
Abstract
Main conclusion
Our transcriptomic analysis expanded the repertoire of nitrate-responsive genes/processes in rice and revealed their phenotypic association with root/shoot, stomata, tiller, panicle/flowering and yield, with agronomic implications for nitrogen use efficiency.
Abstract
Nitrogen use efficiency (NUE) is a multigenic quantitative trait, involving many N-responsive genes/processes that are yet to be fully characterized. Microarray analysis of early nitrate response in excised leaves of japonica rice revealed 6688 differentially expressed genes (DEGs), including 2640 hitherto unreported across multiple functional categories. They include transporters, enzymes involved in primary/secondary metabolism, transcription factors (TFs), EF-hand containing calcium binding proteins, hormone metabolism/signaling and methytransferases. Some DEGs belonged to hitherto unreported processes viz. alcohol, lipid and trehalose metabolism, mitochondrial membrane organization, protein targeting and stomatal opening. 1158 DEGs were associated with growth physiology and grain yield or phenotypic traits for NUE. We identified seven DEGs for shoot apical meristem, 66 for leaf/culm/root, 31 for tiller, 70 for heading date/inflorescence/spikelet/panicle, 144 for seed and 78 for yield. RT-qPCR validated nitrate regulation of 31 DEGs belonging to various important functional categories/traits. Physiological validation of N-dose responsive changes in plant development revealed that relative to 1.5 mM, 15 mM nitrate significantly increased stomatal density, stomatal conductance and transpiration rate. Further, root/shoot growth, number of tillers and grain yield declined and panicle emergence/heading date delayed, despite increased photosynthetic rate. We report the binding sites of diverse classes of TFs such as WRKY, MYB, HMG etc., in the 1 kb up-stream regions of 6676 nitrate-responsive DEGs indicating their role in regulating nitrate response/NUE. Together, these findings expand the repertoire of genes and processes involved in genomewide nitrate response in rice and reveal their physiological, phenotypic and agronomic implications for NUE.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fertilizers used in agriculture are the largest source of environmental losses of reactive nitrogen (N) compounds, contributing to N-pollution globally (Sutton et al. 2013) as well as in South Asia (Raghuram et al. 2021) and India (Abrol et al. 2017). The UN Environment Program recently identified reactive N as one of the emerging threats to planet Earth (Sutton et al. 2019). Further, a UN resolution on ‘sustainable nitrogen management’ (UNEP/EA.4/L.16) underscored the urgency to improve nitrogen use efficiency (NUE). There is a limit to its improvement through agronomic practices, beyond which only biological crop improvement can break the barrier for further enhancement (Pathak et al. 2008; Chakraborty and Raghuram 2011). This understanding has fuelled research on the biological basis of N-response and NUE in various crops and model plants (Li et al. 2017; Wang et al. 2018; Raghuram and Sharma 2019; Vidal et al. 2020; Souza and Tavares 2021; Móring et al. 2021; Udvardi et al. 2021). Rice is an ideal target crop for improving NUE because of its lowest NUE among cereals, its excellent germplasm diversity and the opportunities it offers as a post-genomic model crop (Li et al. 2017). Moreover, the NUE phenotype in rice was delineated recently (Sharma et al. 2018, 2021).
Transcriptomic studies have identified thousands of N-responsive differentially expressed genes (DEGs) in rice under different conditions, including different subspecies, stages/tissues, treatment methods, N-doses and N-forms such as nitrate or ammonium or both, and urea. However, there is considerable variation in the genes/processes involved, even with a single N-source like nitrate (Sawaki et al. 2013; Coneva et al. 2014; Misyura et al. 2014; Pathak et al. 2020). Similarly, there are significant differences in the genes reported in response to ammonium (Yang et al. 2015a; Chandran et al. 2016; Sun et al. 2017, 2020), or ammonium nitrate (Yang et al. 2017; Wang et al. 2020) or urea (Reddy and Ulaganathan 2015).
Many more important nitrate-responsive genes/processes are expected in rice than reported so far, based on the lessons learnt from Arabidopsis (Li et al. 2017; Meyer et al. 2019). Their further shortlisting for NUE also remains a challenge due to divergent transcriptomic conditions, despite a recent attempt at their prediction (Kumari et al. 2021). Leaves are the primary sites of nitrate assimilation and subsequent N-remobilization for yield and NUE, whereas root transcriptomes are predominant in literature. Similarly, nitrate is the preferred form of N for all plants including rice, even though it may tolerate ammonium accumulation in flooded paddy fields (Kronzucker et al. 2000). Thus, there is ample scope for further studies to obtain a clearer genomewide understanding of N-responsive gene expression and NUE in rice. Therefore, we explored nitrate-responsive leaf transcriptome in japonica rice (cv. Nipponbare), using our previously standardized experimental conditions in indica rice (Pathak et al. 2020). This is a part of the broader aim to identify the genes/processes that contribute to the phenotypic traits of N-response and N use efficiency, so as to find suitable targets for genetic improvement of crop NUE. We hypothesized that while all the genes that respond to N may not contribute to NUE, the genes that drive NUE should be a subset of N-responsive transcriptome associated with the NUE phenotype.
Accordingly, we report many hitherto unreported nitrate-responsive genes/processes associated with important phenotypic/agronomic roles. We also explore their reported expression patterns in relation to stages, tissues and traits to find novel components that may regulate N-response/NUE.
Materials and methods
Plant material, growth conditions and nitrate treatments for microarray
Seeds of rice (O. sativa japonica Nipponbare) were procured from the Faculty of Agriculture, Kyushu University, Japan. Their surface sterilization was performed using 70% ethanol and 0.01% Triton-X 100 and germinated hydroponically in ultrapure water in a plant growth chamber at 25 °C, with 75% relative humidity and light intensity of 2800 ± 100 lx with 12/12 h photoperiod (warm white LED tubes). Germinated seeds were transferred to 6-inch pots containing 1:1 mixture of soilrite and vermiculite. They were grown with ultrapure water for the first 10 days, followed by fertilization every alternate day with 30 ml of Arnon-Hoagland (AH) nutrient solution (pH 5.6–5.8) (Hoagland and Arnon 1950) per seedling for the next 20 days. It was composed of 5 mM each of KNO3 and Ca(NO3)2, 1 mM KH2PO4, 2 mM MgSO4, 46 µM H3BO3, 9 µM MnCl2·4H2O, 0.765 µM ZnSO4·7H2O, 0.32 µM CuSO4·5H2O, 0.111 µM H2MoO4·H2O, 0.1 mM FeSo4·7H2O and 0.1 mM Na2EDTA. After 30 days, the pots were flushed with ultrapure water and fertilized with N-free 1X Arnon-Hoagland solution for the next two days to create N-depleted condition. On the 32nd day, leaf tissues were harvested and treated with 120 mM KNO3 and 120 mM KCl (control) in Petri plates for 90 min, as standardized earlier (Pathak et al. 2020) and reconfirmed here by evaluating the transcript abundance of nitrate reductase gene after 0, 45 and 90 min of N treatment. The treated tissues from three independent biological replicate experiments were frozen in liquid N2 and stored at − 80 °C till further use. Each biological replicate meant leaves derived from plants grown independently at a different time. The treatments with nitrate/control were always separate for every biological replicate and their microarray analyses were performed under MIAME compliant conditions.
Total RNA extraction and microarray
Total RNA was isolated from frozen independent biological triplicates of control and N-treated leaf tissues using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) as per manufacturer’s instructions and processed for microarray analysis at Genotypic Technologies (Bengaluru, India). RNA was quantified using a NanoDrop spectrophotometer (ND-2000, Thermo Fisher Scientific, Waltham, MA, USA). The integrity of the isolated RNA samples was determined by the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) as per the manufacturer’s instructions. The ratio of 18S and 28S rRNA was obtained from 2100 Expert software (Agilent Technologies) and RNA integrity number was obtained from RIN Beta Version Software (Agilent Technologies). The RNA samples used for microarray hybridization had RIN values of above 6. They were reverse transcribed using 500 ng of each RNA sample into double stranded cDNA using MMLV-RT enzyme and random primer tagged to a T7 polymerase promoter. The double stranded cDNA was then used as a template to generate Cy3-labeled cRNA by in-vitro transcription using RNaseOUT, inorganic pyrophosphatase, and T7 RNA polymerase at 40 °C as per the manufacturer’s instructions (Agilent Quick-Amp labeling Kit p/n5190-0442). Labeled cRNA was purified using Qiagen RNeasy columns (Qiagen, Cat No: 74106) and assessed for yields and specific activity. Agilent Rice Gene Expression 8 × 60 K microarrays were customized to include nuclear and organellar gene probes. Labeled cRNA samples of 600 ng each were fragmented and hybridized onto microarrays using the gene expression hybridization kit (Agilent) in a Agilent’s Surehyb hybridization chamber at 65 °C for 16 h. The hybridized slides were washed and scanned using Agilent microarray scanner.
Microarray data analysis
Scanned images were processed using Agilent Feature Extraction Software (Version-11.5) to obtain raw data, which were analyzed using Agilent GeneSpring GX software (Version-12.6.1). The data were normalized using the 75th percentile shift method, a global normalization that adjusts the locations of all the spot intensities in an array and provides fold expression values with respect to specific control samples. The raw data were deposited in the NCBI-GEO database (GSE62164). Transcripts showing geometric mean fold change value ± 1.0 (log2FC) with statistically significant cut-off (FDR ≤ 0.05) were considered as differentially expressed genes (DEGs) in the nitrate-treated samples with respect to the control samples. Student’s T-test was used to calculate P value among the replicates.
Functional classification, pathway analyses and subcellular localization of DEGs
Gene Ontology (GO) based functional annotation was performed using AgriGO 2.0 and EXPath. Mapping of DEGs onto various cellular pathways was performed using MapMan, KEGG (https://www.genome.jp/kegg/tool/map_pathway3.html), RAP-DB database (https://rapdb.dna.affrc.go.jp/), Rice Genome Annotation Project database (http://rice.plantbiology.msu.edu/) and Gramene (http://pathway.gramene.org/). DEGs involved in the regulation of various morphological and agronomic traits like leaf, culm, panicle, yield and productivity were picked from the Oryzabase database (https://shigen.nig.ac.jp/rice/oryzabase/) and literature. Protein subcellular localization was predicted using cropPAL database (https://crop-pal.org/) (Hooper et al. 2016) using default parameters for rice plants.
Construction of protein–protein interaction networks
Using the gene IDs of the nitrate-responsive DEGs analyzed in the present study, STRING (https://string-db.org/) database was searched for protein–protein interactions (PPI). Only those interactions which were either experimentally validated and/or retrieved from curated databases were used to construct PPI networks using Cytoscape version 8.0.
In silico promoter analyses of nitrate-responsive genes
Sequences of 1000 bp up-stream of DEGs were downloaded from RAP-DB (https://rapdb.dna.affrc.go.jp/tools/dump) and searched for enriched motifs (6–8 bases oligonucleotides) using AME (http://meme-suite.org/tools/ame) tools (McLeay and Bailey 2010). We used 7th major release of the JASPAR CORE (NON-REDUNDANT) motif database as the background database to search for experimentally defined transcription factor binding sites along with default parameters.
RT-qPCR validation of nitrate-responsive expression of DEGs
Leaf tissues for RT-qPCR experiments were either obtained after in vitro nitrate treatment as described above for microarray experiments, or after in vivo N-treatments in intact plants as described for physiological experiments below [except that nitrate doses were either 15 mM (normal) or 1.5 mM (low)]. Leaves were harvested at the active tillering and grain filling stage and used for RNA isolation as described above. 5 µg each of total RNA was reverse transcribed into cDNA using PrimeScript 1st strand cDNA synthesis kit (Takara, Kusatsu, Shiga, Japan). To avoid non-specific amplification from genomic DNA, we designed the primers spanning exon-exon junctions. To validate the differential expression of selected genes, RT-qPCR reaction was carried out in 10 µl volume using 1 µl of undiluted cDNA, 0.2 μl of forward and reverse primers (10 µM) and 5 μl of KAPA SYBR FAST Master Mix (2X) Universal (Kapa Biosystems, Wilmington, MA, USA) in an Agilent AriaMx Real-Time PCR System. The relative changes in gene expression were quantified by 2–△△CT method (Livak and Schmittgen 2001) using actin genes (LOC_Os01g64630 and LOC_Os03g50885) as internal controls. Melting curve analyses of the amplicons were used to determine the specificity of RT-qPCR reactions. The data were statistically analyzed by unpaired t-test using the GraphPad Prism 6 software. Experiments were performed using three technical replicates and three independent biological replicates.
Phenotypic and physiological measurements
For phenotypic measurements, germinated seedlings were grown hydroponically in ultrapure water for 10 days in a growth chamber as described under growth conditions above. They were treated for 10 more days with 1X AH nutrient solution with nitrate concentrations 10–100% of the recommended N (1.5 mM—15 mM N). These doses were achieved with Ca(NO3)2 as the only source of nitrate upto 7.5 mM N, while 15 mM concentration in the original AH medium was achieved using a combination of Ca(NO3)2 and KNO3. All the remaining ionic concentrations were balanced using K2SO4 and CaCl2. Length of the longest root of each seedling and shoot lengths were measured before transplanting them into separate 2-l cylindrical pots with 1:1 mixture of soilrite and vermiculite to monitor shoot length, number of tillers and yield in a growth chamber at 25 °C, with 75% relative humidity and average light intensity of 7200 lx (warm white LED tubes) and 12/12 h photoperiod. A randomized design was used for the experiments. The nutrient solutions were replenished every 48 h till panicle emergence and every 72 h thereafter. At the active tillering stage, photosynthesis, stomatal conductance and transpiration rate were measured using LI-6400XT Portable Photosynthesis System (LI-COR® Biosciences, Lincoln, NE, USA). Net photosynthetic rate was measured in terms of net CO2 assimilated (μmol CO2 m−2 s−1) with an average light intensity of 7200 lx (98 µmol m−2 s−1) falling on the leaf from the LED tubelights. The reference CO2 concentration was 410 ± 20 μmol mol−1 during the measurements. All LI-COR measurements were carried out at the time of maximal photosynthetic activity, which in our case was between 12:00 noon till 4:00 p.m IST. Photosynthetic leaf area and leaf chlorophyll content were measured as described earlier (Lichtenthaler 1987). Yield parameters such as the number of panicles and the weight of filled grains per plant were recorded at harvest. All the measurements were done in three independent experimental replicates grown at different times (between October 2019 and March 2021), each involving four internal replicates.
Scanning electron microscopy
Stomatal density was measured by scanning electron microscopy (SEM) at the UNESCO Regional Centre for Biotechnology, Faridabad, India, with leaf samples from three of the plants (used for phenotypic experiments mentioned above) at the reproductive stage and stored at − 80 °C. Each plant was sampled twice. The frozen leaves were immediately fixed for 2 h in 2% Karnovsky solution (2% glutaraldehyde and 2% paraformaldehyde in 0.1 M phosphate buffer). They were washed thrice for 15 min using 0.1 M phosphate buffer followed by dehydration for 15 min each, using gradually increasing concentration of AR-grade acetone (30–99.5%). Final dehydration was carried out using 100% acetone twice for 30 min each and critical point drying (CPD) was carried using CO2 followed by sputter coating with gold (18 mA for 15 s). The SEM was carried out using Apreo VS FESEM, FEI and the final images were obtained using xT Microscope Server 13.5.0 software.
Data analysis and statistics
The data were analyzed statistically by unpaired t-test and one-way ANOVA using GraphPad Prism ver. 6 (GraphPad Software, San Diego, CA, USA). All the results were expressed as mean ± SE of at least three replicates. The significance levels are represented as *for P ≤ 0.05, **for P ≤ 0.01 and ***for P ≤ 0.001.
Results
Nitrate-responsive leaf transcriptome of japonica rice
To understand early genomewide nitrate (N)-response in japonica rice, microarray analyses were carried out using 32-day-old rice plants under MIAME compliant conditions. Excised leaves were treated with nitrate in vitro to ensure that the measured N-response was only to locally supplied N, uninfluenced by soil conversions, in planta downstream metabolism or root-shoot translocation parameters. The concentration of N and duration of treatment (120 mM KNO3 for 90 min with KCl as control) were as optimized earlier in indica rice (Pathak et al. 2020). We reconfirmed N-induced accumulation of nitrate reductase transcript at 45 and 90 min for the current study by RT-qPCR (Supplementary Fig. S1).
Transcripts showing geometric mean fold change value ± 1.0 (log2FC) with statistically significant cut-off (FDR ≤ 0.05) were considered as differentially expressed genes (DEGs). In total, 6688 genes showed differential expression, including 3225 upregulated and 3463 downregulated genes (Supplementary Tables S1 and S2). Many of the well-known nitrogen-regulated genes figured among the DEGs identified in this study, confirming the reliability of our transcriptome data. In addition, there were many hitherto unreported DEGs, taking their total to a higher level, as detailed below.
Meta-analysis and functional annotation reveal many novel DEGs and processes
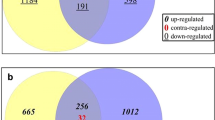
Nitrate-responsive DEGs from rice transcriptomes of any rice tissue at any developmental stage were retrieved for meta-analysis from all four published microarrays (Sawaki et al. 2013; Coneva et al. 2014; Misyura et al. 2014; Pathak et al. 2020) and two of the RNA-Seq datasets for which gene lists were available (Yang et al. 2015a; Shin et al. 2018). Their Venn selection with the 6688 nitrate-responsive genes identified in our microarrays revealed 2640 hitherto unreported DEGs in rice (Fig. 1a). A more detailed comparison at the subspecies level revealed 2675 DEGs over those previously reported in japonica (Sawaki et al. 2013; Coneva et al. 2014; Misyura et al. 2014; Yang et al. 2015a; Shin et al. 2018) and 6528 DEGs over those reported in indica (Pathak et al. 2020). These novel genes expand the repertoire of nitrate-responsive transcriptome known in rice in general and japonica in particular.

N-responsive genes in rice leaves and their functional annotation. a Venn diagram showing the common and exclusive nitrate-responsive genes among this study and those reported earlier by microarray (Sawaki et al. 2013; Coneva et al. 2014; Misyura et al. 2014; Pathak et al. 2020) or RNA-Seq (Yang et al. 2015a, b; Shin et al. 2018). b AgriGO based significant GO terms (P value < 0.05) associated with total up- and down-regulated DEGs identified in this study
To understand the functional roles of all the 6688 DEGs found in this study, functional annotations were performed using the software tools KEGG, EXPath and AgriGO 2.0. The over-represented GO terms for upregulated DEGs were carbon (C) metabolism, auxin-activated signaling pathway, lipid biosynthetic process, response to oxidative stress, primary cell wall biogenesis, alcohol metabolic process and regulation of transcription, among others (Fig. 1b, Supplementary Table S3). The most enriched GO terms associated with downregulated DEGs were related to transport, localization, protein synthesis and degradation pathways such as ribosome biogenesis and proteasome, regulation of transcription and translation, among others (Supplementary Table S3). Detailed comparison of functional categories across the above-mentioned published nitrate-responsive transcriptomes revealed 376 unreported biological processes in our study, such as alcohol catabolic process, lipid catabolic process, trehalose metabolic process, regulation of stomatal opening and, mitochondrial membrane organization and protein targeting, among others (Supplementary Table S4).
More DEGs found even from known N-responsive processes
Most of the other hitherto unreported DEGs found in the present study belonged to known processes or functional categories, revealing more extensive involvement in certain processes than previously reported, even in japonica. They include cellular protein metabolic process (265 additional DEGs), cellular nitrogen compound metabolic process (49 additional DEGs), intracellular transport (36 additional DEGs), sucrose metabolic process (9 additional DEGs), and starch biosynthetic process (8 additional DEGs) common across transcriptomes (Supplementary Table S4).
MapMan analysis revealed that 501 N-responsive DEGs were involved in transcriptional regulation and 135 in hormone signaling (Fig. 2 and Supplementary Table S4). In addition, there were 192 DEGs for development, 69 for calcium regulation, 500 for protein modification and degradation, 61 for redox as well, highlighting their N-dependent regulation in rice. Further, pathway analyses using EXPath and KEGG tools revealed significant enrichment of upregulated genes in the pentose phosphate pathway, photosynthesis, glycolysis/gluconeogenesis, starch and sucrose metabolism and biosynthesis of various secondary metabolites, among others. The downregulated genes were enriched in proteasome and ribosome biogenesis and protein processing in endoplasmic reticulum, indicating nitrate regulation of protein assembly and turnover (Supplementary Table S5).

MapMan based annotation of DEGs involved in regulation (a), cellular responses (b) and metabolism (c). Each box represents a DEG and color-coded according to their expression value (log2FC). Gradient of Red indicates upregulated DEGs and gradient of blue represents downregulated DEGs according to the color scale
Further analysis of the DEGs using the MapMan as well as KEGG and Gramene databases revealed the involvement of 228 DEGs in the metabolism and signaling of plant hormones like cytokinin, auxin, ethylene and jasmonic acid with log2FC values ranging from + 7.91 to − 6.6 (Fig. 3). They include many hitherto unreported N-responsive DEGs, including 18 related to auxin (and 46 known) and 11 related to ethylene (and 16 known), five related to cytokinin (and 25 known) and six known DEGs related to jasmonic acid (Fig. 3). These results clearly show more extensive nitrate-regulation of hormone signaling than understood so far (Krouk 2016; Lacuesta et al. 2018).

Enrichment of DEGs involved in development, hormone metabolism and signaling, calcium binding and DNA/RNA methylation. DEGs-associated data were retrieved from KEGG, RGAP, RAP-DB, and Gramene databases. Numbers in each block represent the log2FC values of the DEGs. Red and green color gradient represent the up- and down-regulated genes, respectively, according to the color scale. Expression value (log2FC) of each DEG is shown in its respective color gradient box
Calcium binding proteins such as calmodulins (CaMs), calcium-dependent protein kinases (CDPKs) and calcineurin B-like proteins (CBLs) act as sensors of temporal changes in cytosolic concentration of Ca2+ in response to many environmental changes, including nutrient availability (Ma et al. 2013). Therefore, we searched for DEGs possessing Ca2+-binding helix-loop-helix structure, known as the EF-hand motif, in RAP-DB, Gramene and RGAP databases. We found 89 DEGs (42 down and 47 up) with such motifs (Fig. 3), with fold change ranging from + 9.8 to − 3.11. Thirty three of them are hitherto unreported as N-responsive (Supplementary Table S6), indicating more extensive links between nitrate and calcium signaling.
In view of the emerging evidence on the role of methylation-mediated epigenetic and post-transcriptional regulation of N-responsive gene expression (Séré and Martin 2020), we searched for all RNA/DNA methyltransferases among our DEGs in RAP-DB, RGAP, KEGG and Gramene databases. We found 20 RNA/DNA methyltransferases to be N-responsive (13 down and 7 up, Fig. 3), 10 of which for the first time, indicating the prominent role of epigenetic and post-transcriptional regulation of N-response in rice. Similarly, our analysis of DEGs in the KEGG database revealed 223 additional genes of primary and secondary metabolism than reported so far (Supplementary Table S6). Indeed, biosynthesis of secondary metabolites was one of the most significantly enriched metabolic pathway in KEGG with 101 additional DEGs. This was followed by 36 additional DEGs in carbon metabolism (18 up, 18 down), 36 in synthesis of amino acids (18 up, 18 down), 29 in RNA transport (3 up, 29 down), 16 in glycolysis and gluconeogenesis (10 up, 6 down) and 8 in starch and sucrose metabolism (7 up, 1 down). Other significantly enriched functional categories were plant hormone signal transduction, protein processing in endoplasmic reticulum, plant-pathogen interacting proteins and fatty acid metabolism (Supplementary Table S6).
Our search with the DEGs using Oryzabase revealed their association with many agronomically important traits, not only for the 372 DEGs hitherto unreported in japonica or 368 unreported in any rice, but also for the 790 DEGs identified in the 6 previously reported nitrate-transcriptomes in any rice (Supplementary Table S7). These include 7 DEGs in controlling shoot apical meristem, including 1 hitherto unreported (5 up- and 2 down-regulated). In addition, there were 66 DEGs for leaf and/or culm and/or root, including 22 unreported (35 up- and 31 down-regulated) and 31 DEGs for tiller development, of which 18 were unreported (21 up- and 10 down-regulated) (Fig. 4 and Supplementary Table S7). There were also 70 DEGs for heading date and/or inflorescence and/or spikelet and/or panicle, including 34 unreported (20 up- and 50 down-regulated) and 2 validated by RT-qPCR.

Venn selection of nitrate-responsive DEGs with functional groups and agronomic traits. The functional categories of DEGs for development, hormone metabolism and signaling, calcium signaling, methyltransferases and TFs were identified using MapMan and/or RGAP and/or RAP-DB and/or Gramene, PlantTFDB and/or STIFDB2. They were subjected to Venn selection with all nitrate-responsive DEGs and the genes associated with agronomic traits (from Oryzabase database). It revealed the DEGs involved in the regulation of shoot apical meristem (a), leaf and/or culm and/or root (b), heading date and/or inflorescence and/or spikelet and/or panicle (c), seed development (d), tolerance and/or resistance (e), and yield and/or productivity (f)
In addition, 144 DEGs were for seed development, including 53 unreported (89 up- and 55 down-regulated) and 3 validated by RT-qPCR; 981 DEGs for tolerance and/or resistance, including 299 unreported (503 up- and 478 down-regulated) and 7 validated; 78 DEGs for yield and productivity, including 30 unreported (43 up- and 35 down-regulated) and 3 validated (Figs. 4 and 5). Considering that many of these traits such as root/shoot length, time to flowering/heading and yield parameters are known to constitute the phenotype for NUE in rice (Misyura et al. 2014; Sharma et al. 2021), our findings aid in narrowing the gap between N-responsive gene expression and its role in the NUE phenotype. The role of many DEGs in modulating gene expression is evident from the observation that the proteins encoded by 18% of all the DEGs were localized to the nucleus (Supplementary Fig. S2 and Supplementary Table S8). This also corroborated our functional annotation of 814 of all DEGs (12%) as encoding TFs (Supplementary Table S9 and Supplementary Table S10).

Validation of expression profile of nitrate-responsive genes by RT-qPCR. a RNA from excised leaves of 32-day-old rice seedlings treated in vitro with control (120 mM KCl) and nitrate (120 mM KNO3). b RNA from leaves of potted intact plants at active tillering stage, treated in vivo by growing them throughout with AH nutrient solution containing 1.5 mM (control) or 15 mM nitrate. c RNA from leaves of potted intact plants at grain filling stage, treated in vivo by growing them throughout with AH nutrient solution containing 1.5 mM (control) or 15 mM nitrate. Relative change in the gene expression was calculated by comparative Ct method and actin genes were used for data normalization. The control values were taken as zero and only the test values are shown as averages of three technical and three independent biological replicates (± SE). Statistical unpaired t-test analyses (control vs. nitrate) were performed in the GraphPad Prism 6 software using all replicates (*P value ≤ 0.05, **P value ≤ 0.01, ***P value ≤ 0.001, ****P value ≤ 0.0001). OsMADS1, OsMADS1-MADS-box family gene with MIKCc type-box; OsMADS58, MADS-box family gene with MIKCc type-box; OsIAA30, Aux/IAA gene family member; OsIAA8, Aux/IAA gene family member; BRCA2 repeat family protein; AK-HD2, aspartokinase/homoserine dehydrogenase; OsRH16, DEAD-box ATP-dependent RNA helicase; DDX47or OsRH10, DEAD-box ATP-dependent RNA helicase; NYC3, hydrolase alpha/beta fold family domain containing protein; DUF248, dehydration response related protein; USP family protein, universal stress protein domain containing protein; DUF1771, DUF1771 domain containing protein; TrpH, bifunctional trpH, putative; OsXTH11, glycosyl hydrolases family; OsNRT1, peptide transporter PTR2; OsMFS5, nodulin; IAA-amino acid hydrolase, OsAH8 or Similar to Auxin amidohydrolase; OsSULTR3;3, sulfate transporter; YELLOW-LEAF1; RNA methyltransferases; tRNA methyltransferase; Methyladenine glycosylase domain containing protein, OsMET1-2, DNA methyltransferase; OsERF062, APETALA2/ethylene-responsive element binding protein; OsEFCAX1, EF-Hand type domain containing protein; LTPL43-protease inhibitor, LTPL43—Protease inhibitor/seed storage/LTP family protein precursor; OsLhcb2, Similar to Photosystem II type II chlorophyll a/b binding protein; OsFBP1, Cytosolic fructose-1,6-bisphosphatase; OsWD40-125, Similar to Coatomer protein complex; OsNiR, Ferredoxin-nitrite reductase
RT-qPCR validates DEGs across categories in excised leaves and intact plants
We validated nitrate-regulation of the expression of 9 upregulated and 7 downregulated genes (DEGs) belonging to 6 functional categories by RT-qPCR (Fig. 5a) using primers provided in Supplementary Table S11. They were selected from MapMan/KEGG based annotations such as transcriptional regulation and/or hormone signaling (2 up and 2 downregulated), development and/or stress and/or metabolism (5 up and 5 down) and nutrient-related (both upregulated).
We additionally validated few selected DEGs identified in excised leaves with their N-response in intact plants used for physiological/phenotypic experiments. For this purpose, plants were continuously grown with AH nutrient solution containing N at 1.5 mM (control) or 15 mM (test) till active tillering stage. There were 7 up- and 8 down-regulated genes belonging to various N-responsive pathways. There were 1 up- and 4 down-regulated genes in the stress and/or transcriptional regulation and/or development and/or hormone signaling category, 2 up- and 1 down-regulated genes in the nutrient-related category, 2 up- and 2 -down-regulated genes in RNA/DNA methyltransferases and 2 up- and 1 down-regulated gene in the calcium binding category. We also validated the nitrate-responsive downregulation of 5 and upregulation of 2 DEGs at 15 mM N in the leaf tissues of the plants at the grain filling stage. These DEGs belonged to functional categories like hormone signaling and/or yield and/or development and/or tolerance and/or metabolism. Their data shown in Fig. 5b, and c clearly validate the overall microarray results, both in excised leaves under conditions identical to microarray experiments, as well as in potted intact plants. Six DEGs were deliberately kept common between cut-leaves and intact plants, which confirmed the consistency of our results and interpretations under in vitro and in vivo nitrate treatments at the whole plant level.
Phenotypic changes in response to N
To relate genomewide N-response found in leaves to the different associated organs, traits and overall agronomic performance in whole plants, phenotypic data were generated in the same plants used for RT-qPCR validation of some associated DEGs. For this purpose, the root and shoot lengths of 10 day old seedlings grown hydroponically in different doses of N (1.5, 3.75, 7.5 and 15 mM) were recorded (Supplementary Fig. S3), and then transferred to pots to complete their life cycle. Under contrasting doses of 1.5 and 15 mM nitrate N, significant differences were found in phenotypic and physiological responses of japonica rice plants in three independent experimental replicates (Fig. 6). The datasets for different experimental replicates and parameters are shown in Supplementary Table S12. Photosynthetic parameters were measured at tillering stage, shoot length and number of tillers at heading stage and filled grain weight at harvest. Chlorophyll content did not show significant difference under any treatment (Supplementary Fig. S3). However, plants grown under 15 mM N showed maximum photosynthetic rate per unit area (Fig. 6e) despite narrow leaf width (Supplementary Fig. S3).

Phenotypic and physiological measurements of rice plants in response to nitrate in rice. a Representative image of 20-day-old rice seedlings grown hydroponically at two different concentrations of nitrate. b Representative image of rice plants grown in pots with two different concentrations of nitrate. c Bar graph showing the root length of 20-day-old seedlings grown hydroponically at two different nitrate concentrations. d Bar graph representing the shoot length of rice plants at heading stage grown in pots with two different concentrations of nitrate. e Photosynthetic rate of the most active leaf at tillering stage at two different levels of nitrate. f Number of days to panicle emergence under two different levels of nitrate. g Number of productive tillers with panicles under two different levels of nitrate. h Weight of filled grains per plant in different nitrate concentrations. Each bar graph represents mean ± SE of the data from three independent experiments, each containing four biological replicates. Student’s t-test was used to calculate the statistically significant difference among treated samples. (**P value ≤ 0.01,***P value ≤ 0.001, ****P value ≤ 0.0001)
Panicle emergence was delayed with increasing N concentration (Fig. 6f). Conversely, the number of productive tillers (Fig. 6g) and the weight of filled grains (yield) per plant (Fig. 6h) decreased drastically with increased concentration of N. We observed that early root (Fig. 6c) and shoot growth (Supplementary Fig. S4) declined with increase in N concentration. Plants showed reduced shoot length in 15 mM N at the tillering and grain filling stages (Fig. 6). This trend began as early as 10 days after transfer to nutrient solution (Supplementary Fig. S4). Overall, these findings on nitrate-responsive phenotypic traits must have a basis in gene expression, as many of the nitrate-responsive DEGs found in this study are functionally linked to such phenotypic traits (Fig. 4, Supplementary Fig. S5 and Supplementary Fig. S6). At least 13 of them have been validated as nitrate responsive by RT-qPCR (Fig. 5), clearly indicating their role in regulating the agronomic response to nitrate.
N regulates stomatal density, conductance and transpiration
Nutrient delivery from root to shoot and subsequent utilization depends on transpirational pull, which in turn depends on stomatal density and activity. Therefore, we evaluated them on potted plants grown till active tillering stage with AH nutrient solution, containing either 1.5 mM (control) or 15 mM (test) nitrate N. Scanning electron microscopy revealed that stomatal density on the adaxial surfaces of fully developed leaves increased from 128 µm−2 with 1.5 mM to 145 µm−2 with 15 mM N (Fig. 7a, b), a significant increase of about 13% (Fig. 7c). Further, LI-COR-based measurements revealed that this small difference in stomatal density translated into about 35% increase in both stomatal conductance and transpiration rate (Fig. 7d, e and Supplementary Table S12), indicating the role of N in regulating stomatal physiology in rice.

Measurements of stomatal density and physiology at different levels of nitrate. Stomatal density on the adaxial leaf surfaces (a–c), stomatal conductance (d) and transpiration rate (e) measurements of photosynthetically active fully developed leaf at active tillering stage grown with 1.5 mM or 15 mM nitrate in the AH nutrient solution. SEM analyses were performed using three independent replicate plants and two technical replicates from each plant. Stomatal density was analyzed in frames of 550.56 × 825.43 (H × W) µm2 area on the leaf surfaces. Bar graphs (d and e) represent mean ± SE of the data from three independent experiments each containing four replicate plants. The statistical significance is calculated using unpaired t-test in GraphPad Prism 6.0 and is represented as *for P value ≤ 0.05, **for P value ≤ 0.01 and ***for P value ≤ 0.001
Protein interaction networks reveal links between DEGs and traits
PPI networks constructed using the STRING database helped in identifying TFs that may mediate the nitrate regulation of many important NUE traits like plant growth, flowering, yield and tolerance (Fig. 8a). We then searched for cis-regulatory elements in the promoter regions of all N-responsive DEGs at the AME database using JASPAR2018 CORE plants non-redundant as the background database with default parameters. It revealed binding sites for 345 TFs, such as members of MYB, bHLH, bZIP, ERF, MYC. TCP, TGA, NAC and WRKY families were enriched in 6676 DEGs (Fig. 8b and Supplementary Table S10).

Nitrate-responsive protein–protein interaction (PPI) networks and the most enriched cis-regulatory elements in the 1000 bp up-stream sequence of nitrate-responsive DEGs. a The PPI networks were constructed using information retrieved from the STRING database. The nodes in red and green color represent the up- and downregulated nitrate-responsive DEGs, respectively, some of which are validated by RT-qPCR. Interactors that are not DEGs are shown in gray color. The gray edges represent the experimentally validated interactions while pink edges represent those picked from curated databases. Some of the validated TFs (OsIAA8, IAA30, MADS1 and MADS58), methyltransferases and helicases (RNA helicase 10 and 16) and their expression profile were mapped onto the PPI networks to understand their role in the regulation of important NUE traits according to previous research (Tsuji et al. 2011; Wang et al. 2016; Jin et al. 2019; Qin et al. 2020). b Ten most enriched motifs for binding sites of TFs in our DEG list using AME tool (http://meme-suite.org/tools/ame) with JASPAR plant core as the background
Discussion
Studies on genomewide N-response yielded different gene sets, depending on the N-form(s) used and whether the response was to the addition or removal of N, apart from plant species/subspecies/cultivar, growth conditions, age and tissue, method of N treatment etc. Transcriptomics in field crops can be particularly challenging, as farm soils tend to have a dynamic mix of N-forms, not only due to different fertilizers or their doses used (urea, ammonium, nitrate, etc.) but also due to microbial conversions. Laboratory treatments can be more precise and specific to understand the molecular basis of N-response, which can be further confirmed under natural conditions. Therefore, the present study was specifically designed to analyze the short-term transcriptomic response to nitrate treatment in excised leaves of japonica rice. Some of the findings were revalidated in intact plants, both for physiological/phenotypic changes and associated gene expression simultaneously under controlled conditions, for final validation under field conditions.
Expansion of N-responsive genes/process in rice
Our microarray analysis yielded 6688 DEGs, and meta-analysis revealed that about 40% of them were hitherto unreported to be nitrate responsive in rice. Literature searches using the keywords ‘rice’ AND ‘nitrate’/‘ammonium’/‘nitrogen’/‘nitr*’ AND transcriptome” revealed 34 published research articles, of which Venn selections using available data from 15 transcriptomes yielded only 41 common DEGs (data not shown). These transcriptomes used different N sources, such as only ammonium (Beatty et al. 2013; Midorikawa et al. 2014; Chandran et al. 2016; Sun et al. 2017), ammonium nitrate (Obertello et al. 2015; Takehisa and Sato 2019; Xin et al. 2019) and nitrite (Wang et al. 2014), N-starvation or N-limitation (Cai et al. 2012; Yang et al. 2015b; Takehisa et al. 2015) or only nitrate (Sawaki et al. 2013; Coneva et al. 2014; Misyura et al. 2014; Pathak et al. 2020). But even these four nitrate-transcriptomes had reported different numbers of DEGs in the range of hundreds, which were too small, relative to the thousands known in Arabidopsis (Wang et al. 2004). Even after including available RNA-seq data (Yang et al. 2015a; Shin et al. 2018) along with the above 4 nitrate-responsive rice microarray datasets for Venn selection, 2675 DEGs from our microarray emerged as hitherto unreported in japonica rice. Despite significant overlaps with all published microarray and RNA-seq data, their differences could be due to the subspecies-based microarray, array version, sequence assembly, or experimental conditions of transcriptome analyses, including tissue/stage and N-treatment conditions. Venn selection with our earlier indica transcriptome data (Pathak et al. 2020) revealed only 160 common DEGs, despite using similar nitrate concentration and treatment method. The overall number of DEGs found in indica was also relatively very low at 519, probably due to the reasons mentioned above, including the fact that the microarrays used for indica were designed with japonica gene sequences. Conversely, this could have been an advantage for the current study on japonica, which yielded 6528 unique DEGs not common with indica.
We found many hitherto unreported DEGs across known functional categories/processes (Supplementary Table S4) and/or agronomically important traits (Supplementary Fig. S5 and Supplementary Table S7), indicating that previous studies significantly under-estimated the sheer number of genes involved in nitrate-responsive processes/traits in rice in general and japonica in particular. More importantly, we found several entirely unreported nitrate-responsive functional categories/processes associated with the DEGs such as mRNA transcription, positive regulation of response to salt stress, cell wall biogenesis, auxin biosynthetic process and auxin-activated signaling pathway. Some of them have important molecular functions such as TFs, hormone metabolism and signaling (Fig. 4), stress proteins and helicases as discussed below.
We found 814 DEGs encoding transcriptional regulators and/or TFs belonging to 36 families, representing about 34% of all TFs in the rice genome (Supplementary Table S9). One-third of these TFs are hitherto unreported to be nitrate-responsive in the 5 published nitrate-transcriptomes of japonica rice (Sawaki et al. 2013; Coneva et al. 2014; Misyura et al. 2014; Yang et al. 2015a; Shin et al. 2018). This increased to 800 DEGs when compared with the TFs reported in indica (Pathak et al. 2020). They include 7 out of the 75 MADS box TFs known in the rice genome, of which 2 hitherto unreported members have been validated by RT-qPCR, viz. OsMADS1 and OsMADS58 (Fig. 5a). Very few other members of the MADS family TFs are validated for their nitrate-responsive role in rice (Yu et al. 2015; Huang et al. 2019).
Universal Stress Proteins (USPs) play important roles in initiating plant responses to various abiotic stresses (Bhowmick 2019). There are 43 non-redundant OsUSP genes reported in rice and most of them showed differential regulation under various stress conditions (Bhowmick 2019). We found 16 OsUSP and 11 OsXTH (glycosyl hydrolases family) genes as nitrate responsive and validated one each of them by RT-qPCR. We also found 39 helicases as DEGs, most of which are unknown to be nitrate-responsive. Helicases regulate plant’s response to various abiotic stresses. RNA helicases regulate RNA metabolism, splicing, ribosome biogenesis, nucleo-cytoplasmic transport and organellar gene expression (Umate et al. 2010). Validation of OsRH16 and OsRH10 expression by RT-qPCR, indicate the role of helicases in nitrate response.
Association of DEGs with phenotypic/agronomic traits
In line with our finding that as many as 1158 DEGs had annotations associating them with important phenotypic/agronomic traits, our physiological experiments revealed nitrate-responsive changes in such traits. They include early root/shoot development, heading date, number of productive tillers and yield. RT-qPCR validation of few hitherto unreported nitrate-responsive DEGs and PPI network associated with them helped understand the pathways/processes underlying the phenotypic basis for nitrate response and/or NUE as discussed below.
Our physiological finding that nitrate delays panicle emergence and reduces yield (Fig. 6 and Supplementary Table S12) may be explained in part by our microarray findings. Out of the 70 DEGs related to heading date and/or panicle, as many as 50 are downregulated by nitrate. Downregulated DEGs also dominate the experimentally validated PPI networks with only two upregulated ones (Supplementary Fig. S6), indicating the role of these interactions in connecting N-responsive gene expression with the associated phenotypic traits. One of them is a transcription factor OsMADS1 involved in panicle development, disease resistance and yield improvement in rice (Lim et al. 2000; Agrawal et al. 2005; Yu et al. 2015). Its downregulation by nitrate at tillering and grain filling stages (Fig. 5) in our study may explain the delayed panicle emergence and reduced yield (Fig. 6). Similarly, OsMADS58 (LOC_Os05g11414), which was also downregulated during the grain filling stage, is known to play important role(s) in floral organ identity (Hong et al. 2017). Two other important genes OsFBP1 (LOC_Os01g64660) and OsOAT (LOC_Os03g44150) were also found to be downregulated. They have previously been shown to be involved in growth retardations in rice (Lee et al. 2008) and floral development and seed setting through mediating nitrogen reutilization in rice (Liu et al. 2018). Flowering time has been identified as a phenotypic trait for NUE in rice, as cultivars with contrasting NUE exhibited contrasting flowering times (Sharma et al. 2021). Our findings on these TFs suggest their involvement in the underlying mechanisms that mediate nitrate-regulation of flowering in japonica (Fig. 8a). Even though flowering-associated TFs have been identified in rice and Arabidopsis (Bluemel et al. 2015; Wei et al. 2020) their nitrate regulation is not well known in rice.
The expression of the helicase OsRH10, known as DDX47 (LOC_Os03g46610), increases with temperature to enhance thermotolerance and plant biomass, whereas its mutant exhibits a thermosensitive dwarf phenotype (Wang et al. 2016). Similarly, OsWD40 (LOC_Os06g05180) and OsERF062 (LOC_Os03g08470) is also related to ‘tolerance and/or resistance’ (Oryzabase). Downregulation of these DEGs (Fig. 5c) in response to 15 mM nitrate in our study indicates the roles of nitrate in thermosensitivity/tolerance, flowering and yield (Fig. 8a). These are agronomically important aspects that needs further investigation.
Among other hitherto unknown nitrate-regulated genes validated by RT-qPCR in this study, Yellow Leaf 1 (LOC_Os01g17170), involved in chloroplast development (Li et al. 2019). Another gene, OsSULTR3;3 (LOC_Os04g55800) is involved in metabolism in rice grains and modulates phytic acid and phosphorus concentration (Zhao et al. 2016), which are important from human/animal nutritional point of view. The third is an IAA-amino acid hydrolase (LOC_Os01g37960) involved in auxin synthesis/degradation. In our study, Yellow Leaf 1 and IAA-amino acid hydrolase were upregulated and OsSULTR3;3 was downregulated by 15 mM nitrate at active tillering stage (Fig. 5b). The expression of three more DEGs related to metabolism was checked at the grain filling stage. OsNiR (LOC_Os01g25484) as well as OsLhcb2 (LOC_Os03g39610) were upregulated whereas OsFBP1 (LOC_Os01g64660) was downregulated. The downregulation of OsFBP1 and OsSULTR3;3 may have a role in reducing the number of productive tillers and yield (Oryzabase) at higher N level (Fig. 6 and Supplementary Table S12). Recently, axillary bud outgrowth was shown to be inhibited at 15 mM N (Wang et al. 2020). The involvement of these genes in important processes such as growth and seed development indicate the agronomic link to our findings on their regulation by nitrate.
Methyltransferases are known to epigenetically regulate yield and related agronomic traits in rice (Li et al. 2018). One of them, the Polycomb Repressive Complex 2(PRC2) has been implicated in nitrate-responsive chromatin modification (Wu et al. 2020). We found 20 methyltransferases as nitrate responsive in our microarray, of which 7 were upregulated and 13 were downregulated (Fig. 3). A homolgue of tRNA methyltransferase (LOC_Os01g29409) in Arabidopsis has an important role in vegetative growth (Jin et al. 2019). Its downregulation along with its interacting member (LOC_Os07g40700) can explain the reduced tiller and plant development at higher N dose (Figs. 6 and 8a). Further elucidation of the extent of their involvement in N-response/NUE may open up options for epigenetic interventions to improve NUE.
Increase in the photosynthetic rate at 15 mM N (Fig. 6e and Supplementary Table S12) may be explained in part by our microarray finding that out of the 44 DEGs related to photosynthesis, as many as 32 are upregulated by nitrate (Supplementary Table S6). This include OsLhcb2 (LOC_Os03g39610) which was upregulated at higher N (Fig. 5c). Four of the nitrate-responsive DEGs (Os05g0140100, Os03g0858800, Os05g0373300 and Os03g0364500) are hitherto unreported to be associated with stomatal movement. Stomatal development and density control gaseous exchange as well as transpiration in plants. Our results show that stomatal density increased by 13% with increase in nitrate dose from 1.5 to 15 mM, which translates into about threefold larger increase in stomatal conductance and transpiration (Fig. 7). Nitrate has been shown to regulate stomatal activity in maize (Qi et al. 2018), but its role in regulating stomatal density is not well known, at least in rice.
During rice tiller development, strigolactone, gibberellic acid and auxin negatively regulate axillary bud outgrowth (Fig. 4), whereas cytokinin has a positively role (Zhuang et al. 2019). We found 228 genes related to hormones in our microarray data, including 56 hitherto unreported as nitrate-responsive. Among them, a majority of 64 genes were involved in auxin metabolism and signaling (42 up-, 22 down-regulated). The upregulation of two of them, OsIAA30 and OsIAA8 was validated by RT-qPCR (Fig. 5). Their downregulation was reported to be associated with tillering and grain yield, respectively (Paul et al. 2012; He et al. 2018). OsTIR1 (LOC_Os05g05800) is an auxin receptor and positively regulates several stress responses in rice (Qin et al. 2020). Its nitrate mediated downregulation can negatively affect plant development (Figs. 6d and 8a). Our comparison of growth and yield in pot experiment with two different doses of nitrate showed increase in root length, shoot length and yield at lower dose (1.5 mM) (Fig. 6). This pattern seems to have been established since early stage of growth (10 days) and remained so till seed setting. This could be due to better adaptation at low nitrate dose (Supplementary Fig. S4) and involvement of nitrate-associated hormone signaling in the regulation of early meristematic growth (Krouk 2016; Li et al. 2018). The exact doses that promote or inhibit yield may vary under field conditions, but overdose of N is known to inhibit crop yields as elaborated in Sharma et al. (2021). We also found many of the genes associated with root and shoot growth and development to be downregulated in our study. These include FPLP4 (LOC_Os04g21350), 1CYSPRXB (LOC_Os07g44440), OsDOF15(LOC_Os03g55610), OsSCR (LOC_Os12g02870), OsARD4(LOC_Os10g28360) with their expression levels between − 2.4 and − 6.6 in our microarray. They have been implicated in the modulation of root development, regulation of primary root elongation, and modulation of root growth pattern (Kamiya et al. 2003; Ramanathan et al. 2018; Qin et al. 2019; Guo et al. 2020; Yang et al. 2020).
Overall, we found many hitherto unreported nitrate-responsive genes in rice in general and japonica in particular, apart from corroborating many known DEGs in rice. We also found their associated processes, functional categories and agronomic traits relevant to N-response/NUE.
Author contribution statement
APJ performed the microarray experiment. VKM re-analyzed the microarray data, performed in silico analyses, RT-qPCR validation and prepared the draft manuscript; VKM and NC did phenotypic, physiological and metadata analyses. NR conceived, planned and supervised the transcriptome analyses, data interpretation, edited and finalized the manuscript. All authors have reviewed the manuscript.
Data availability
Our raw microarray data that support the findings of this study have been deposited in NCBI-GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE62164) with the accession number GSE62164 (GSM1520726, GSM1520727, GSM1520730, GSM1520731, GSM1520734 and GSM1520735). Additional transcriptome datasets were obtained from either the published supplementary materials and/or their authors cited in the article. All other datasets pertaining to the analyses are included in the supplementary information files.
Abbreviations
- AH:
-
Arnon-Hoagland nutrient solution
- DEG:
-
Differentially expressed gene
- NUE:
-
Nitrogen use efficiency
- PPI:
-
Protein–protein interaction
- TF:
-
Transcription factor
References
Abrol YP, Adhya TK, Aneja VP, Raghuram N, Pathak H, Kulshrestha U, Sharma C, Singh B (2017) The Indian nitrogen assessment: Sources of reactive nitrogen, environmental and climate effects, management options, and policies. Elsevier, Cambridge, MA, USA
Agrawal GK, Abe K, Yamazaki M, Miyao A, Hirochika H (2005) Conservation of the E-function for floral organ identity in rice revealed by the analysis of tissue culture-induced loss-of-function mutants of the OsMADS1 gene. Plant Mol Biol 59(1):125–135
Beatty PH, Carroll RT, Shrawat AK, Guevara D, Good AG (2013) Physiological analysis of nitrogen-efficient rice overexpressing alanine aminotransferase under different N regimes. Botany 91(12):866–883
Bhowmick R (2019) Comprehensive analysis of universal stress proteins and their promoter sequences in rice. Int J Curr Microbiol App Sci 8(7):1279–1286
Bluemel M, Dally N, Jung C (2015) Flowering time regulation in crops—what did we learn from Arabidopsis? Curr Opin Biotechnol 32:121–129
Cai H, Lu Y, Xie W, Zhu T, Lian X (2012) Transcriptome response to nitrogen starvation in rice. J Biosci 37(4):731–747
Chakraborty N, Raghuram N (2011) Nitrate sensing and signaling in genomewide plant N response. In: Jain V, Anandakumar P (eds) Nitrogen use efficiency in plants. New India Publishing Agency, New Delhi, pp 45–62
Chandran AKN, Priatama RA, Kumar V, Xuan Y, Je BI, Kim CM, Jung K-H, Han C-D (2016) Genome-wide transcriptome analysis of expression in rice seedling roots in response to supplemental nitrogen. J Plant Physiol 200:62–75
Coneva V, Simopoulos C, Casaretto JA, El-kereamy A, Guevara DR, Cohn J, Zhu T, Guo L, Alexander DC, Bi Y-M (2014) Metabolic and co-expression network-based analyses associated with nitrate response in rice. BMC Genomics 15(1):1056
Guo Y, Wu Q, Xie Z, Yu B, Zeng R, Min Q, Huang J (2020) OsFPFL4 is involved in the root and flower development by affecting auxin levels and ROS accumulation in rice (Oryza sativa). Rice 13(1):2. https://doi.org/10.1186/s12284-019-0364-0
He Y, Zhou J, Shan L, Meng X (2018) Plant cell surface receptor-mediated signaling–a common theme amid diversity. J Cell Sci 131(2):jcs209353. https://doi.org/10.1242/jcs.209353
Hoagland DR, Arnon DI (1950) The water-culture method for growing plants without soil, 2nd edn. Circular California agricultural experiment station, p 347
Hong W-J, Chandran AKN, Jeon J-S, Jung K-H (2017) Construction and application of functional gene modules to regulatory pathways in rice. J Plant Biol 60(4):358–379
Hooper CM, Castleden IR, Aryamanesh N, Jacoby RP, Millar AH (2016) Finding the subcellular location of barley, wheat, rice and maize proteins: the compendium of crop proteins with annotated locations (cropPAL). Plant Cell Physiol 57(1):e9–e9
Huang S, Liang Z, Chen S, Sun H, Fan X, Wang C, Xu G, Zhang Y (2019) A transcription factor, OsMADS57, regulates long-distance nitrate transport and root elongation. Plant Physiol 180(2):882–895
Jin X, Lv Z, Gao J, Zhang R, Zheng T, Yin P, Li D, Peng L, Cao X, Qin Y, Persson S, Zheng B, Chen P (2019) AtTrm5a catalyses 1-methylguanosine and 1-methylinosine formation on tRNAs and is important for vegetative and reproductive growth in Arabidopsis thaliana. Nucl Acids Res 47(2):883–898
Kamiya N, Itoh JI, Morikami A, Nagato Y, Matsuoka M (2003) The SCARECROW gene’s role in asymmetric cell divisions in rice plants. Plant J 36(1):45–54
Kronzucker H, Glass A, Siddiqi M, Kirk G (2000) Comparative kinetic analysis of ammonium and nitrate acquisition by tropical lowland rice: implications for rice cultivation and yield potential. New Phytol 145(3):471–476
Krouk G (2016) Hormones and nitrate: a two-way connection. Plant Mol Biol 91(6):599–606
Kumari S, Sharma N, Raghuram N (2021) Meta-analysis of yield-related and N-responsive genes reveals chromosomal hotspot, key processes and candidate genes for nitrogen use efficiency (NUE) in rice. Front Plant Sci 12:1006
Lacuesta M, Saiz-Fernández I, Podlešáková K, Miranda-Apodaca J, Novák O, Doležal K, De Diego N (2018) The trans and cis zeatin isomers play different roles in regulating growth inhibition induced by high nitrate concentrations in maize. Plant Growth Regul 85(2):199–209
Lee SK, Jeon JS, Boernke F, Voll L, Cho JI, Goh CH, Jeong SW, Park YI, Kim SJ, Choi SB (2008) Loss of cytosolic fructose-1,6-bisphosphatase limits photosynthetic sucrose synthesis and causes severe growth retardations in rice (Oryza sativa). Plant Cell Environ 31(12):1851–1863
Li H, Hu B, Chu C (2017) Nitrogen use efficiency in crops: lessons from Arabidopsis and rice. J Exp Bot 68(10):2477–2488
Li Y, Xiao J, Chen L, Huang X, Cheng Z, Han B, Zhang Q, Wu C (2018) Rice functional genomics research: past decade and future. Mol Plant 11(3):359–380
Li C, Ma F, Jiao R, Chen C, Wang Q, Xiao F, Sun C, Deng X, Dong C, Wang P (2019) Mutation in Mg-protoporphyrin IX monomethyl ester cyclase causes yellow and spotted leaf phenotype in rice. Plant Mol Biol Rep 37(4):253–264
Lichtenthaler HK (1987) Chlorophylls and carotenoids: pigments of photosynthetic biomembranes. Methods Enzymol 148:350–382
Lim J, Moon Y-H, An G, Jang SK (2000) Two rice MADS domain proteins interact with OsMADS1. Plant Mol Biol 44(4):513–527
Liu C, Xue Z, Tang D, Shen Y, Shi W, Ren L, Du G, Li Y, Cheng Z (2018) Ornithine δ-aminotransferase is critical for floret development and seed setting through mediating nitrogen reutilization in rice. Plant J 96(4):842–854
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 25(4):402–408
Ma P, Liu J, Yang X, Ma R (2013) Genome-wide identification of the maize calcium-dependent protein kinase gene family. Appl Biochem Biotechnol 169(7):2111–2125
McLeay RC, Bailey TL (2010) Motif enrichment analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 11(1):1–11
Meyer RC, Gryczka C, Neitsch C, Müller M, Bräutigam A, Schlereth A, Schön H, Weigelt-Fischer K, Altmann T (2019) Genetic diversity for nitrogen use efficiency in Arabidopsis thaliana. Planta 250(1):41–57. https://doi.org/10.1007/s00425-019-03140-3
Midorikawa K, Kuroda M, Terauchi K, Hoshi M, Ikenaga S, Ishimaru Y, Abe K, Asakura T (2014) Additional nitrogen fertilization at heading time of rice down-regulates cellulose synthesis in seed endosperm. PLoS One 9(6):e98738
Misyura M, Guevara D, Subedi S, Hudson D, McNicholas PD, Colasanti J, Rothstein SJ (2014) Nitrogen limitation and high density responses in rice suggest a role for ethylene under high density stress. BMC Genomics 15(1):681. https://doi.org/10.1186/1471-2164-15-681
Móring A, Hooda S, Raghuram N, Adhya TK, Ahmad A, Bandyopadhyay SK, Barsby T, Beig G, Bentley A, Bhatia A (2021) Nitrogen challenges and opportunities for agricultural and environmental science in India. Front Sustain Food Syst 5:505347
Obertello M, Shrivastava S, Katari MS, Coruzzi GM (2015) Cross-species network analysis uncovers conserved nitrogen-regulated network modules in rice. Plant Physiol 168(4):1830–1843
Pathak RR, Ahmad A, Lochab S, Raghuram N (2008) Molecular physiology of plant nitrogen use efficiency and biotechnological options for its enhancement. Curr Sci 94:1394–1403
Pathak RR, Jangam AP, Malik A, Sharma N, Jaiswal DK, Raghuram N (2020) Transcriptomic and network analyses reveal distinct nitrate responses in light and dark in rice leaves (Oryza sativa Indica var. Panvel1). Sci Rep 10(1):1–17
Paul P, Awasthi A, Rai AK, Gupta SK, Prasad R, Sharma T, Dhaliwal H (2012) Reduced tillering in Basmati rice T-DNA insertional mutant OsTEF1 associates with differential expression of stress related genes and transcription factors. Func Integr Genomics 12(2):291–304
Qi G-N, Yao F-Y, Ren H-M, Sun S-J, Tan Y-Q, Zhang Z-C, Qiu B-S, Wang Y-F (2018) The S-type anion channel ZmSLAC1 plays essential roles in stomatal closure by mediating nitrate efflux in maize. Plant Cell Physiol 59(3):614–623
Qin H, Li Y, Huang R (2020) Advances and challenges in the breeding of salt-tolerant rice. Int J Mol Sci 21(21):8385
Qin H, Wang J, Chen X, Wang F, Peng P, Zhou Y, Miao Y, Zhang Y, Gao Y, Qi Y (2019) Rice OsDOF 15 contributes to ethylene-inhibited primary root elongation under salt stress. New Phytol 223(2):798–813
Raghuram N, Sutton MA, Jeffery R, Ramachandran R, Adhya TK (2021) From South Asia to the world: embracing the challenge of global sustainable nitrogen management. One Earth 4(1):22–27
Raghuram N, Sharma N (2019) Improving crop nitrogen use efficiency. In: Moo-Young M (ed) Comprehensive biotechnology, vol 4. Elsevier, pp 211–220. https://doi.org/10.1016/B978-0-444-64046-8.00222-6
Ramanathan V, Rahman H, Subramanian S, Nallathambi J, Kaliyaperumal A, Manickam S, Ranganathan C, Muthurajan R (2018) OsARD4 encoding an acireductone dioxygenase improves root architecture in rice by promoting development of secondary roots. Sci Rep 8(1):15713. https://doi.org/10.1038/s41598-018-34053-y
Reddy MM, Ulaganathan K (2015) RNA-Seq analysis of urea nutrition responsive transcriptome of Oryza sativa elite indica cultivar RP Bio 226. Genom Data 6:112–113
Sawaki N, Tsujimoto R, Shigyo M, Konishi M, Toki S, Fujiwara T, Yanagisawa S (2013) A nitrate-inducible GARP family gene encodes an auto-repressible transcriptional repressor in rice. Plant Cell Physiol 54(4):506–517
Séré D, Martin A (2020) Epigenetic regulation: another layer in plant nutrition. Plant Signal Behav 15(1):1686236
Sharma N, Sinha VB, Gupta N, Rajpal S, Kuchi S, Sitaramam V, Parsad R, Raghuram N (2018) Phenotyping for nitrogen use efficiency: rice genotypes differ in N-responsive germination, oxygen consumption, seed urease activities, root growth, crop duration, and yield at low N. Front Plant Sci 9:1452
Sharma N, Sinha VB, Prem Kumar NA, Subrahmanyam D, Neeraja C, Kuchi S, Jha A, Parsad R, Sitaramam V, Raghuram N (2021) Nitrogen use efficiency phenotype and associated genes: roles of germination, flowering, root/shoot length and biomass. Front Plant Sci 11:2329
Shin S-Y, Jeong JS, Lim JY, Kim T, Park JH, Kim J-K, Shin C (2018) Transcriptomic analyses of rice (Oryza sativa) genes and non-coding RNAs under nitrogen starvation using multiple omics technologies. BMC Genomics 19(1):532. https://doi.org/10.1186/s12864-018-4897-1
Souza LA, Tavares R (2021) Nitrogen and stem development: a puzzle still to be solved. Front Plant Sci 12:181
Sun L, Di D, Li G, Kronzucker HJ, Shi W (2017) Spatio-temporal dynamics in global rice gene expression (Oryza sativa L.) in response to high ammonium stress. J Plant Physiol 212:94–104
Sun Y, Wang M, Mur LAJ, Shen Q, Guo S (2020) Unravelling the roles of nitrogen nutrition in plant disease defences. Int J Mol Sci 21(2):572
Sutton MA, Bleeker A, Howard C, Erisman J, Abrol Y, Bekunda M, Datta A, Davidson E, De Vries W, Oenema O (2013) Our nutrient world. The challenge to produce more food & energy with less pollution. Centre for Ecology & Hydrology, https://edepot.wur.nl/249094. Accessed 3 May 2021
Sutton M, Raghuram N, Adhya TK, Baron J, Cox C, de Vries W, Hicks K, Howard C, Ju X, Kanter D (2019) The nitrogen fix: from nitrogen cycle pollution to nitrogen circular economy. Frontiers 2018/19: Emerging Issues of Environmental Concern. United Nations Environment Programme, Nairobi
Takehisa H, Sato Y, Antonio B, Nagamura Y (2015) Coexpression network analysis of macronutrient deficiency response genes in rice. Rice 8(1):24. https://doi.org/10.1186/s12284-015-0059-0
Takehisa H, Sato Y (2019) Transcriptome monitoring visualizes growth stage-dependent nutrient status dynamics in rice under field conditions. Plant J 97(6):1048–1060
Tsuji H, Taoka K-i, Shimamoto K (2011) Regulation of flowering in rice: two florigen genes, a complex gene network, and natural variation. Curr Opin Plant Biol 14(1):45–52
Udvardi M, Below FE, Castellano MJ, Eagle A, Giller KE, Ladha JK, Liu X, Mcclellan Maaz T, Nova-Franco B, Raghuram N (2021) A research road map for responsible use of agricultural nitrogen. Front Sustain Food Syst 5:165
Umate P, Tuteja R, Tuteja N (2010) Genome-wide analysis of helicase gene family from rice and Arabidopsis: a comparison with yeast and human. Plant Mol Biol 73(4–5):449–465
Vidal EA, Alvarez JM, Araus V, Riveras E, Brooks MD, Krouk G, Ruffel S, Lejay L, Crawford NM, Coruzzi GM (2020) Nitrate in 2020: thirty years from transport to signaling networks. Plant Cell 32(7):2094–2119
Wang R, Tischner R, Gutiérrez RA, Hoffman M, Xing X, Chen M, Coruzzi G, Crawford NM (2004) Genomic analysis of the nitrate response using a nitrate reductase-null mutant of Arabidopsis. Plant Physiol 136(1):2512–2522
Wang Y-Y, Cheng Y-H, Chen K-E, Tsay Y-F (2018) Nitrate transport, signaling, and use efficiency. Annu Rev Plant Biol 69:85–122
Wang R, Qian J, Fang Z, Tang J (2020) Transcriptomic and physiological analyses of rice seedlings under different nitrogen supplies provide insight into the regulation involved in axillary bud outgrowth. BMC Plant Biol 20:197. https://doi.org/10.1186/s12870-020-02409-0
Wang X, Li Y, Fang G, Zhao Q, Zeng Q, Li X, Gong H, Li Y (2014) Nitrite promotes the growth and decreases the lignin content of indica rice calli: a comprehensive transcriptome analysis of nitrite-responsive genes during in vitro culture of rice. PLoS One 9(4):e95105
Wang D, Qin B, Li X, Tang D, Ye Z, Cheng Z, Xue Y (2016) Nucleolar DEAD-box RNA helicase TOGR1 regulates thermotolerant growth as a pre-rRNA chaperone in rice. PLoS Genet 12(2):e1005844
Wei H, Wang X, Xu H, Wang L (2020) Molecular basis of heading date control in rice. aBIOTECH 1:219–232
Wu K, Wang S, Song W, Zhang J, Wang Y et al (2020) Enhanced sustainable green revolution yield via nitrogen-responsive chromatin modulation in rice. Science 367(6478):eaaz2046. https://doi.org/10.1126/science.aaz2046
Xin W, Zhang L, Zhang W, Gao J, Yi J, Zhen X, Li Z, Zhao Y, Peng C, Zhao C (2019) An integrated analysis of the rice transcriptome and metabolome reveals differential regulation of carbon and nitrogen metabolism in response to nitrogen availability. Inter J Mol Sci 20(9):2349
Yang SY, Hao DL, Song ZZ, Yang GZ, Wang L, Su YH (2015a) RNA-Seq analysis of differentially expressed genes in rice under varied nitrogen supplies. Gene 555(2):305–317
Yang W, Yoon J, Choi H, Fan Y, Chen R, An G (2015b) Transcriptome analysis of nitrogen-starvation-responsive genes in rice. BMC Plant Biol 15:31. https://doi.org/10.1186/s12870-015-0425-5
Yang HC, Kan CC, Hung TH, Hsieh PH, Wang SY, Hsieh WY, Hsieh MH (2017) Identification of early ammonium nitrate-responsive genes in rice roots. Sci Rep 7:16885. https://doi.org/10.1038/s41598-017-17173-9
Yang S, Hao D, Jin M, Li Y, Liu Z, Huang Y, Chen T, Su Y (2020) Internal ammonium excess induces ROS-mediated reactions and causes carbon scarcity in rice. BMC Plant Biol 20:143. https://doi.org/10.1186/s12870-020-02363-x
Yu C, Liu Y, Zhang A, Su S, Yan A, Huang L, Ali I, Liu Y, Forde BG, Gan Y (2015) MADS-box transcription factor OsMADS25 regulates root development through affection of nitrate accumulation in rice. PLoS One 10(8):e0135196
Zhao H, Frank T, Tan Y, Zhou C, Jabnoune M, Arpat AB, Cui H, Huang J, He Z, Poirier Y (2016) Disruption of OsSULTR 3; 3 reduces phytate and phosphorus concentrations and alters the metabolite profile in rice grains. New Phytol 211(3):926–939
Zhuang L, Ge Y, Wang J, Yu J, Yang Z, Huang B (2019) Gibberellic acid inhibition of tillering in tall fescue involving crosstalks with cytokinins and transcriptional regulation of genes controlling axillary bud outgrowth. Plant Sci 287:110168
Acknowledgements
This work was supported by research grants to NR from ICAR (F. No. 2-2(60)/10-11/NICRA), Department of Biotechnology (DBT) [BT/IN/UK-VNC/44/NR/2015-16], UKRI GCRF South Asian Nitrogen Hub (SANH) [NE/S009019/1] GGSIPU [GGSIPU/DRC/Ph.D/Adm/2016/1549], [GGSIPU/DRC/FRGS/2018/22] and [GGSIPU/DRC/FRGS/2019/1553/24]. Fellowships were paid to VKM from DBT (DBT/JRF/14/AL/445) and GGSIPU (STRF:GGSIPU/DRC/2020/2049), APJ from CSIR (09/806(013)2008-EMR-I) and NC from UKRI GCRF-SANH [NE/S009019/1]. We thank Prof. T. Kumamaru from Kyushu University, Japan, for providing the rice seeds and the Regional Centre for Biotechnology (RCB), Faridabad for help with the scanning electron microscopy. We thank Dr. Dinesh Kumar Jaiswal for his assistance in early data analysis and initial draft of the manuscript. We also thank to Prof. Chanseok Shin from Seoul National University, Korea and Dr. Rumei Chen from Chinese Academy of Agricultural Sciences, China for providing their RNA-seq data for our meta-analyis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Communicated by Dorothea Bartels.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mandal, V.K., Jangam, A.P., Chakraborty, N. et al. Nitrate-responsive transcriptome analysis reveals additional genes/processes and associated traits viz. height, tillering, heading date, stomatal density and yield in japonica rice. Planta 255, 42 (2022). https://doi.org/10.1007/s00425-021-03816-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00425-021-03816-9