, Feng YY, Shah NM, Bonakdar M, Trani L, Matlock M, Ramu A, Campbell KM, Spies GC, Graubert AP, Gangavarapu K, Eldred JM, Larson DE, Walker JR, Good BM, Wu C, Su AI, Dienstmann R, Margolin AA, Tamborero D, Lopez-Bigas N, Jones SJ, Bose R, Spencer DH, Wartman LD, Wilson RK, Mardis ER, Griffith OL (2017) CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet 49:170–174. doi:10.1038/ng.3774
Gupta S, Chaudhary K, Dhanda SK, Kumar R, Kumar S, Sehgal M, Nagpal G, Raghava GP (2016) A platform for designing genome-based personalized immunotherapy or vaccine against cancer. PLoS One 11:e0166372. doi:10.1371/journal.pone.0166372
Harrison SM, Dolinsky JS, Knight Johnson AE, Pesaran T, Azzariti DR, Bale S, Chao EC, Das S, Vincent L, Rehm HL (2017) Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet Med. doi:10.1038/gim.2017.14
Hofman P, Popper HH (2016) Pathologists and liquid biopsies: to be or not to be? Virchows Arch 469:601–609. doi:10.1007/s00428-016-2004-z
Hyman DM, Solit DB, Arcila ME, Cheng DT, Sabbatini P, Baselga J, Berger MF, Ladanyi M (2015) Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discov Today 20:1422–1428. doi:10.1016/j.drudis.2015.08.005
Jones S, Anagnostou V, Lytle K, Parpart-Li S, Nesselbush M, Riley DR, Shukla M, Chesnick B, Kadan M, Papp E, Galens KG, Murphy D, Zhang T, Kann L, Sausen M, Angiuoli SV, Diaz LA, Velculescu VE (2015) Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med 7:283ra253. doi:10.1126/scitranslmed.aaa7161
Kline CN, Joseph NM, Grenert JP, van Ziffle J, Talevich E, Onodera C, Aboian M, Cha S, Raleigh DR, Braunstein S, Torkildson J, Samuel D, Bloomer M, Campomanes AG, Banerjee A, Butowski N, Raffel C, Tihan T, Bollen AW, Phillips JJ, Korn WM, Yeh I, Bastian BC, Gupta N, Mueller S, Perry A, Nicolaides T, Solomon DA (2016) Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro-Oncology. doi:10.1093/neuonc/now254
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann Y, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blöcker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowki J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ, Consortium IHGS (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921. doi:10.1038/35057062
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, Maglott DR (2017) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44:D862–D868. doi:10.1093/nar/gkv1222
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, Tsimberidou AM, Vnencak-Jones CL, Wolff DJ, Younes A, Nikiforova MN (2017) Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 19:4–23. doi:10.1016/j.jmoldx.2016.10.002
Luthra R, Patel KP, Routbort MJ, Broaddus RR, Yau J, Simien C, Chen W, Hatfield DZ, Medeiros LJ, Singh RR (2016) A targeted high-throughput next-generation sequencing panel for clinical screening of mutations, gene amplifications, and fusions in solid tumors. J Mol Diagn. doi:10.1016/j.jmoldx.2016.09.011
Martin L, Chang HY (2012) Uncovering the role of genomic “dark matter” in human disease. J Clin Invest 122:1589–1595. doi:10.1172/JCI60020
Mody RJ, Wu YM, Lonigro RJ, Cao X, Roychowdhury S, Vats P, Frank KM, Prensner JR, Asangani I, Palanisamy N, Dillman JR, Rabah RM, Kunju LP, Everett J, Raymond VM, Ning Y, Su F, Wang R, Stoffel EM, Innis JW, Roberts JS, Robertson PL, Yanik G, Chamdin A, Connelly JA, Choi S, Harris AC, Kitko C, Rao RJ, Levine JE, Castle VP, Hutchinson RJ, Talpaz M, Robinson DR, Chinnaiyan AM (2015) Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA 314:913–925. doi:10.1001/jama.2015.10080
Myers RE, Wolf T, Shwae P, Hegarty S, Peiper SC, Waldman SA (2016) A survey of physician receptivity to molecular diagnostic testing and readiness to act on results for early-stage colon cancer patients. BMC Cancer 16:766. doi:10.1186/s12885-016-2812-1
Nakagawa H, Wardell CP, Furuta M, Taniguchi H, Fujimoto A (2015) Cancer whole-genome sequencing: present and future. Oncogene 34:5943–5950. doi:10.1038/onc.2015.90
Ritter DI, Roychowdhury S, Roy A, Rao S, Landrum MJ, Sonkin D, Shekar M, Davis CF, Hart RK, Micheel C, Weaver M, Van Allen EM, Parsons DW, McLeod HL, Watson MS, Plon SE, Kulkarni S, Madhavan S, Group CSCW (2016) Somatic cancer variant curation and harmonization through consensus minimum variant level data. Genome Med 8:117. doi:10.1186/s13073-016-0367-z
Schrader KA, Cheng DT, Joseph V, Prasad M, Walsh M, Zehir A, Ni A, Thomas T, Benayed R, Ashraf A, Lincoln A, Arcila M, Stadler Z, Solit D, Hyman DM, Hyman D, Zhang L, Klimstra D, Ladanyi M, Offit K, Berger M, Robson M (2016) Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol 2:104–111. doi:10.1001/jamaoncol.2015.5208
Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, Garcia EP, Jia Y, Davineni P, Abo RP, Pugh TJ, van Hummelen P, Thorner AR, Ducar M, Berger AH, Nishino M, Janeway KA, Church A, Harris M, Ritterhouse LL, Campbell JD, Rojas-Rudilla V, Ligon AH, Ramkissoon S, Cleary JM, Matulonis U, Oxnard GR, Chao R, Tassell V, Christensen J, Hahn WC, Kantoff PW, Kwiatkowski DJ, Johnson BE, Meyerson M, Garraway LA, Shapiro GI, Rollins BJ, Lindeman NI, MacConaill LE (2016) Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 1:e87062. doi:10.1172/jci.insight.87062
Singh RR, Patel KP, Routbort MJ, Reddy NG, Barkoh BA, Handal B, Kanagal-Shamanna R, Greaves WO, Medeiros LJ, Aldape KD, Luthra R (2013) Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J Mol Diagn 15:607–622. doi:10.1016/j.jmoldx.2013.05.003
Yen JL, Garcia S, Montana A, Harris J, Chervitz S, Morra M, West J, Chen R, Church DM (2017) A variant by any name: quantifying annotation discordance across tools and clinical databases. Genome Med 9:7. doi:10.1186/s13073-016-0396-7
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, Hellmann MD, Barron DA, Schram AM, Hameed M, Dogan S, Ross DS, Hechtman JF, DeLair DF, Yao J, Mandelker DL, Cheng DT, Chandramohan R, Mohanty AS, Ptashkin RN, Jayakumaran G, Prasad M, Syed MH, Rema AB, Liu ZY, Nafa K, Borsu L, Sadowska J, Casanova J, Bacares R, Kiecka IJ, Razumova A, Son JB, Stewart L, Baldi T, Mullaney KA, Al-Ahmadie H, Vakiani E, Abeshouse AA, Penson AV, Jonsson P, Camacho N, Chang MT, Won HH, Gross BE, Kundra R, Heins ZJ, Chen H-W, Phillips S, Zhang H, Wang J, Ochoa A, Wills J, Eubank M, Thomas SB, Gardos SM, Reales DN, Galle J, Durany R, Cambria R, Abida W, Cercek A, Feldman DR, Gounder MM, Hakimi AA, Harding JJ, Iyer G, Janjigian YY, Jordan EJ, Kelly CM, Lowery MA, Morris LGT, Omuro AM, Raj N, Razavi P, Shoushtari AN, Shukla N, Soumerai TE, Varghese AM, Yaeger R, Coleman J, Bochner B, Riely GJ, Saltz LB, Scher HI, Sabbatini PJ, Robson ME, Klimstra DS, Taylor BS, Baselga J, Schultz N, Hyman DM, Arcila ME, Solit DB, Ladanyi M, Berger MF (2017) Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. doi:10.1038/nm.4333 http://www.nature.com/nm/journal/vaop/ncurrent/abs/nm.4333.html#supplementary-information
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
The original version of this article was revised: Entries were incorrectly aligned in Table 2.
An erratum to this article is available at https://doi.org/10.1007/s00428-017-2193-0.
Rights and permissions
About this article

Cite this article
Nguyen, D., Gocke, C.D. Managing the genomic revolution in cancer diagnostics. Virchows Arch 471, 175–194 (2017). https://doi.org/10.1007/s00428-017-2175-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-017-2175-2