
Abstract
Main conclusion
Deep learning is a promising technology to accurately select individuals with high phenotypic values based on genotypic data.
Abstract
Genomic selection (GS) is a promising breeding strategy by which the phenotypes of plant individuals are usually predicted based on genome-wide markers of genotypes. In this study, we present a deep learning method, named DeepGS, to predict phenotypes from genotypes. Using a deep convolutional neural network, DeepGS uses hidden variables that jointly represent features in genotypes when making predictions; it also employs convolution, sampling and dropout strategies to reduce the complexity of high-dimensional genotypic data. We used a large GS dataset to train DeepGS and compared its performance with other methods. The experimental results indicate that DeepGS can be used as a complement to the commonly used RR-BLUP in the prediction of phenotypes from genotypes. The complementarity between DeepGS and RR-BLUP can be utilized using an ensemble learning approach for more accurately selecting individuals with high phenotypic values, even for the absence of outlier individuals and subsets of genotypic markers. The source codes of DeepGS and the ensemble learning approach have been packaged into Docker images for facilitating their applications in different GS programs.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Genomic selection (GS), originally proposed by Meuwissen et al. (2001) for animal breeding, is regarded as a promising breeding paradigm to better predict the plant or crop phenotypes of polygenic traits using genome-wide markers (Desta and Ortiz 2014; Bhat et al. 2016; Poland and Rutkoski 2016). Unlike both phenotypic and traditional marker-based selection, GS has the inherent advantages of predicting phenotypic trait values of individuals before planting, of estimating the breeding values of individuals before crosses are made, and, notably, of reducing the time length of the breeding cycle (Jannink et al. 2010; Jonas and de Koning 2013; Desta and Ortiz 2014; Yu et al. 2016). Recently, several GS projects have been launched for crop species, namely wheat, maize, rice and cassava (Spindel et al. 2015; Guzman et al. 2016; Marulanda et al. 2016; Poland and Rutkoski 2016). However, the application of GS in the field of practical crop breeding is still nascent, largely because it must overcome the requirement of robust approaches for making accurate predictions in a high-dimensional marker data, where the number of genotypic markers (p) is much larger than the population size (n) (p > > n) (Jannink et al. 2010; Desta and Ortiz 2014; Schmidt et al. 2016; Crossa et al. 2017).
Up to date, various GS prediction models have been developed with traditional statistical algorithms, including BLUP (best linear unbiased prediction)-based algorithms, such as the ridge regression BLUP (RR-BLUP) (Endelman 2011) and the genomic relationship BLUP (GBLUP) (VanRaden 2008), and Bayesian-based algorithms, such as Bayes A, Bayes B, Bayes Cπ and Bayes LASSO (Meuwissen et al. 2001; de los Campos et al. 2009). However, among the different GS prediction models, not much variation in prediction accuracy was frequently observed (Varshney 2016; Roorkiwal et al. 2016). In addition, the GS prediction models typically make strong assumptions and perform linear regression analysis. A representative example is the commonly used RR-BLUP model, which assumes that all the marker effects are normally distributed with a small but non-zero variance, and predicts phenotypes from a linear function of genotypic markers (Xu and Crouch 2008). Thus, these GS models face statistical challenges related to the high dimensionality of marker data, and have difficulty capturing complex relationships within genotypes (e.g., multicollinearity among markers), and between genotypes and phenotypes (e.g., genotype-by-environment-by-trait interaction) (van Eeuwijk et al. 2010; Crossa et al. 2017). Therefore, novel algorithms are urgently needed to augment GS and its potential in plant breeding.
Deep learning (DL) is a recently developed machine-learning technique that builds multi-layered neural networks containing a large number of neurons to model complex relationships in big data (large datasets) (LeCun et al. 2015). DL has proven capable of improved prediction performance over traditional models for speech recognition, image identification and natural language processing (LeCun et al. 2015). Most recently, however, DL has drawn the attention of systems biologists, who have successfully applied it to several prediction problems: the inference of gene expression (Chen et al. 2016; Singh et al. 2016), the functional annotation of genetic variants (Quang et al. 2015; Xiong et al. 2015; Zhou and Troyanskaya 2015; Quang and Xie 2016), the recognition of protein folds (Jo et al. 2015; Wang et al. 2016) and the prediction of genome accessibility (Kelley et al. 2016), of enhancers (Kim et al. 2016; Liu et al. 2016), and of DNA- and RNA-binding proteins (Alipanahi et al. 2015; Zeng et al. 2016; Zhang et al. 2016). These successful applications in the fields of systems biology and computational biology have demonstrated that DL has a powerful capability of learning complex relationships from biological data (Angermueller et al. 2016; Min et al. 2017). However, to the best of our knowledge, the application of DL to GS in plants and other organisms has not yet been investigated.
In this study, we present a DL method, named DeepGS, to predict phenotypes from genotypes using a deep convolutional neural network (CNN). Unlike the conventional statistical models, DeepGS can automatically “learn” complex relationships between genotypes and phenotypes from the training dataset, without pre-defined rules (e.g., normal distribution, non-zero variance) for various variables in the neural network. To avoid overfitting of CNN, DeepGS also takes the advantages of DL technologies to reduce the complexity of high-dimensional marker data through dimensionality reduction using convolution, sampling and dropout strategies. We performed cross-validation experiments on a large GS data of wheat [2000 individuals × 33,709 DArT (Diversity Array Technology) markers; eight phenotypic traits], to evaluate the prediction performance of DeepGS, two BLUP-based GS models (GBLUP and RR-BLUP), and three conventional feed-forward neural network-based GS models. We also proposed an ensemble learning approach to combine the predictions of DeepGS and RR-BLUP for further improving the prediction performance. The effect of outlier individuals and subsets of genotypic markers on the performance of DeepGS, RR-BLUP and the ensemble GS model was also explored. The source codes, user manuals of DeepGS and the ensemble learning approach have been packaged as Docker images for public use.
Materials and methods
GS dataset
The GS dataset used in this study contains 2000 Iranian bread wheat (Triticum aestivum) landrace accessions, each of which was genotyped with 33,709 DArT markers. For the DArT markers, an allele was encoded by either 1 or 0, to indicate its presence or absence, respectively. This dataset includes the phenotypes of eight measured traits, including grain length (GL), grain width (GW), grain hardness (GH), thousand-kernel weight (TKW), test weight (TW), sodium dodecyl sulphate sedimentation (SDS), grain protein (GP), and plant height (PHT). These genotypic and phenotypic data can be obtained from the CIMMYT (The International Maize and Wheat Improvement Center) wheat gene bank (http://genomics.cimmyt.org/mexican_iranian/traverse/iranian/standarizedData_univariate.RData). More details for this GS dataset can be found in (Crossa et al. 2016).
Tenfold cross-validation
Cross-validation is a widely used approach to evaluate the prediction performance of GS models (Resende et al. 2012; Crossa et al. 2016; Gianola and Schön 2016; Qiu et al. 2016). In a tenfold cross-validation experiment, individuals in the whole GS dataset were first randomly partitioned into ten groups with approximately equal size. The GS model was trained and validated using genotypic and phenotypic data of individuals from nine groups (90% individuals for the training set; 10% individuals for the validation set). The trained GS model was subsequently applied to predict phenotypic trait values of individuals from the remaining group (testing set) using only genotypic data. This process was repeated ten times until each group was used once for testing; the predicted phenotypic trait values were finally combined for performance evaluation using the mean normalized discounted cumulative gain value (MNV) (Blondel et al. 2015).
The MNV measures the performance of GS models in selecting the top-ranked k individuals (Blondel et al. 2015). Given n individuals, the predicted and observed phenotypic values form an n × 2 matrix of score pairs (X, Y). The MNV can be calculated in an iterative manner:
where \(d\left( i \right) = 1/(\log_{2} i + 1)\) is a monotonically decreasing discount function at position i; y(i, Y) is the ith value of observed phenotypic values Y sorted in descending order, here y(1, Y) \(\ge y\left( {2,Y} \right) \ge \ldots \; y\left( {n,Y} \right); y\left( {i,X} \right)\) is the corresponding value of Y in the score pairs (X, Y) for the ith value of predicted scores X sorted in descending order. Thus, MNV has a range of 0 to 1 when all the observed phenotypic values are larger than zero; a higher MNV(k, X, Y) indicates a better performance of the GS model to select the top-ranked k (α = k/2000, 1 ≤ k ≤ 2000, 1% ≤ α ≤ 100%) individuals with high phenotypic values.
The entire tenfold cross-validation experiment was repeated ten times with different seeds used to shuffle the order of individuals in the original GS dataset. Thus, for each given level of α, this procedure produced ten different MNV values, and the average was used as the final result.
GS models
Ridge regression-based linear unbiased prediction (RR-BLUP)
RR-BLUP is one of the most extensively used and robust regression models for GS (Wimmer et al. 2013; Bhering et al. 2015; Huang et al. 2016). Given the genotype matrix Z (n × p; n individuals, p markers) and the corresponding phenotype vector Y (n ×1), the GS model is built using the standard linear regression formula:
where μ is the mean of phenotype vector Y, \(g\left( {p \times 1} \right)\) is a vector of marker effects, and ε (n × 1) is the vector of random residual effects. The ridge regress algorithm is used to simultaneously estimate the effects of all genotypic markers, under the assumption that marker effects in \(g\left( {p \times 1} \right)\) follow a normal distribution norm (\(g\sim N ( {0, I\sigma_{g}^{2} } )\)) with a small but non-zero variance (\(I\sigma_{g}^{2}\)) (Whittaker et al. 2000; Endelman 2011; Riedelsheimer et al. 2012; Desta and Ortiz 2014). I is the identity matrix; \(\sigma_{g}^{2}\) is the variance of \(g\). The RR-BLUP model was implemented using the function “mixed.solve” in the R package “rrBLUP” (https://cran.r-project.org/web/packages/rrBLUP).
DeepGS model
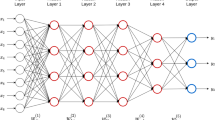
The DeepGS model was built using the DL technique-deep convolutional neural network (CNN) with an 8–32–1-architecture; this included one input layer, one convolutional layer (eight neurons), one sampling layer, three dropout layers, two fully connected layers (32 and one neurons) and one output layer (Fig. 1). The input layer receives the genotypic matrix (1 × p) of a given individual. The first convolutional layer filters the input matrix with eight kernels that are each 1 × 18 in size with a stride size of 1 × 1, followed by a 1 × 4 max-pooling layer with a stride size of 1 × 4. The output of the max-pooling layer is passed to a dropout layer with a rate of 0.2 for reducing overfitting (Srivastava et al. 2014). The first fully connected layer with 32 neurons is used after the dropout layer to join together the convolutional characters with a dropout rate of 0.1. A nonlinearity active function-rectified linear unit (ReLU) is applied in the convolutional and first fully connected layers. The output of the first fully connected layer is then fed to the second fully connected layer with one neural and a dropout rate of 0.05. Using a linear regression model, the output of the second fully connected layer is finally connected to the output layer which presents the predicted phenotypic value of the analyzed individual.

The DeepGS model is a deep convolutional neural network that has an 8–32–1 architecture. ‘ReLU’ indicates the rectified linear unit; Dropout indicates the dropout conduct layer
To avoid overfitting, the DeepGS was trained on the training set and validated on the validation set during each fold of cross-validation. Parameters in the DeepGS were optimized with the back propagation algorithm (Rumelhart et al. 1986), by setting the number of epochs to 6000, the learning rate to 0.01, the momentum to 0.5, and the WD to 0.00001. The back propagation process was prematurely stopped, if the mean absolute difference between predicted and observed phenotypic values on the validation dataset became stable. DeepGS was implemented using the graphics processing unit (GPU)-based DL framework MXNet (version 0.9.3; https://github.com/dmlc/mxnet); it was run on a GPU server that was equipped with four NVIDIA GeForce TITAN-XGPUs, each of which has 12 GB of memory and 3072 CUDA (Compute Unified Device Architecture) cores.
An ensemble GS model based on RR-BLUP and DeepGS
An ensemble GS model (E) was constructed using the ensemble learning approach by linearly combining the predictions of DeepGS (D) and RR-BLUP (R), using the formula.
For each fold of cross-validation, parameters (\(W_{D}\) and \(W_{R}\)) were optimized on the corresponding validation dataset using the particle swarm optimization (PSO) algorithm, which was developed by inspiring from the social behavior of bird flocking or fish schooling (Kennedy and Eberhart 1995). PSO has the capability of parallel searching on very large spaces of candidate solutions, without making assumptions about the problem being optimized. Details of the parameter optimization using the PSO algorithm are given in Online Resource S1.
The source codes and user manuals of DeepGS and the ensemble learning approach have been packaged into two Docker images: one is for central processing unit (CPU) computing (https://hub.docker.com/r/malab/deepgs_cpu), and the other is for GPU computing (https://hub.docker.com/r/malab/deepgs_gpu). These two Docker images provide a smooth and quick way to run DeepGS on a local image since they integrate all dependencies into a standardized software image, overcoming issues related to code changes, dependencies and backward compatibility over time. The project homepage of DeepGS is available at GitHub (https://github.com/cma2015/DeepGS).
Statistical analysis in this study
The Pearson’s correlation coefficient (PCC) was calculated with the function “cor.test” in R programming language (https://www.r-project.org). The Student’s t test was performed using the R function “t.test” to examine the significance level of the difference between paired samples.
Results
Performance comparison between DeepGS and other five GS models
To perform the regression-based GS using neural network algorithms, we were interested in whether or not the DL-based neural network model (DeepGS) was more powerful than the traditional neural network-based GS models and BLUP-based GS models (RR-BLUP and GBLUP). To address this task, three fully connected, feed-forward neural networks (FNNs) with different architectures were built using the matlab function “feedforwardnet”: FNN1 with an 8–32–1 architecture (i.e., eight nodes in the first hidden layer, 32 nodes in the second hidden layer, and one node in the output layer), FNN2 with an 8-1 architecture and FNN3 with an 8–32–10–1 architecture. In each of these three FNNs, nodes in one layer were fully connected to all nodes in the next layer. Two BLUP-based GS models were constructed using RR-BLUP and GBLUP, respectively. The RR-BLUP was implemented using the function “mixed.solve” in the R package “rrBLUP” (https://cran.r-project.org/web/packages/rrBLUP). The GBLUP was implemented using the function “BGLR” in the R package “BGLR” (https://cran.r-project.org/web/packages/BGLR). The tenfold cross-validation with ten replicates was performed to evaluate the performance of DeepGS and FNN for predicting the phenotypic values of the eight tested traits using 33,709 DArT markers.
Figure 2 illustrates the performance of these six GS models (DeepGS, FNN1, FNN2, FNN3, RR-BLUP, GBLUP) in predicting grain length (GL) in one tenfold cross-validation experiment. The experimental results show that DeepGS, RR-BLUP and GBLUP yielded a high correlation between predicted and observed grain lengths, corresponding to a Pearson’s correlation coefficient (PCC) value of 0.742, 0.737 and 0.731, respectively (Fig. 2a). The other three GS models (FNN1, FNN2, and FNN3) yielded relatively low PCC values, corresponding to 0.409, 0.363, and 0.428, respectively. Correspondingly, the predictions of DeepGS, RR-BLUP and GBLUP had markedly lower absolute differences (paired samples t test; p value < 1.0E-59) between observed and predicted phenotypic values compared with those for the FNN1, FNN2, and FNN3 (Fig. 2b). The MNV was further used to evaluate the performance of these six GS models for selecting individuals with long grain length. With top-ranked α increasing from 1% to 100%, the MNV of the DeepGS model (0.42–0.68) was significantly higher than that of RR-BLUP (0.35–0.66) and GBLUP (0.34–0.66), FNN1 (0.26–0.41), FNN2 (0.16–0.34), and FNN3 (0.19–0.41) (Fig. 2c). Online Resource S2 depicts the average MNV curves obtained using the tenfold cross-validation with ten replicates for each of these six GS models. We observed the superiority of DeepGS, RR-BLUP and GBLUP over FNNs (FNN1, FNN2, and FNN3) in terms of MNV for the prediction of GL and the other seven traits under study (Online Resource S2). Of note, a different architecture for the FNN might lead to different results (Fig. 2; Online Resource S2). In addition, due to the comparable performance of GBLUP, RR-BLUP was used as a representative BLUP-based GS model in the following sections.

Performance comparison of six GS models for the prediction grain length using a tenfold cross-validation. a Pearson’s correlation coefficient (PCC) analysis between observed and predicted phenotypic values for different GS models. The analysis results from different GS models are shown in different colors. b Box plot of the absolute errors between the observed and predicted phenotypic values. c MNV curves for different GS models with top-ranked α increasing from 1 to 100%
DeepGS is a complement to RR-BLUP for selecting individuals with high phenotypic values
As reported in the aforementioned section, PCC analysis between observed and predicted phenotypic values showed that DeepGS and RR-BLUP yielded comparable PCC values for each of the eight tested traits (Online Resource S3). Further PCC analysis showed that the PCC value of DeepGS and RR-BLUP strongly decreased in the phenotype prediction for individuals with high phenotypic values (Online Resource S3). When focusing on the top-ranked 400 individuals with the longest grain length, the PCC value of DeepGS and RR-BLUP was decreased to ~ 0.275 (Online Resource S3). These results indicated that neither DeepGS nor RR-BLUP performed particularly well on the individuals with high phenotypic values. Therefore, in the prediction of phenotypes from genotypes, more efforts need to be focused on the individuals with high phenotypic values.
Considering that DeepGS and RR-BLUP used different algorithms to build regression-based GS models, we suspected that they may capture different aspects of the relationships between genotypes and phenotypes. As expected, we observed the differences between these two GS models in their ranking of individuals with different orders. For instance, among the top-ranked 400 (α = 20%) individuals with the longest grain length (purple dots in Fig. 3a), 198 were ranked to be top 400 by both of these two GS models (purple dots in region III in Fig. 3b, c), 32 (purple dots in region I in Fig. 3b, c) and 27 (purple dots in region II in Fig. 3b, c) of which were specifically ranked to be top 400 by DeepGS and RR-BLUP, respectively. This result indicated that DeepGS and RR-BLUP have different strengths in ranking individuals with high phenotypic values for the trait of grain length. This difference was also evident for the prediction of grain length when α varied from 1 to 100% (Fig. 3b), indicating the complementarity between DeepGS and RR-BLUP. Besides grain length, the complementarity was also found in the prediction of the other seven traits under study (Online Resource S4).

Performance comparison of RR-BLUP, DeepGS, and the ensemble GS model for the prediction of grain length. a Comparison of predicted grain length for RR-BLUP and DeepGS. Dots in purple represent the top 400 ranked individuals with the longest grain length. Dots in region I and II denote individuals predicted to be top 400 ranked by DeepGS. Dots in region II and III indicate individuals predicted to be top 400 ranked by RR-BLUP. b Percentages of individuals with the longest grain length identified by RR-BLUP and DeepGS, for which the dashed line represents the α of 20% (i.e., the top-ranked 400 individuals). c Numbers and percentages of top-ranked 400 individuals with longest grain lengths predicted to be top 400 ranked by RR-BLUP and DeepGS. d The absolute increases in MNV of DeepGS and the ensemble GS models over RR-BLUP evaluated using tenfold cross-validation with ten replicates
To make full use of the complementarity between DeepGS and RR-BLUP, we introduced an ensemble GS model that simultaneously considered the predicted phenotypic values from these two GS models based on particle swarm optimization (PSO) algorithms. Experimental results of tenfold cross-validation with ten replicates showed that the ensemble GS model yielded significant higher MNV values than RR-BLUP for all tested traits (except PHT) when top-ranked α ranged from 1 to 100% (paired samples t test; Online Resource S5–S6). Obviously, the ensemble GS model substantially improved the prediction performance over RR-BLUP and DeepGS for GH, TKW, TW, SDS, and PHT (Fig. 3d; Online Resource S5–S6). Compared with RR-BLUP, DeepGS improved the MNVs by 0.46–1.55 × 1.0E-02 for TW, while the ensemble GS model improved the MNVs by 1.13–3.39 × 1.0E-02 for TW (Online Resource S5).
These results indicated that the DeepGS can be used as a supplementary to the RR-BLUP model in selecting individuals with high phenotypic values for all of the eight tested traits.
Outlier individuals and their effects on prediction performance
An outlier individual is one with an extremely high or low phenotypic value for a particular trait under study. These outlier individuals are valuable for breeding programs and for identifying trait-related genes in the bulked sample analysis (Zou et al. 2016). We were interested in how the respective performance of DeepGS and RR-BLUP models might be affected by outlier individuals. For each of the eight traits, the outlier individuals were defined as above 75% quartile (Q3) plus 1.5 times the interquartile range (IQR = Q3–Q1) and below 25% quartile (Q1) minus 1.5 times IQR of phenotypic values. As a result, there were 50, 22, 40, 19, and 65 outlying individuals detected for GL, GW, TW, GP and PHT, respectively (Online Resource S7a). We re-evaluated the performance of RR-BLUP, DeepGS, and the ensemble GS model using the tenfold cross-validation with ten replicates, in which outlying individuals were omitted from the training analysis.
We observed that RR-BLUP and DeepGS are differentially sensitive to outlier individuals (Online Resource S7b). After the removal of outlier individuals, DeepGS still yielded a higher prediction performance than it did by RR-BLUP for all tested five traits at some levels of \(\alpha\) (Fig. 4; Online Resource S7c). As expected, the ensemble GS model always yielded a higher prediction performance than it did by RR-BLUP for all tested five traits at all possible levels of \(\alpha\) (\(1{\text{\% }} \le \alpha \le 100{\text{\% }}\)) (Fig. 4; Online Resource S7c). The corresponding absolute increase in MNV was as high as 14.1 × 1.0E-02 for trait PHT at the level of α = 1% (Online Resource S7c–S8).

MNV curves of RR-BLUP, DeepGS and the ensemble GS models evaluated using the tenfold cross-validation with ten replicates. Outlying individuals were omitted in the training process. a–e Prediction performance for grain length (GL), grain width (GW), thousand-kernel weight (TW), grain protein (GP), and plant height (PHT), respectively
These results indicate that, even after omitting the outlier individuals from the training set, DeepGS and the ensemble GS models outperform RR-BLUP in selecting individuals with high phenotypic values for all tested traits.
Marker number effect on prediction performance
Various technology platforms have been developed to generate genotypic markers with different size. The number of genotypic markers has been reported to have significant influences on the prediction performance of GS models (Heffner et al. 2011). In this analysis, we examined the effect of marker number on prediction performance of RR-BLUP, DeepGS, and the ensemble GS model. For each of the eight tested traits, the tenfold cross-validation experiment was performed using a different number of randomly selected markers at 5000, 10,000, and 20,000. This process was repeated ten times to generate ten MNVs of a given α for each marker number. Their average served as the final prediction performance of the GS models.
When the marker number decreased from 20,000 to 5000, the advantage of DeepGS over RR-BLUP in selecting individuals with high phenotypic values was consistently observed for five of eight tested traits (GL, GW, TKW, GP and PHT), while for the ensemble GS model, the MNV improvement over RR-BLUP could be observed for all tested traits except SDS and GP (Fig. 5). Interestingly, when 5000 markers were used, DeepGS yielded a relatively lower MNV than RR-BLUP at all possible levels of \(\alpha\) ranging from 1 to 100% for GH and SDS (Fig. 5c). However, by combining predictions of DeepGS and RR-BLUP, the ensemble GS model generated higher MNV values than RR-BLUP.

The absolute increase of MNV for DeepGS (red) and the ensemble GS model (green) over RR-BLUP, when a different number of randomly selected markers were used. a 20,000 markers. b 10,000 markers. c 5000 markers
These results indicated that DeepGS outperforms RR-BLUP even when a subset of 33,709 markers was used and could be used as a supplementary to RR-BLUP in selecting individuals with high phenotypic values for most of the tested traits.
Discussion
GS is currently revolutionizing the applications of plant breeding, and novel prediction models are crucial for accurately predicting phenotypes from genotypes (Jannink et al. 2010; Jonas and de Koning 2013; Desta and Ortiz 2014). DL is a recently developed machine-learning technique, which has the capability of capturing complex relationships hidden in big data. In this study, we explored the application of DL in the field of GS. The main contributions are the following: (1) We successfully applied the DL technique to build a novel and robust GS model for predicting phenotypes from genotypes. (2) We implemented the DeepGS model as an open source R package “DeepGS”, thus providing a flexible framework to ease the application of DL techniques in GS. This R package also provides functions to calculate the MNV and to implement the RR-BLUP model as well as the cross-validation procedure. (3) We proposed an ensemble learning approach to get a better performance through combining the predictions of DeepGS and RR-BLUP.
There are two caveats that we should mention for the application of DL in the GS. First, the design of appropriate network architectures requires considerable knowledge of DL and neural network. In this study, the genotyping markers are DArT markers encoded with a binary (1 or 0) allele call. In the convolutional layer, eight neurons were expected to be used to capture the information from paired alleles deriving from four basic features (00, 01, 10, and 11). While in the first fully connected layer, 32 neurons were used to capture information from these four basic features and their high-ordered features (e.g., six features between two of them, three features among three of them, and one feature among all of them). In the second fully connected layer, the input features were summarized to be a feature for the regression output. Therefore, we designed the DeepGS model with an 8–32–1 architecture. We note that a different architecture designed for DeepGS may lead to comparable or superior performance on the GS dataset used in this study (Online Resource S9).
Second, the convolutional, sampling, dropout, and fully connected layers have different sets of hyper-parameters each and thus handle different parts of the data characteristics, resulting in a challenge of understanding the inherent biological significances in the CNN (Angermueller et al. 2016; Chen et al. 2016; Min et al. 2017). We performed a primary analysis to explore the marker effects in the CNN for the prediction of grain length (Online Resource S10), and found that both DeepGS and RR-BLUP identified a proportion of markers with relatively high effects (Online Resource S11). For the top-ranked 1000 markers with the highest effect (absolute value), 24 are common in both GS models. Further investigation of these DeepGS-specific markers with high effects would be helpful in elucidating their contributions to phenotypic variations.
In summary, this research work opens up a new avenue for the application of the DL technique in the field of GS. In the future, we will cooperate with population geneticists and continue to amend our DeepGS to enable it to explain the detected relationships between phenotypes and genotypes. In addition, we will cooperate with crop breeders and carry out practical applications of DeepGS in the GS-based breeding programs of wheat and other vital crops.
Author contribution statement
Designed the experiments: CM. Performed the experiments: WM, JS, ZQ, JL, QC and JZ. Analyzed the data: WM, ZQ, JL, QC and CM. Wrote the paper: CM and WM. All authors read and approved the final manuscript.
Abbreviations
- CNN:
-
Deep convolutional neural network
- DL:
-
Deep learning
- GS:
-
Genomic selection
- MNV:
-
Mean normalized discounted cumulative gain value
- (RR)-BLUP:
-
(Ridge regression)-Best linear unbiased prediction
References
Alipanahi B, Delong A, Weirauch MT, Frey BJ (2015) Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol 33(8):831–838. https://doi.org/10.1038/nbt.3300
Angermueller C, Pärnamaa T, Parts L, Stegle O (2016) Deep learning for computational biology. Mol Syst Biol 12(7):878. https://doi.org/10.15252/msb.20156651
Bhat JA, Ali S, Salgotra RK, Mir ZA, Dutta S, Jadon V, Tyagi A, Mushtaq M, Jain N, Singh PK, Singh GP, Prabhu KV (2016) Genomic selection in the era of next generation sequencing for complex traits in plant breeding. Front Genet 7:221. https://doi.org/10.3389/fgene.2016.00221
Bhering LL, Junqueira VS, Peixoto LA, Cruz CD, Laviola BG (2015) Comparison of methods used to identify superior individuals in genomic selection in plant breeding. Genet Mol Res 14(3):10888–10896. https://doi.org/10.4238/2015.September.9.26
Blondel M, Onogi A, Iwata H, Ueda N (2015) A ranking approach to genomic selection. PLoS One 10(6):e0128570. https://doi.org/10.1371/journal.pone.0128570
Chen Y, Li Y, Narayan R, Subramanian A, Xie X (2016) Gene expression inference with deep learning. Bioinformatics 32(12):1832–1839. https://doi.org/10.1093/bioinformatics/btw074
Crossa J, Jarquín D, Franco J, Pérez-Rodríguez P, Burgueño J, Saint-Pierre C, Vikram P, Sansaloni C, Petroli C, Akdemir D, Sneller C, Reynolds M, Tattaris M, Payne T, Guzman C, Peña RJ, Wenzl P, Singh S (2016) Genomic prediction of gene bank wheat landraces. G3 (Bethesda) 6(7):1819–1834. https://doi.org/10.1534/g3.116.029637
Crossa J, Pérez-Rodríguez P, Cuevas J, Montesinos-López O, Jarquín D, de los Campos G, Burgueño J, Camacho-González JM, Pérez-Elizalde S, Beyene Y, Dreisigacker S, Singh R, Zhang X, Gowda M, Roorkiwal M, Rutkoski J, Varshney RK (2017) Genomic selection in plant breeding: methods, models, and perspectives. Trends Plant Sci 22(11):961–975. https://doi.org/10.1016/j.tplants.2017.08.011
de los Campos G, Naya H, Gianola D, Crossa J, Legarra A, Manfredi E, Weigel K, Cotes JM (2009) Predicting quantitative traits with regression models for dense molecular markers and pedigree. Genetics 182(1):375–385. https://doi.org/10.1534/genetics.109.101501
Desta ZA, Ortiz R (2014) Genomic selection: genome-wide prediction in plant improvement. Trends Plant Sci 19(9):592–601. https://doi.org/10.1016/j.tplants.2014.05.006
Endelman JB (2011) Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 4(3):250. https://doi.org/10.3835/plantgenome2011.08.0024
Gianola D, Schön CC (2016) Cross-validation without doing cross-validation in genome-enabled prediction. G3 (Bethesda) 6(10):3107–3128. https://doi.org/10.1534/g3.116.033381
Guzman C, Peña RJ, Singh R, Autrique E, Dreisigacker S, Crossa J, Rutkoski J, Poland J, Battenfield S (2016) Wheat quality improvement at CIMMYT and the use of genomic selection on it. Appl Transl Genom 11:3–8. https://doi.org/10.1016/j.atg.2016.10.004
Heffner EL, Jannink JL, Sorrells ME (2011) Genomic selection accuracy using multifamily prediction models in a wheat breeding program. Plant Genome 4(1):65–75. https://doi.org/10.3835/plantgenome2010.12.0029
Huang M, Cabrera A, Hoffstetter A, Griffey C, Van Sanford D, Costa J, McKendry A, Chao S, Sneller C (2016) Genomic selection for wheat traits and trait stability. Theor Appl Genet 129(9):1697–1710. https://doi.org/10.1007/s00122-016-2733-z
Jannink JL, Lorenz AJ, Iwata H (2010) Genomic selection in plant breeding: from theory to practice. Brief Funct Genomics 9(2):166–177. https://doi.org/10.1093/bfgp/elq001
Jo T, Hou J, Eickholt J, Cheng J (2015) Improving protein fold recognition by deep learning networks. Sci Rep 5:17573. https://doi.org/10.1038/srep17573
Jonas E, de Koning DJ (2013) Does genomic selection have a future in plant breeding? Trends Biotechnol 31(9):497–504. https://doi.org/10.1016/j.tibtech.2013.06.003
Kelley DR, Snoek J, Rinn JL (2016) Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res 26(7):990–999. https://doi.org/10.1101/gr.200535.115
Kennedy J, Eberhart R (1995) Particle swarm optimization. ICNN 4:1942–1948. https://doi.org/10.1109/icnn.1995.488968
Kim SG, Harwani M, Grama A, Chaterji S (2016) EP-DNN: a deep neural network-based global enhancer prediction algorithm. Sci Rep 6:38433. https://doi.org/10.1038/srep38433
LeCun Y, Bengio Y, Hinton G (2015) Deep learning. Nature 521(7553):436–444. https://doi.org/10.1038/nature14539
Liu F, Li H, Ren C, Bo X, Shu W (2016) PEDLA: predicting enhancers with a deep learning-based algorithmic framework. Sci Rep 6:28517. https://doi.org/10.1038/srep28517
Marulanda JJ, Mi X, Melchinger AE, Xu JL, Würschum T, Longin CF (2016) Optimum breeding strategies using genomic selection for hybrid breeding in wheat, maize, rye, barley, rice and triticale. Theor Appl Genet 129(10):1901–1913. https://doi.org/10.1007/s00122-016-2748-5
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157(4):1819–1829
Min S, Lee B, Yoon S (2017) Deep learning in bioinformatics. Brief Bioinform 18(5):851–869. https://doi.org/10.1093/bib/bbw068
Poland J, Rutkoski J (2016) Advances and challenges in genomic selection for disease resistance. Annu Rev Phytopathol 54:79–98. https://doi.org/10.1146/annurev-phyto-080615-100056
Qiu Z, Cheng Q, Song J, Tang Y, Ma C (2016) Application of machine learning-based classification to genomic selection and performance improvement. In: Huang DS, Bevilacqua V, Premaratne P (eds) Intelligent computing theories and applicaton. Proceedings of the 12th international conference on intelligent computing (ICIC 2016), Lecture notes in computer science, vol 9771, pp 412–421. https://doi.org/10.1007/978-3-319-42291-6_41
Quang D, Xie X (2016) DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res 44(11):e107. https://doi.org/10.1093/nar/gkw226
Quang D, Chen Y, Xie X (2015) DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31(5):761–763. https://doi.org/10.1093/bioinformatics/btu703
Resende MF Jr, Muñoz P, Resende MD, Garrick DJ, Fernando RL, Davis JM, Jokela EJ, Martin TA, Peter GF, Kirst M (2012) Accuracy of genomic selection methods in a standard data set of loblolly pine (Pinus taeda L.). Genetics 190(4):1503–1510. https://doi.org/10.1534/genetics.111.137026
Riedelsheimer C, Technow F, Melchinger AE (2012) Comparison of whole-genome prediction models for traits with contrasting genetic architecture in a diversity panel of maize inbred lines. BMC Genomics 13:452. https://doi.org/10.1186/1471-2164-13-452
Roorkiwal M, Rathore A, Das RR, Singh MK, Jain A, Srinivasan S, Gaur PM, Chellapilla B, Tripathi S, Li Y, Hickey JM, Lorenz A, Sutton T, Crossa J, Jannink JL, Varshney RK (2016) Genome-enabled prediction models for yield related traits in chickpea. Front Plant Sci 7:1666. https://doi.org/10.3389/fpls.2016.01666
Rumelhart DE, Hinton GE, Williams RJ (1986) Learning representations by back-propagating errors. Nature 323(6088):533–536. https://doi.org/10.1038/323533a0
Schmidt M, Kollers S, Maasberg-Prelle A, Großer J, Schinkel B, Tomerius A, Graner A, Korzun V (2016) Prediction of malting quality traits in barley based on genome-wide marker data to assess the potential of genomic selection. Theor Appl Genet 129(2):203–213. https://doi.org/10.1007/s00122-015-2639-1
Singh R, Lanchantin J, Robins G, Qi Y (2016) DeepChrome: deep-learning for predicting gene expression from histone modifications. Bioinformatics 32(17):i639–i648. https://doi.org/10.1093/bioinformatics/btw427
Spindel J, Begum H, Akdemir D, Virk P, Collard B, Redoña E, Atlin G, Jannink JL, McCouch SR (2015) Genomic selection and association mapping in rice (Oryza sativa): effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet 11(2):e1004982. https://doi.org/10.1371/journal.pgen.1004982
Srivastava N, Hinton G, Krizhevsky A, Sutskever I, Salakhutdinov R (2014) Dropout: a simple way to prevent neural networks from overfitting. JMLR 15:1929–1958
van Eeuwijk FA, Bink MC, Chenu K, Chapman SC (2010) Detection and use of QTL for complex traits in multiple environments. Curr Opin Plant Biol 13(2):193–205. https://doi.org/10.1016/j.pbi.2010.01.001
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91(11):4414–4423. https://doi.org/10.3168/jds.2007-0980
Varshney RK (2016) Exciting journey of 10 years from genomes to fields and markets: some success stories of genomics-assisted breeding in chickpea, pigeonpea and groundnut. Plant Sci 242:98–107. https://doi.org/10.1016/j.plantsci.2015.09.009
Wang S, Peng J, Ma J, Xu J (2016) Protein secondary structure prediction using deep convolutional neural fields. Sci Rep 6:18962. https://doi.org/10.1038/srep18962
Whittaker JC, Thompson R, Denham MC (2000) Marker-assisted selection using ridge regression. Genet Res 75(2):249–252. https://doi.org/10.1017/S0016672399004462
Wimmer V, Lehermeier C, Albrecht T, Auinger HJ, Wang Y, Schön CC (2013) Genome-wide prediction of traits with different genetic architecture through efficient variable selection. Genetics 195(2):573–587. https://doi.org/10.1534/genetics.113.150078
Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, Morris Q, Barash Y, Krainer AR, Jojic N, Scherer SW, Blencowe BJ, Frey BJ (2015) The human splicing code reveals new insights into the genetic determinants of disease. Science 347(6218):1254806. https://doi.org/10.1126/science.1254806
Xu Y, Crouch JH (2008) Marker-assisted selection in plant breeding: from publications to practice. Crop Sci 48(2):391. https://doi.org/10.2135/cropsci2007.04.0191
Yu X, Li X, Guo T, Zhu C, Wu Y, Mitchell SE, Roozeboom KL, Wang D, Wang ML, Pederson GA, Tesso TT, Schnable PS, Bernardo R, Yu J (2016) Genomic prediction contributing to a promising global strategy to turbocharge gene banks. Nat Plants 2:16150. https://doi.org/10.1038/nplants.2016.150
Zeng H, Edwards MD, Ge L, Gifford DK, Zeng H, Edwards MD, Ge L, Gifford DK (2016) Convolutional neural network architectures for predicting DNA–protein binding. Bioinformatics 32(12):i121–i127. https://doi.org/10.1093/bioinformatics/btw255
Zhang S, Zhou J, Hu H, Gong H, Chen L, Cheng C, Zeng J (2016) A deep learning framework for modeling structural features of RNA-binding protein targets. Nucleic Acids Res 44(4):e32. https://doi.org/10.1093/nar/gkv1025
Zhou J, Troyanskaya OG (2015) Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods 12(10):931–934. https://doi.org/10.1038/nmeth.3547
Zou C, Wang P, Xu Y (2016) Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnol J 14(10):1941–1955. https://doi.org/10.1111/pbi.12559
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31570371), the Agricultural Science and Technology Innovation and Research Project of Shaanxi Province, China (2015NY011), the Youth 1000-Talent Program of China, the Hundred Talents Program of Shaanxi Province of China, the Innovative Talents Promotion Project of Shaanxi Province of China (2017KJXX-67), and the Fund of Northwest A&F University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
We declare that we have no competing interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ma, W., Qiu, Z., Song, J. et al. A deep convolutional neural network approach for predicting phenotypes from genotypes. Planta 248, 1307–1318 (2018). https://doi.org/10.1007/s00425-018-2976-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-018-2976-9