
Abstract
Main conclusion
The Albizia julibrissin chloroplasts have a classical chloroplast genome structure, containing 93 coding genes and 34 non-coding genes. Our research provides basic data for plant phylogenetic evolutionary studies.
Abstract
There is limited genomic information available for the important Chinese herb Albizia julibrissin Durazz. In this study, we constructed the chloroplast (Cp) genome of A. julibrissin. The length of the assembled Cp genome was 175,922 bp consisting of four conserved regions: a 5145 bp small single-copy (SSC) region, a 91,323 bp large single-copy (LSC) region, and two identical length-inverted repeat (IR) regions (39,725 bp). This Cp genome included 34 non-coding RNAs and 93 unique genes, the former contains 30 transfer and 4 ribosomal RNA genes. Gene annotation indicated some of the coding genes (82) in the A. julibrissin Cp genome classified in the Leguminosae family, with some to other related families (11). The results show that low GC content (36.9%) and codon bias towards A- or T-terminal codons may affect the frequency of gene codon usage. The sequence analysis identified 30 forward, 18 palindrome, and 1 reverse repeat > 30 bp length, and 149 simple sequence repeats (SSR). Fifty-five RNA editing sites in the Cp of A. julibrissin were predicted, most of which are C-to-U conversions. Analysis of the reverse repeat expansion or contraction and divergence area between several species, including A. julibrissin, was performed. The phylogenetic tree revealed that A. julibrissin was most closely related to Albizia odoratissima and Albizia bracteata, followed by Samanea saman, forming an evolutionary branch with Mimosa pudica and Leucaena trichandra. The research results are helpful for breeding and genetic improvement of A. julibrissin, and also provide valuable information for understanding the evolution of this plant.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Albizia julibrissin Durazz., also called “He Huan” in traditional Chinese medicine practice, is a large tree found on hillsides or that is cultivated in fields. It is naturally from the northeast to the southern parts of China, including Southwest China (Han et al. 2011), as well as Korea, India, other Asian regions, Africa, and North America (Li and Yang 2020). The medicinal parts of A. julibrissin are the flowers and bark (China-Pharmacopoeia-Committee 2020). The flowers of A. julibrissin may ameliorate memory loss induced by sleep deprivation in the Drosophila model (Chang et al. 2019), and the bark of A. julibrissin could reduce the risk of developing dementia (Chen et al. 2021).
A Cp of plants has a circular genome outside the nucleus and mitochondria, which plays a key role in photosynthesis, development, and physiology (Neuhaus and Emes 2000; Howe et al. 2003). The Cp genome is generally a tetrad ring structure (120–160 kb in size) and contains about 110–130 unique genes (Jarvis and Lopez-Juez 2013). Most angiosperms have a tetrad ring structure, with a LSC region and a SSC region, separated by a pair of IR regions of the same length (Wicke et al. 2011; Shetty et al. 2016). The evolution rate of the Cp genome is much lower than that of the nuclear genome (Wolfe et al. 1987), due to the high difficulty of mutation, the low nucleotide substitution rate, and maternal genetic characteristics. Cp genomes are generally more conserved and less mutable than genes in the nucleus, which is of vital importance to plant phylogeny and species identification (Wang et al. 2020). Therefore, the Cp genome is not only used to analyse genes and their encoded proteins but also to examine the evolutionary relationship between the pedigrees of different species (Moore et al. 2010; Shaw et al. 2014; Zhu et al. 2019). At present, ~ 30 thousand records of the complete Cp genome are accessible in the database of the National Centre for Biotechnology Information (NCBI).
Although studies have reported transcriptome sequencing, the coding sequence (CDS) of genes in Cps of A. julibrissin (Wojciechowski et al. 2004; Wang et al. 2009) is not available. In this study, the complete Cp genome of A. julibrissin was de novo assembled from short Illumina reads, and the detailed features of the Cp genome structure and organization were analysed. In addition, the phylogenetic analysis on the five genomes previously annotated in the Leguminosae family and 21 Cp genomes from other related species from GenBank and A. julibrissin were performed to determine genealogical relationships. Our results could provide valuable basic information for the conservation and sustainable utilization of A. julibrissin.
Materials and methods
Plant materials
Fresh and healthy leaves of A. julibrissin were collected from a single individual from Anhui University of Chinese Medicine, Anhui, China (N31°56′17″; E117°23′25″). Species identity was confirmed by experts in our lab. All tissues were collected from different parts of the same plant. A voucher specimen of this plant is stored in the Center of Herbarium, Anhui University of Chinese Medicine (Hefei, China) with a depository No. of AHTCM2020yxy04ZJ.
DNA extraction and library construction
An optimized protocol previously used by our team was taken to extract the complete genomic DNA using a commercial DNAsecure Plant Kit (TIANGEN Biotech Co., Ltd, Beijing, China) (Meng et al. 2021; Yao et al. 2021). Total DNA of A. julibrissin was checked for integrity and quality with BioPhotometer Plus (Nucleic Acid and Protein Detector, Eppendorf, Germany) and 1.0% agarose gel electrophoresis, and high-quality DNA was used to construct gene libraries. DNA libraries were constructed using the VAHTSTM Universal DNA Library Prep Kit for Illumina® V3 (Vazyme Biotech Co., Ltd, Nanjing, China) following the manufacturer’s protocol (Slatko et al. 2018). The total DNA was sonically fragmented into < 500 bp (Covaris S220), end-repaired, 5'-phosphorylated, and detailed with End Prep, followed by T-A ligation with splices at both ends. Fragments of ~ 470 bp were then recovered for PCR amplification. PCR products were purified and validated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA), and quantified by a Qubit 3.0 Fluorometer (Invitrogen, Carlsbad, CA, USA). The library with different indexes was multiplexed and loaded onto the Illumina HiSeq instrument according to the manufacturer's instructions (Illumina, San Diego, CA, USA).
Illumina sequencing and genome assembly
To obtain the sequence raw data, Genewiz Biotechnology Co. Ltd. (Suzhou, China) was commissioned to perform the next-generation sequencing using the Illumina Hiseq platform (Illumina, San Diego CA, USA) with a 2 × 150 paired-end (PE) configuration. Image analysis and base calling were executed by HiSeq Control Software (HCS) + OLB + GAPipeline-1.6 (Illumina) on the HiSeq instrument, and raw reads on the Cp genome of A. julibrissin were subsequently obtained. Trimmomatic software (version 0.39) (Bolger et al. 2014) was used to filter low-quality reads of the raw data, and the Q value was defined by Sanger. Ends were cut if the base quality was less than Q30 and the reads were excluded from further analysis if they had a length less than 100 bp or with an average Q value less than 30. The reads that passed QC were assembled to contigs using Velvet (version 1.2.10) (Zerbino and Birney 2008), and gap-filling was performed by SSPACE (version 3.0) (Boetzer et al. 2011) and GapFiller (version 1–10) (Boetzer and Pirovano 2012). The Cp genome of A. julibrissin was assembled using NOVOPlasty 2.7.2 (Dierckxsens et al. 2017) and auxiliary software Spades (Bankevich et al. 2012) on all the contigs, and the complete Cp sequences of A. odoratissima (NC_034987) were used as the reference genome. The assembled complete Cp genome sequence of A. julibrissin was submitted to NCBI, with the accession number MW539046.
Genome annotation
Based on the assembled clean data, the coding gene, tRNA, rRNA, and other ncRNAs were predicted by GoSeq (version 1.78) (Tillich et al. 2017), and the programs tRNAscan-SE (Lowe and Eddy 1997) and RNAmmer (Lagesen et al. 2007). The predicted protein sequence of the coding genes was subjected to database (NR, KEGG, and GO) searches using BLAST (version 2.2.31 +) (Wixon and Kell 2000; Harris et al. 2004; Boratyn et al. 2013; Yu and Zhang 2013), and the best matching result was selected as the gene annotation.
Comparative genomics
The Cp genome was outlined using Organellar Genome DRAW (Lohse et al. 2013) (http://ogdraw.mpimp-golm.mpg.de/index.shtml), with manual adjustment when necessary. The sequence divergence and LSC/IRb/SSC/IRa junction regions were compared and analysed among A. julibrissin, Arabidopsis thaliana, and four other Leguminosae species by mVISTA (Frazer et al. 2004) (http://genome.lbl.gov/vista/mvista/submit.shtml) and IRscope (Amiryousefi et al. 2018) (https://irscope.shinyapps.io/irapp/).
Repeat sequence analysis
The REPuter program (Kurtz et al. 2001) (https://bibiserv.cebitec.uni-bielefeld.de/reputer) was used to identify the four repetitive sequences of the Cp genome including the forward, reverse, palindrome, and complement sequences. The detection length and accuracy of repeated sequences were set to ≥ 30 bp and > 90%, respectively (Vieira Ldo et al. 2014; Chen et al. 2015). The Cp SSRs were detected using MISA (Thiel et al. 2003) with the minimum repeats of single, two, three, four, five, and hexanucleotides set to 10, 5, 4, 3, 3, and 3, respectively.
Codon usage analysis
The program CodonW1.4.2 (http://downloads.fyxm.net/CodonW-76666.html) (Du et al. 2020) was used to analyse the synonymous codon usage of 93 protein-coding genes (PCGs) in the Cp genome of A. julibrissin and to calculate two related parameters number of effective codons (Nc) and relative synonymous codon usage (RSCU). Nc is often used to evaluate codon bias at the level of the individual gene (Frank 1990). RSCU is the observed codon frequency divided by the expected frequency. An RSCU value close to 1.0 indicates that the deviation is not significant (Sharp et al. 1986). The AA frequency was calculated as the percentage of codons encoding the same amino acid divided by the total codons.
Prediction of RNA editing sites
Application of Prep-Cp (Mower 2009) (http://prep.unl.edu/) and CURE (Du et al. 2009) (http://bioinfo.au.tsinghua.edu.cn/pure/) were used to predict the RNA editing sites of 93 PCGs, and the threshold (cut-off value) was set to 0.8 to ensure the accuracy of the editing site prediction.
Phylogenomic analyses
To understand the phylogenetic relationship between A. julibrissin and related species, a total of 26 available Cp genomes records (in addition to A. julibrissin) were downloaded from NCBI, with four species, Penthorum chinese, Nicotiana tabacum, Schizophragma hydrangeoides, and Hydrangea petiolaris, as outgroups. The 27 plastome sequences were aligned and Maximum Likelihood (ML) phylogenetic analysis was conducted with MEGA-11 (version: 11) (Tamura et al. 2021). The percentage of replicate trees in which the related taxa clustered together in the bootstrap (BS) test (1000 replicates) are shown aside from the branches (Fig. 7).
Statistical analysis
All experimental tissues were collected from different parts of the same plant. In the DNA extraction experiment, three repetitions of independent biological experiments were used to ensure the reproducibility of the results. In the sequencing process, a sufficiently high threshold was set to eliminate unreliable sequencing results. In the subsequent analysis, at least three calculations were conducted to prevent result deviation caused by uncontrollable reasons, such as network or computer performance.
Results and discussion
Raw data analysis
Raw data of the complete Cp genome of A. julibrissin from Illumina sequencing contained 24,165,368 reads representing 3,624,805,200 bp, which were deposited at https://www.ncbi.nlm.nih.gov/sra/SRX11034648. The sequencing quality distribution (Fig. S1 A), base type percentage (Fig. S1 B), and mean quality distribution (Fig. S1 C) of raw reads were evaluated. These assessments indicated the sequencing quality met standard requirements and was suitable for further analysis. After optimizing the raw data, the effective ratio of Q20 and Q30 were as high as 99.82% and 99.31%, respectively.
Basic characteristics of the chloroplast genome of A. julibrissin
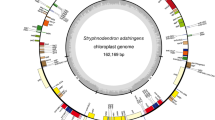
The complete Cp genome size of A. julibrissin was 175,922 bp according to the de novo genome assembly (Fig. S1 D). Four distinct regions, which were similar to other reported Cp genome structures, were also contained in the A. julibrissin Cp genome (Howe et al. 2003; Vieira Ldo et al. 2014): the LSC region, SSC region, and a pair of IR regions, with lengths of 91,323 bp, 5145 bp, 39,725 bp, and 39,725 bp, respectively (Fig. 1). The GC content in the A. julibrissin cp genome was 35.48% with 8.34% GC bias.

Plastid genome map of A. julibrissin. Genes inside the circle are transcribed clockwise, genes outside are transcribed counter-clockwise. Genes are color-coded to indicate functional groups. The dark grey area in the inner circle corresponds to GC content while the light grey corresponds to the adenine–thymine (AT) content of the genome. The small (SSC) and large (LSC) single-copy regions and inverted repeat (IRa and IRb) regions are noted in the inner circle
The Cp genome of A. julibrissin encoded a total of 34 ncRNAs and 93 unique genes, including 93 PCGs, 30 tRNAs, and four rRNA genes (rrn5, rrn4.5, rrn16, rrn23), as annotated by the software Prodigal (version 3.02) (Hyatt et al. 2010) and database of Rfam (version 14) (Kalvari et al. 2021) (Table 1). Eighteen contained introns, of which 15 (six tRNA genes and nine protein-coding genes) contained one intron, while the other three (rps12, clpP1, and ycf3) contained two (Table 2). The rps12 is a trans-spliced gene with three exons located in the LSC and the IR regions. The average length of the 93 PCGs was 959 bp (N50 was 1533 bp) with a 36.90% GC content. The genome data of the A. julibrissin Cp genome was submitted to the database of NCBI GenBank with a registration number of MW539046.
Only the complete Cp genome of A. odoratissima and A. bracteata has been reported for the genus Albizia of Leguminosae (Wang et al. 2017a, b; Zhang et al. 2020), with a Cp genome size of 174,816 bp and 176,054 bp in total and GC content of 36% and 35.4%, respectively. Similar to A. julibrissin, four different regions were characterized in A. odoratissima: the SSC region (4928 bp), LSC region (90,169 bp), and a pair of IR regions (each 39,882 bp). There were also 93 coding genes in the A. odoratissima Cp genome, and the total ncRNA, rRNA, and tRNA were 45, 8, and 37, respectively. The complete Cp genome of A. bracteata also had four different regions, the SSC region (5033 bp), LSC region (91,245 bp), and a pair of IR regions (each 39,888 bp), with the number of coding genes, total ncRNA, rRNA, and tRNA being 92, 46, 9, and 37, respectively. According to these data, the Cp genomes of A. julibrissin, A. bracteata, and A. odoratissima are highly similar.
The gene function annotation of A. julibrissin
Results gleaned from the NR database (Non-Redundant Protein Sequence Database) revealed 18 protein transcription genes that are similar to those in Acacia ligulata. In addition, about 8, 11, 10, and 4 genes were homologous to those in A. odoratissima, Inga leiocalycina, S. saman, and Staphylococcus aureus, respectively (Table S1). The annotation suggested that 82 of 93 coding genes in the A. julibrissin Cp genome had been classified in the Cp of Leguminosae, and some to other related families (11).
Based on the KEGG (Kyoto Encyclopedia of Genes and Genomes) biological pathway annotation, the Cp genes of A. julibrissin were distributed to four primary classes (cellular processes, genetic information processing, metabolism, organic systems). Among the four primary classes, the number of genes belonging to metabolism was the largest (especially genes related to energy metabolism processes) (Fig. 2). This embodied the biological function of Cp energy synthesis. The detailed gene annotation is presented in Table S2.

The KEGG pathway categories of A. julibrissin. The table of contents within the diagram is the second classification of biological pathways; the numbers are the number of genes; the different colors are used to distinguish the primary classification of biological pathways. KEGG divides biological metabolic pathways into six categories, and each category is divided into secondary classifications by the system and each of the statistical secondary classifications
With different GO (Gene Ontology) distributions, the second-level GO terms that target genes are reflected in the Cp genome of A. julibrissin (Fig. 3). The annotations indicated that the gene encoding proteins are involved in the regulation of a variety of cellular biological processes. Among them, 63 genes involved in metabolic processes belonged to the biological process followed by the cellular process. The detailed gene annotations are in Table S3.

Histogram of target gene distribution of A. julibrissin in GO terms
Codon usage analysis
Codon usage bias is affected by a variety of factors including gene mutation and selective evolution (Ermolaeva 2001; Wong et al. 2002). To check the codon usage, we calculated the Nc values of the 93 PCGs from the complete Cp genome of A. julibrissin (Table S4). The Nc value for each PCG indicated that the number of codons in all 93 PCGs ranged from 23.41 (rpl36) to 61 (petG, petL). The Nc value of rpl36 indicated that it had the most biased codon usage, with an average value of 46.21. Figure 4 and Table S5 show the status of codon usage and RSCU of the A. julibrissin Cp genome, where 31 codons in the 93 PCGs showed codon usage preference. Twenty-nine of thirty-one codons with preference are A or T-ending codons. Similar conclusions have been found in forsythia, rice, and other plants (Liu and Xue 2004; Wang et al. 2017a, b; Li et al. 2019). Correspondingly, the G or C terminal codons did not show codon preference (RSCU value < 1), while the stop codons are more biased towards the use of TAA.

The RSCU values of the 20 amino acids in the 93 PCGs of the A. julibrissin Cp genome and their different codon usages
Our results indicated that the GC content and codon usage bias at the A- and T-ends might be the main factors that determine the codon usage of the A. julibrissin Cp gene. The 93 unique PCGs contain 89,190 bp and encode 29,730 codons. The amino acid frequency of the A. julibrissin Cp genome was also calculated. Among all the codons used in the PCGs, 3133 (10.54%) encode leucine, which is the most commonly used in the A. julibrissin Cp genome. Cysteine was rarest, which is encoded by only 367 (1.23%) codons.
SSRs and LSRs of the A. julibrissin Cp genome
The software REPuter (Kurtz et al. 2001) was used to analyse the repeat sequences in the A. julibrissin Cp genome. Among 30 forward repeats, 18 palindrome repeats, and one reverse repeat were found with lengths ≥ 30 bp (identity > 90%), and without the detection of complement repeats (Table S6). Among 49 repetitions, 10 repetitions (20.4%) were 30–39 bp, 23 repetitions (46.9%) were 40–49 bp, 11 repetitions (22.4%) were 50–59 bp, 2 repetitions (4.1%) were 60–69 bp, 3 repeats (6.1%) were 70–81 bp, and the longest was 81 bp. Generally, duplications occur in non-coding regions (Nazareno et al. 2015; Yao et al. 2015), but 36.7% of the duplications were found in coding regions of the A. julibrissin Cp genome (Table S6).
Playing an important role in gene mutations, simple sequence repeats (SSRs) are widely distributed in the genomes of most species (Cavalier-Smith 2002). Previous studies have used them as valuable molecular markers for the study of species polymorphisms and population genetics (Xue et al. 2012; Hu et al. 2015). In this study, we analysed the occurrence, type, and distribution of SSRs in the Cp genome of A. julibrissin. The results showed that there were 149 SSRs in the complete genome (Table S7), which accounts for 4131 bp (2.35%) of the total sequence.
A large proportion of these SSRs consisted of mono-repeats that were found in 98 cases. Di-nucleotide-(7), Tri-(4), tetra-(3), and pentanucleotide repeat sequences-(2) occurred at lower levels. There were also 35 compound SSRs. The results showed that most of the repetitive sequences consist of A and T nucleotides instead of tandem G or C repetitive sequences. Other studies have obtained similar results (Kuang et al. 2011; Qian et al. 2013). A total of 31 SSR repeats occurred in coding gene regions, including ndhA, ndhF, petB, petD, rpl16, rpoC1, rpoC2, rps16, rps19, trnV-UAC, ycf1, ycf3, and ycf4. We found that ycf1 (12) had the most repetitions. Almost all SSRs were distributed across the entire A. julibrissin Cp genome, including SSC, LSC, and double IR. These SSRs could be used as lineage-specific markers, which would guide evolution and genetic diversity studies.
Prediction of RNA editing sites
Among the 93 PCGs of the A. julibrissin Cp genome, we predicted 55 RNA editing sites in 20 of them (Table 3). The ndhB gene contains the most editing sites (13), a similar finding to previous studies. (Freyer et al. 1995; Kahlau et al. 2006; Chateigner et al. 2007; Wang et al. 2017a, b). The ndhD genes were predicted to have eight editing sites, with five in ndhF; four in matK and rpoB; three in ropC2 and ndhG; two in rps14 and clpP1; and one each in accD, atpA, atpF, matK, ndhD, ndhF, ndhA, ndhB, ndhG, petB, psbE, psbF, rpoA, rpoB, rpoC1, rpoC2, rps2, rps14, and rps16. All predicted editing sites involve the conversion from C to U. The phenomenon of editing is also common in the Cps and mitochondria of Spermatophyta (Bock 2000). The predicted results showed that editing sites were only in the first position of the codon, with no editing sites found in the second and third codons. The amino acid with 32 editing sites changed can lead to variations in the acidity and polarity of proteins, such as serine, phenylalanine, leucine, and tyrosine. The conversion from serine to leucine is the most abundant in the prediction of editing sites. In addition, we also compared the predicted RNA editing sites of A. julibrissin with two other species of the same genus (A. bracteate and A. odoratissima). This showed that their features were highly similar, including both the predicted site locations and amino acid shifts (Table S8). As a form of post-transcriptional regulation of gene expression, previous studies also have revealed this characteristic in most RNA editing research (Jiang et al. 2011).
Comparative genome and phylogenetic analysis of A. julibrissin
To detect potential divergence among the A. julibrissin complete Cp genome and its associated species, we downloaded five additional Cp genome sequences already reported (A. odoratissima, S. saman, L. trichandra, Pyrus flexicaule, A. bracteata) in the Leguminosae family, as well as the model plant A. thaliana from the NCBI. As shown in Table S9, these Cp genome sequences ranged from 154,478 bp (A. thaliana) to 178,887 bp (P. flexicaule) in length, and every part of the quadrilateral cycle is comparable in the selected Cp genomes. The total GC content of these Cp genomes was also similar (35–36%).
The highly conserved IR region plays an essential role in stabilizing the structure of the Cp genome (Maréchal and Brisson 2010; Fu et al. 2016). For the IR and SC boundary regions, their expansion and contraction are usually viewed as the chief mechanisms behind the variation of Cp genome length in angiosperms (Chumley et al. 2006; Lei et al. 2016). The adjacent genes and boundaries of LSC/IRb/SSC/IRa of A. julibrissin Cp genomes were compared with the five other species in Leguminosae family, and the model plant A. thaliana (Fig. 5). Moreover, the Cp genome composition of the seven species was compared (Table S9), and the expansions and contractions in IR boundary regions were observed.

Comparison of the borders of the LSC, SSC, and IR regions in Cp genomes of six species of Leguminosae and Arabidopsis thaliana. The number above the gene represents the distance between the ends of the genes and the junction sites. The arrows indicate the location of the distance
Different from A. thaliana, the rps19 gene of the five species of legumes (S. saman, L. trichandra, A. odoratissima, A. julibrissin, A. bracteate) all existed at the LSC/IRb boundary, while five of them have rpl2 gene in the IRb region. There was an expansion of the ndhF gene at the IRb/SSC boundary in A. julibrissin, and the ccsA gene is 188 bp away from the SSC/IRa boundary. This was similar to S. saman but differed from A. odoratissima and A. bracteate. This indicated that the IRa region of A. julibrissin is partially contracted. In addition, the rps19 gene was found in A. bracteate, S. saman, and A. odoratissima, but not at the same position as in A. julibrissin. It is speculated that the presence of the rps19 gene in other genomes may be the result of gene duplication. The trnH gene existed in all six plants of Leguminosae, located in the LSC region, except for A. thaliana. The distance from trnH to the LSC/IRa boundary ranges from 0 to 30 bp among these six same family species, among which, it is closer to the boundary of LSC/IRa in S. saman, A. bracteate, A. odoratissima, and A. julibrissin. In general, the IR/SC junctions among these five species in the family Leguminosae are similar, but there are certain differences when compared with A. thaliana. Our results again suggested that the Cp genomes of related species are conserved, whereas greater diversity may exist between species of different families (Fig. 5).
To further detect the differences in the Cp genome among associated species and identify whether gene rearrangements are also present in the A. julibrissin Cp genome, we compared the homology of entire Cp sequences in five species from the family Leguminosae and A. thaliana using mVISTA (Frazer et al. 2004). Among them, the A. julibrissin Cp genome was used as the reference genome (Fig. 6). The results indicated no occurrence of genomic structural rearrangements in the selected Cp genomes, except for A. thaliana; the genome similarity of the other six Cp genomes was all higher than 90% and highly conserved.

Sequence alignment of five Cp genomes of Leguminosae and one Cp genome of Arabidopsis by mVISTA, with the annotation of A. julibrissin as the reference. The vertical scale indicates the percentage of identity, ranging from 50 to 100%. The horizontal axis indicates the coordinates within the chloroplast genome. Genome regions are color-coded as protein-coding, rRNA, tRNA, intron, and conserved non-coding sequences
To understand the phylogenetic relationships among A. julibrissin and other 26 related species (Table S10), sequences of the Cp genomes were aligned by MEGA-11 (version 11) (Tamura et al. 2021). The percentage of replicate trees where the relevant taxa are clustered together is shown next to the branches in bootstrap (BS) tests (1000 replicates). As can be expected, A. julibrissin was distributed to genus Albizia, the Leguminosae subfamily, and was closely related to A. bracteate and A. odoratissima, with a 100% BS value (Fig. 7).

Maximum likelihood phylogenetic tree of A. julibrissin with 26 Cp genomes of previously reported species as constructed by Cp genome sequences. Numbers on the nodes are bootstrap values from 1000 replicates
Conclusions
In this research, we constructed the complete Cp genome of A. julibrissin using Illumina HiSeq reads and other technical approaches. The results show that the Cp genome of A. julibrissin had a classical tetrameric structure of 175,922 bp in length, consisting of one LSC of 91,327 bp, one SSC of 5,145 bp, and two copies of IR regions of 39,725 bp. Overall, the GC content of Cp genome in this plant was 35.48%. The Cp genome contains 127 unique genes, which are 93 PCGs, 30 tRNA genes, and 4 rRNA genes.
A total of 93 coding genes of the A. julibrissin Cp genome were classified as Cp genes in the Leguminosae family. In the codon usage analysis, most of the Nc values were greater than 44, suggesting that the gene codon usage bias in the A. julibrissin Cp genome is weak. In addition, we detected 149 SSRs. There is no denying that SSRs found in this study are significant in studying the evolution and diversities of genomes in other species if they can be used as specific lineage markers. Most of the repetitive sequences are filled with A and T nucleotides, while tandem G or C repetitive sequences are not common. Among the coding genes, the amino acid changes at 32 editing sites can lead to changes in the acidity and polarity of the proteins. The overall GC content, boundaries of LSC/IRb/SSC/IRa, and homology of the entire Cp sequence, were also similar among different Cp genomes included in the current research (A. odoratissima, S. saman, L. trichandra, P. flexicaule, and A. thaliana). The phylogenetic relationship of A. julibrissin and another 26 related species shows that A. julibrissin was placed within genus Albizia, the Leguminosae subfamily, and closely related to A. bracteate and A. odoratissima in the same genus with a 100% BS value.
These results do not only contribute to offering valuable evidence to clarify the evolutionary history of A. julibrissin at the genetic level but are also beneficial to explore more genetic information and better breeding of A. julibrissin.
Author Contributions Statement
SX, JZ, HH, CQ, XM, and BH designed the project and/or conducted aspects of the experimental work. JZ, HH, FM, XY, JW, and XG conducted the experiments and the collection of electronic resources. SX and HH supported this work financially and participated in its planning. JZ, SX, and HH wrote the manuscript. All authors edited the manuscript and approve its submission in the current form.
Data availability
The genome sequence data that support the findings of this study are available in GenBank of NCBI (https://www.ncbi.nlm.nih.gov/nuccore/MW539046) under the access number MW539046.1. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA734110, SSR16696562, and SAMN19471633 respectively.
References
Amiryousefi A, Hyvönen J, Poczai P (2018) IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34:3030–3031. https://doi.org/10.1093/bioinformatics/bty220
Bankevich A, Nurk S, Antipov D et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. https://doi.org/10.1089/cmb.2012.0021
Bock R (2000) Sense from nonsense: How the genetic information of chloroplastsis altered by RNA editing. Biochimie 82:549–557. https://doi.org/10.1016/S0300-9084(00)00610-6
Boetzer M, Pirovano W (2012) Toward almost closed genomes with GapFiller. Genome Biol 13:R56. https://doi.org/10.1186/gb-2012-13-6-r56
Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W (2011) Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27:578–579. https://doi.org/10.1093/bioinformatics/btq683
Bolger A, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Boratyn GM, Camacho C, Cooper PS et al (2013) BLAST: a more efficient report with usability improvements. Nucleic Acids Res 41:W29-33. https://doi.org/10.1093/nar/gkt282
Cavalier-Smith T (2002) Chloroplast evolution: secondary symbiogenesis and multiple losses. Curr Biol 12:R62–R64. https://doi.org/10.1016/s0960-9822(01)00675-3
Chang J, Liu H, Cheng J, Chen C, Hwang S, Tseng C, Hsu L, Lin W (2019) Albizia julibrissin ameliorates memory loss induced by insomnia in Drosophila. Evid Based Complement Alternat Med 2019:7395962. https://doi.org/10.1155/2019/7395962
Chateigner B, Anne L, Small I (2007) A rapid high-throughput method for the detection and quantification of RNA editing based on high-resolution melting of amplicons. Nucleic Acids Res 35:e114. https://doi.org/10.1093/nar/gkm640
Chen J, Hao Z, Xu H, Yang L, Liu G, Sheng Y, Zheng C, Zheng W, Cheng T, Shi J (2015) The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Front Plant Sci 6:447. https://doi.org/10.3389/fpls.2015.00447
Chen C, Liu X, Chiou J, Hang L, Li T, Tsai F, Ko C, Lin T, Liao C, Huang S, Liang W, Lin Y (2021) Effects of Chinese herbal medicines on dementia risk in patients with sleep disorders in Taiwan. J Ethnopharmacol 264:113267. https://doi.org/10.1016/j.jep.2020.113267
China-Pharmacopoeia-Committee (2020) Pharmacopoeia of the People’s Republic of China. China Chemical Industry Press, Beijing
Chumley TW, Palmer JD, Mower JP, Fourcade MH, Calie PJ, Boore JL, Jansen RK (2006) The complete chloroplast genome sequence of Pelargonium × hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol 23:2175–2190. https://doi.org/10.1093/molbev/msl089
Dierckxsens N, Mardulyn P, Smits G (2017) NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res 45:e18. https://doi.org/10.1093/nar/gkw955
Du P, Jia L, Li Y (2009) CURE-Chloroplast: a chloroplast C-to-U RNA editing predictor for seed plants. BMC Bioinformatics 10:135. https://doi.org/10.1186/1471-2105-10-135
Du X, Zeng T, Feng Q, Hu LJ, Luo X, Weng QB, He J, Zhu B (2020) The complete chloroplast genome sequence of yellow mustard (Sinapis alba L.) and its phylogenetic relationship to other Brassicaceae species. Gene. https://doi.org/10.1016/j.gene.2020.144340
Ermolaeva MD (2001) Synonymous codon usage in bacteria. Curr Issues Mol Biol 3:91–97
Frank W (1990) The ‘effective number of codons’ used in a gene. Gene 87:23–29. https://doi.org/10.1016/0378-1119(90)90491-9
Frazer K, Pachter L, Poliakov A, Rubin E, Dubchak I (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 32:W273-279. https://doi.org/10.1093/nar/gkh458
Freyer R, LoPez C, Maier RM, Martin M, Sabater B, KoSsel H (1995) Editing of the chloroplast ndhB encoded transcript shows divergence between closely related members of the grass family (Poaceae). Plant Mol Biol 29:679–684. https://doi.org/10.1007/bf00041158
Fu J, Liu H, Hu J, Liang Y, Liang J, Wuyun T, Tan X (2016) Five complete chloroplast genome sequences from diospyros: genome organization and comparative analysis. PLoS ONE 11:e0159566. https://doi.org/10.1371/journal.pone.0159566
Han L, Pan G, Wang Y, Song X, Gao X, Ma B, Kang L (2011) Rapid profiling and identification of triterpenoid saponins in crude extracts from Albizia julibrissin Durazz. By ultra high-performance liquid chromatography coupled with electrospray ionization quadrupole time-of-flight tandem mass spectrometry. J Pharm Biomed Anal 55:996–1009. https://doi.org/10.1016/j.jpba.2011.04.002
Harris MA, Clark J, Ireland A et al (2004) The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 32:D258-261. https://doi.org/10.1093/nar/gkh036
Howe CJ, Barbrook AC, Koumandou VL, Nisbet RE, Symington HA, Wightman TF (2003) Evolution of the chloroplast genome. Philos Trans R Soc Lond B Biol Sci 358:99–106. https://doi.org/10.1098/rstb.2002.1176
Hu J, Gui S, Zhu Z, Wang X, Ke W, Ding Y (2015) Genome-wide identification of SSR and SNP markers based on whole-genome re-sequencing of a Thailand wild sacred lotus (Nelumbo nucifera). PLoS ONE 10:e0143765. https://doi.org/10.1371/journal.pone.0143765
Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. https://doi.org/10.1186/1471-2105-11-119
Jarvis P, Lopez-Juez E (2013) Biogenesis and homeostasis of chloroplasts and other plastids. Nat Rev Mol Cell Biol 14:787–802. https://doi.org/10.1038/nrm3702
Jiang Y, Yun H, Fan S, Jia-Ning YU, Song M (2011) The identification and analysis of RNA editing sites of 10 chloroplast protein-coding genes from virescent mutant of Gossypium Hirsutum. Cotton Sci 2011:3–9
Kahlau S, Aspinall S, Gray JC, Bock R (2006) Sequence of the tomato chloroplast DNA and evolutionary comparison of solanaceous plastid genomes. J Mol Evol 63:194–207. https://doi.org/10.1007/s00239-005-0254-5
Kalvari I, Nawrocki EP, Ontiveros-Palacios N et al (2021) Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res 49:D192–D200. https://doi.org/10.1093/nar/gkaa1047
Kuang D, Wu H, Wang Y, Gao L, Zhang S, Lu L (2011) Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome 54:663–673. https://doi.org/10.1139/G11-026
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res 29:4633–4642. https://doi.org/10.1093/nar/29.22.4633
Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108. https://doi.org/10.1093/nar/gkm160
Lei W, Ni D, Wang Y, Shao J, Wang X, Yang D, Wang J, Chen H, Liu C (2016) Intraspecific and heteroplasmic variations, gene losses and inversions in the chloroplast genome of Astragalus membranaceus. Sci Rep 6:21669. https://doi.org/10.1038/srep21669
Li W, Yang HJ (2020) Isolation and identification of lignans and other phenolic constituents from the stem bark of Albizia julibrissin Durazz and evaluation of their nitric oxide inhibitory activity. Molecules. https://doi.org/10.3390/molecules25092065
Li G, Pan Z, Gao S, He Y, Xia Q, Jin Y, Yao H (2019) Analysis of synonymous codon usage of chloroplast genome in Porphyra umbilicalis. Genes Genom 41:1173–1181. https://doi.org/10.1007/s13258-019-00847-1
Liu Q, Xue Q (2004) Codon usage in the chloroplast genome of rice (Oryza sativa L. ssp. japonica). Zuo Wu Xue Bao 30:1220–1224
Lohse M, Drechsel O, Kahlau S, Bock R (2013) OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res 41:W575-581. https://doi.org/10.1093/nar/gkt289
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. https://doi.org/10.1093/nar/25.5.955
Maréchal A, Brisson N (2010) Recombination and the maintenance of plant organelle genome stability. New Phytol 186:299–317. https://doi.org/10.1111/j.1469-8137.2010.03195.x
Meng F, Jiang W, Wu L, Zhang J, Yao X, Wu J, Guo X, Xing S (2021) The complete chloroplast genome of Epilobium hirsutum L. (Onagraceae). Mitochondrial DNA B Resour 6:2174–2176. https://doi.org/10.1080/23802359.2021.1945968
Moore MJ, Soltis PS, Bell CD, Burleigh JG, Soltis DE (2010) Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc Natl Acad Sci U S A 107:4623–4628. https://doi.org/10.1073/pnas.0907801107
Mower JP (2009) The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res 37:W253-259. https://doi.org/10.1093/nar/gkp337
Nazareno AG, Carlsen M, Lohmann LG (2015) Complete chloroplast genome of Tanaecium tetragonolobum: The first Bignoniaceae Plastome. PLoS ONE 10:e0129930. https://doi.org/10.1371/journal.pone.0129930
Neuhaus HE, Emes MJ (2000) Nonphotosynthetic metabolism in plastids. Annu Rev Plant Physiol Plant Mol Biol 51:111–140. https://doi.org/10.1146/annurev.arplant.51.1.111
Qian J, Song J, Gao H, Zhu Y, Xu J, Pang X, Yao H, Sun C, Li X, Li C, Liu J, Xu H, Chen S (2013) The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 8:e57607. https://doi.org/10.1371/journal.pone.0057607
Sharp PM, Tuohy TM, Mosurski KR (1986) Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res 14:5125–5143. https://doi.org/10.1093/nar/14.13.5125
Shaw J, Shafer HL, Leonard OR, Kovach MJ, Schorr M, Morris AB (2014) Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the tortoise and the hare IV. Am J Bot 101:1987–2004. https://doi.org/10.3732/ajb.1400398
Shetty SM, Md Shah MU, Makale K, Mohd-Yusuf Y, Khalid N, Othman RY (2016) Complete chloroplast genome sequence of Musa balbisiana corroborates structural heterogeneity of inverted repeats in wild progenitors of cultivated bananas and plantains. Plant Genome. https://doi.org/10.3835/plantgenome2015.09.0089
Slatko BE, Gardner AF, Ausubel FM (2018) Overview of next-generation sequencing technologies. Curr Protoc Mol Biol 122:e59. https://doi.org/10.1002/cpmb.59
Tamura K, Stecher G, Kumar S (2021) MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38:3022–3027. https://doi.org/10.1093/molbev/msab120
Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422. https://doi.org/10.1007/s00122-002-1031-0
Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S (2017) GeSeq—versatile and accurate annotation of organelle genomes. Nucleic Acids Res 45:W6–W11. https://doi.org/10.1093/nar/gkx391
Vieira Ldo N, Faoro H, Rogalski M, Fraga HP, Cardoso RL, de Souza EM, de Oliveira PF, Nodari RO, Guerra MP (2014) The complete chloroplast genome sequence of Podocarpus lambertii: genome structure, evolutionary aspects, gene content and SSR detection. PLoS ONE 9:e90618. https://doi.org/10.1371/journal.pone.0090618
Wang H, Moore MJ, Soltis PS, Bell CD, Brockington SF, Alexandre R, Davis CC, Latvis M, Manchester SR, Soltis DE (2009) Rosid radiation and the rapid rise of angiosperm-dominated forests. Proc Natl Acad Sci U S A 106:3853–3858. https://doi.org/10.1073/pnas.0813376106
Wang W, Yu H, Wang J, Lei W, Gao J, Qiu X, Wang J (2017a) The complete chloroplast genome sequences of the medicinal plant Forsythia suspensa (Oleaceae). Int J Mol Sci. https://doi.org/10.3390/ijms18112288
Wang Y, Qu X, Chen S, Li D, Yi T (2017b) Plastomes of Mimosoideae: structural and size variation, sequence divergence, and phylogenetic implication. Tree Genet Genomes. https://doi.org/10.1007/s11295-017-1124-1
Wang L, Lu G, Liu H, Huang L, Jiang W, Li P, Lu X (2020) The complete chloroplast genome sequence of Gynostemma yixingense and comparative analysis with congeneric species. Genet Mol Biol. https://doi.org/10.1590/1678-4685-gmb-2020-0092
Wicke S, Schneeweiss GM, dePamphilis CW, Muller KF, Quandt D (2011) The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol 76:273–297. https://doi.org/10.1007/s11103-011-9762-4
Wixon J, Kell D (2000) The kyoto encyclopedia of genes and genomes—KEGG. Yeast 17:48–55. https://doi.org/10.1155/2000/981362
Wojciechowski MF, Lavin M, Sanderson MJ (2004) A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. Am J Bot 91:1846–1862. https://doi.org/10.3732/ajb.91.11.1846
Wolfe KH, Li WH, Sharp PM (1987) Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Natl Acad Sci U S A 84:9054–9058. https://doi.org/10.1073/pnas.84.24.9054
Wong G, Wang J, Tao L, Tan J, Zhang J, Passey D, Yu J (2002) Compositional gradients in Gramineae genes. Genome Res 12:851–856. https://doi.org/10.1101/gr.189102
Xue J, Wang S, Zhou S (2012) Polymorphic chloroplast microsatellite loci in Nelumbo (Nelumbonaceae). Am J Bot 99:e240–e244. https://doi.org/10.3732/ajb.1100547
Yao X, Tang P, Li Z, Li D, Liu Y, Huang H (2015) The first complete chloroplast genome sequences in actinidiaceae: genome structure and comparative analysis. PLoS ONE 10:e0129347. https://doi.org/10.1371/journal.pone.0129347
Yao X, Meng X, Meng F, Zhang J, Wu J, Guo X, Xing S (2021) The complete chloroplast genome sequence of Buxus megistophylla Levl. (Buxaceae Dumort.). Mitochondrial DNA B Resour 6:2695–2696. https://doi.org/10.1080/23802359.2021.1966332
Yu K, Zhang T (2013) Construction of customized sub-databases from NCBI-nr database for rapid annotation of huge metagenomic datasets using a combined BLAST and MEGAN approach. PLoS ONE. https://doi.org/10.1371/journal.pone.0059831
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. https://doi.org/10.1101/gr.074492.107
Zhang R, Wang Y, Jin J, Stull G, Bruneau A, Cardoso D, De Queiroz L, Moore M, Zhang S, Chen S, Wang J, Li D, Yi T (2020) Exploration of plastid phylogenomic conflict yields new insights into the deep relationships of Leguminosae. Syst Biol 69:613–622. https://doi.org/10.1093/sysbio/syaa013
Zhu B, Gao Z, Luo X, Feng Q, Du X, Weng Q, Cai M (2019) The complete chloroplast genome sequence of garden cress (Lepidium sativum L.) and its phylogenetic analysis in Brassicaceae family. Mitochondrial DNA B Resour 4:3601–3602. https://doi.org/10.1080/23802359.2019.1677527
Acknowledgements
We would like to thank Genewiz Biotechnology (Suzhou) Co., Ltd., China, for Cp genome sequencing and bioinformatic analysis. We also thank Dr. Zongyou Lv from Shanghai University of Traditional Chinese Medicine of China and Dr. Weimin Jiang from the Hengyang Normal University of China for revising the manuscript, and Ms. Weiwei Wang from Liaocheng University in China for helpful comments on an earlier version of the manuscript.
Funding
This work was supported by the NSF of Anhui Province (Grant No. 1908085MH268), Anhui Collaborative Innovation Project of Universities (Grant No. GXXT-2019-049), Key Natural Science Research Projects in Anhui Universities (Grant No. KJ2019A0453), Foundation of Hunan Key Laboratory for Conservation and Utilization of Biological Resources in the Nanyue Mountainous Region (Grant No. NY20K04), and Anhui Province Science and Technology Major Special Project (Grant No. 202003a06020020, Grant No. 201904f06020018).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest regarding the publication of this paper.
Additional information
Communicated by Anastasios Melis.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, J., Huang, H., Qu, C. et al. Comprehensive analysis of chloroplast genome of Albizia julibrissin Durazz. (Leguminosae sp.). Planta 255, 26 (2022). https://doi.org/10.1007/s00425-021-03812-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00425-021-03812-z