
Abstract
Main conclusion
Moso bamboo MITEs were genome-wide identified first time, and data shows that MITEs contribute to the genomic diversity and differentiation of bamboo.
Miniature inverted-repeat transposable elements (MITEs) are widespread in animals and plants. There are a large number of transposable elements in moso bamboo (Phyllostachys heterocycla var. pubescens) genome, but the genome-wide information of moso bamboo MITEs is not known yet. Here we identified 362 MITE families with a total of 489,592 MITE-related sequences, accounting for 4.74 % of the moso bamboo genome. The 362 MITE families are clustered into six known and one unknown super-families. Our analysis indicated that moso bamboo MITEs preferred to reside in or near the genes that might be involved in regulation of host gene expression. Of the seven super-families, three might undergo major expansion event twice, respectively, during 8–11 million years ago (mya) ago and 22–28 mya ago; two might experience a long expansion period from 6 to 13 mya. Almost 1/3 small RNAs might be derived from the MITE sequences. Some MITE families generate small RNAs mainly from the terminals, while others predominantly from the central region. Given the high copy number of MITEs, many siRNAs and miRNAs derived from MITE sequences and the preferential insertion of MITE into gene regions, MITEs may contribute to the genomic diversity and differentiation of bamboo.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Miniature inverted-repeat transposable elements (MITEs) are short non-autonomous mobile DNA elements, generally considered to be deletion derivates of autonomous DNA transposable elements. The first MITEs were identified in the mutated maize allele wx-B2 (Bureau and Wessler 1992) and the subsequent studies have revealed that MITEs are predominant in almost all plants and animals. In plants, MITEs are present in tens of thousands of copies throughout the entire genome and influence genomic diversity and differentiation (Wessler et al. 1995; Fattash et al. 2013b). Indeed, MITEs can occupy a major fraction of plant genomes, up to 10 % in rice, 8 % in Medicago, 4 % in Brassica rapa and 0.71 % in Arabidopsis thaliana (Chen et al. 2013).
MITEs often have terminal inverted repeats (TIRs) and target site duplications (TSDs) at the ends of the elements. Based on TIR and TSD sequences, most MITEs are characterized to derive from the autonomous DNA elements, such as PIF/Harbinger (Feschotte and Mouches 2000; Zhang et al. 2004; Zerjal et al. 2009), Tc1/mariner elements (Feschotte et al. 2003), hAT transposons (Moreno-Vazquez et al. 2005; Depra et al. 2012) and Mutator transposons (Yang and Hall 2003). In addition, some MITEs are annotated as unknown super-families due to lack of clear TSD and/or TIR features (Chen et al. 2013). MITEs originated from Tc1/mariner elements and PIF/Harbinger, respectively, named as Stowaway-like MITEs (2-bp, TA) (Bureau and Wessler 1994b) and Tourist-like MITEs (3-bp, TAA) (Bureau and Wessler 1992, 1994a), are especially widespread in grass (Chen et al. 2013; Fattash et al. 2013b).
MITEs are short, typically 70–800 bp in length with rich of AT, They are preferentially inserted into inter-genic, near-genic, intronic regions and exonic regions and play roles in gene regulation and genome evolution. (El Amrani et al. 2002; Santiago et al. 2002; Yang et al. 2005; Oki et al. 2008; Naito et al. 2009; Sampath et al. 2013). A previous study showed that rice mPing elements provide new binding sites for transcription factors or other regulatory proteins, thereby significantly increase the expression level of the genes near mPing insertions (Naito et al. 2009). Many active MITEs have been identified in different species, including mPing (Jiang et al. 2003) and mGing (Dong et al. 2012) in rice, MADE1 (Miskey et al. 2007) in human cell culture, mimp1 in fungus (Dufresne et al. 2007) and dTstu1 in potato (Momose et al. 2010). Some active MITEs have been developed as sources of genetic materials to transfer heterogeneous genes, e.g., Stowaway T7 (Fattash et al. 2013a). Additionally, MITE-related sequences could encode small RNAs which regulate specific target genes at the transcriptional and post-transcriptional levels (Kuang et al. 2009; Cai et al. 2012). In human genome, 20 % of the known miRNAs originate from the transposable elements (Piriyapongsa and Jordan 2007). Small RNAs derived from MITEs through stem-loop structures play an important role in silencing transposable elements (McCue and Slotkin 2012).
Moso bamboo (Phyllostachys heterocycla var. pubescens) is a large woody bamboo that has ecological, economic and cultural values in Asia and accounts for ~70 % of the total bamboo growth area. Moso bamboo genome has the characteristic of a diploid and contains 24 pairs of chromosomes (2n = 48) with a size of 2.075 Gb (Peng et al. 2013). In this study, we performed genome-wide identification of MITEs on the whole genome of moso bamboo. The distribution pattern and polymorphism of MITEs, and their roles in host gene expression regulation were analyzed. Our results provided a solid ground for further understanding and verification of MITE functions in moso bamboo.
Materials and methods
Identification of MITEs in moso bamboo genome
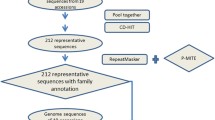
Genomic sequences and gene annotation information of moso bamboo were freely downloaded from the bamboo genome database (BambooGDB, http://www.bamboogdb.org/index.jsp, Zhao et al. 2014a). Considering the large size of moso bamboo genome, MITE Digger was used to identify MITE candidates from moso bamboo genome (Yang 2013) with default parameters. Those elements that share ≥80 % sequence similarity by all vs all Blastn and meet the 80–80–80 rule were grouped into the same MITE family (Wicker et al. 2007). To mine all the remnant copies of each MITE family, the conserved sequences of each family were used as the query to scan the genome sequences by RepeatMasker with a cutoff >250 (http://repeatmasker.org). Cross_Match was served as the search engine. The false results were filtered out based on the criteria of nucleotide identity <80 % and query coverage <80 %.
All MITE families were then searched against the RepBase (version 21.02) (Jurka et al. 2005), P-MITE database (Chen et al. 2013) and Genbank database, respectively, and classified as known and unknown families. The MITE families were also assigned into super-families based on their similarities of TIR and TSD sequences. For the sake of clarity, individual MITE family identified here was designated as PhXY#, where Ph, XY and # represent Moso bamboo (P. heterocycla var. pubescens), the name of the superfamily, and the number of the family, respectively. Word St stands for Stowaway-like, To for Tourist-like, hA for hAT-like, Mi for Micron-like, Mu for Mutator-like, CA for CACTA-like, and Un for Unknown.
Analysis of insertion time and diversity of MITE families
To estimate MITE age, program Clustal W (Larkin et al. 2007) was used to align the DNA sequences of each MITE family, and subsequently DAMBE (Xia and Xie 2001) used for extraction of the consensus sequences of the family. Kimura 2-parameter distance method (Kimura 1980) was applied to estimate the level of nucleotide substitution (k) between each MITE element and the consensus sequence. MITE age was then estimated via the formula T = k/2r, assuming r = 1.30 × 10−8 (Ma and Jackson 2006).
To display MITE intra-family expansion pattern, we adopted the method previously used in maize and silkworm (Zerjal et al. 2009; Han et al. 2010). That is, after alignment of all the full length sequences of each MITE family member, the program Network 4.6 (Bandelt et al. 1999) was applied to construct the median-joining (MJ) networks. The expansion pattern of every family was then evaluated via the network topology.
Estimation of MITE richness in genes
Files for positions of the predicted genes in scaffolds were downloaded from the bamboo genome database (BambooGDB, http://www.bamboogdb.org/index.jsp, Zhao et al. 2014a). Then, a Perl script was written to scan the files to extract and record the information of MITEs that close to or within the predicted genes (i.e., in 5′UTR, intron, exon, 3′UTR). A computer simulation strategy was adopted to detect if MITEs insert accidently close to gene regions (Naito et al. 2006; Han et al. 2010). In brief, the fragments of up to 10 kb were randomly picked from the moso bamboo genome, and the middle of each 10 kb sequence was presumed as the insertion site. The information of the insertion site close to or in the predicted genes was recorded accordingly.
Detection of insertion polymorphism of MITEs in bamboo plantlets
24 seeds were collected from the same moso bamboo, seeded and cultivated into the plantlets. The genomic DNA was prepared using the modified hexadecyltrimethylammonium bromide (CTAB) method (Doyle and Doyle 1987). Some MITEs close to genes were chosen to test of insertion polymorphism and the primers were designed based on the flanking regions of each of the insertion sites (Supplementary Table S1).
Test of transcription level of MITE-related sequences
To analyze transcription level of MITE-related sequences, a total of 127.0 Gb of RNA-seq data was extracted from 7 libraries covering 5 vegetative tissues [the leaf (LF), 20-cm-long shoot (S20), the tip of a 50-cm-long shoot (S50), the rhizome (RH), and the root (RT)], and 2 reproductive tissues [the panicles at the early stage (P1) and flowering stage (P2)] (http://trace.ncbi.nlm.nih.gov/Traces/sra, accession number ERP001341). The downloaded reads were mapped to the moso bamboo genome by Bowtie (-n 2 -l 28 -e 70, Langmead et al. 2009). Based on the location information of MITE-related sequences in the scaffold generated by RepeatMasker, the RPKM (reads per kilobase of exon model per million mapped reads) of MITE-related sequence of every location was counted and normalized by TopHat 2.1.0 (Kim et al. 2011).
Identification of MITE-related small RNAs
To identify small RNAs derived from the MITEs, moso bamboo small RNA data obtained from the rapid elongation culm were downloaded from NCBI database (http://www.ncbi.nlm.nih.gov/sra, SRX398180-SRX398185, He et al. 2013). The siRNAs were then identified by filtering tRNA, rRNA, snoRNA, snRNA, tRNA and some repeat sequences. Their population was divided into two major classes: 20-22nt and 23-25nt, referring hereafter as 21-nt class and 24-nt class. Each of the small RNA sequences was used as the query sequence to scan MITE sequences via Blastn, limiting mismatch less than 2. These MITE-related small RNAs were mapped to the canonical MITE sequences to evaluate their derived positions in MITEs.
Recently, several studies reported the existence of microRNA in moso bamboo (He et al. 2013; Peng et al. 2013; Gao et al. 2014; Xu et al. 2014; Zhao et al. 2014b). After filtering the microRNA that was repeatedly identified, the reported uni-microRNA were collected and used as the query sequence to scan MITE sequence by Blastn, with a mismatch <2. These MITE derived microRNAs were then mapped to the canonical MITE sequences to evaluate their derived positions in MITEs.
To investigate the small RNA mapping ratios, the number of small RNAs that mapped the five specific regions in the MITEs of every family (5′ TIR region, 5′ TIR blanking region (overlap of 5′ TIR region), internal region (no overlap of 5′ and 3′ TIR region), 3′ TIR blanking region (overlap of 3′ TIR region), and 3′ TIR region) was recorded, respectively (A). The number of small RNA derived from every family MITEs was recorded accordingly (B). The mapping ratios were calculated via A/B. To limit the bias caused by small sample size, only elements with similar or same length in a family or the data with more than 100 small RNAs were included and analyzed.
Results
Mining and characterization of MITEs
MITE Digger (Yang 2013), a program identifying candidates based on the features of MITEs (short, TSDs and TIRs structure) and sequence alignment, was first used to search moso bamboo genome sequence. 369 MITE candidates were then identified. Among them, 7 pseudo-MITEs with the copy number in genome <20 were arbitrarily filtered out. By all vs all Blastn, the remaining 362 MITE families were classed based on 80–80–80 rule (the representative sequences and their structure information were provided as Supplementary Table S2) (Wicker et al. 2007). Based on the TSD patterns and TIR sequences, they were then classified into five super-families including Stowaway-like MITEs, hAT-like MITEs, Tourist-like MITEs, Mutator-like MITEs, and CACTA-like MITEs (Table 1). Besides the five super-families, Micron MITE family, which was inserted specifically into the (TA)n repeats with specific TIR sequences, was considered as an independent type (Akagi et al. 2001). Additionally, due to ambiguous TSD and/or TIR features, some MITEs were annotated as unknown super-families (Table 1). Normally, TSD lengths of moso bamboo MITEs range from 2 to 10 bp and TIR lengths from 8 to 57 bp.
To retrieve partial or intact ones, the elements with typical length and structure from each of the family member were pooled together as a reference library and used to search the moso bamboo genome via RepeatMasker. A total of 489,592 MITE-related sequences were obtained from the moso bamboo genome. Among the six known MITE super-families, the content of hAT-like MITEs in moso bamboo genome is the highest (0.65 %), followed by Tourist-like MITEs (0.61 %) and Stowaway-like MITEs (0.19 %). The total content of all the MITE-related sequences in the moso bamboo genome is close to 4.74 %, lower than Oryza sativa (10 %, Chen et al. 2013).
MITE has intact and truncated form: individual with both complete TIRs (full length) is intact, otherwise truncated. In all the MITE families, we identified and confirmed 129,668 (26.48 %) elements to be intact. The ratio of the intact MITEs to the truncated MITEs in individual superfamily varies from 31 to 52 %. Tourist-like MITE super-family is the only one with the ratio beyond 50 %.
As shown in the Supplementary Table S3, the average AT content for each MITE family varies widely, from 28.25 to 72.13 %, and 66.85 % MITE families have AT content >57 %. As a reference, the average AT content of the moso bamboo genome is approximately 56.1 % (Peng et al. 2013).
Estimation of the insertion date and diversity
We estimated the age of each intact MITE by the method adopted for maize and silkworm MITE families (Zerjal et al. 2009; Han et al. 2010). The results showed that the insertion dates vary greatly among seven super-families, ranging from 0 to 43 million years ago (mya) (Fig. 1). Strikingly, three super-families, Mutator-like, Tourist-like, and hAT-like MITEs, might undergo two major expansion events during 8–11 mya ago and 22–28 mya ago, respectively. Stowaway-like and the Unknown MITEs super-families might experience a long expansion period from 6 to13 mya (Fig. 1).

Distribution pattern of insertion dates of the senven MITE superfamilies in moso bamboo genome. The age of each MITE was calculated using the formula T = k/2r. The number of MITEs in each superfamily with different insertion dates is shown in at the Y-axis. The mya is shown at the X-axis with an interval of one mya. The seven MITE super-families are labeled with different colors
To evaluate the intra-family diversity pattern for each MITE family, a network map was constructed base on the alignment of the full length sequences. In case the map center is covered by scattered numerous nodes separated with long branches, a population amplification may be indicated to occur a long time ago. In contrast, a map center covered by nodes encircled by many short branches suggests a recent amplification (Jobling et al. 2004; Zerjal et al. 2009). In the current study, the topologies of every MITE families demonstrated a consistency with the estimated insertion time (Figs. 2, 3). Indeed, the topologies of both PhhAT1 and PhTo1 showed two main population expansions (Fig. 2). And during the two periods, they accumulated their copies as suggested by the insertion dates of PhhAT1 and PhTo1 (Fig. 3).

Median-joining networks of PhhAT1 (a) and PhTo1 (b) MITE families. The circle, circle area and branch length represent MITE sequence, proportional to the number of identical copies and proportional to the number of nucleotide changes, respectively. The network map with numerous nodes scattered around its center and separated by long branches indicates that this MITE family might experience a population amplification a long time ago. In contrast, a network map with a central node encircled by many short branches, shows an amplification from an ancestral element recently (Zerjal et al. 2009; Han et al. 2010)

Distribution pattern of insertion dates of PhhAT1and PhTo1MITE families in moso bamboo genome. The number of MITEs with different insertion dates is shown at the Y-axis. The mya is indicated at the X-axis with an interval of 1 mya
Estimation of MITE richness in genes
We next examined whether the insertion site of each MITE preferentially was in or close to genes. If a MITE inserts into within the 5 kb flanking regions of a gene, it is regarded as close to the gene (Naito et al. 2006; Saito et al. 2006; Kawaoka et al. 2008; Zerjal et al. 2009). In the current case, a larger number of MITEs are inserted into the gene regions (exon, intron) or flanking regions (Table 2). The ratio of predicted MITEs inserted into gene regions (30.94 %) is significant higher than the control (random insertion, 19.44 %, P < 0.05, Chi-square test, same below). In detail, the ratios of MITEs located in the upstream of the closest genes (<2000 bp, 6.317 %), or in the downstream of the closest genes (<2000 bp, 5.160 %), are significant higher than these of the control (1.88, 1.76 %, respectively, P < 0.001). It appears that most moso bamboo MITEs are preferentially inserted in the promoter regions and 3′-flanking regions rather than in the introns and exons. Among the seven super-families, the first three super-families that prefer to be inserted into gene regions are hAT-like (40.564 %), Mutator-like (39.247 %), Stowaway-like MITEs (38.803 %).
Transcription pattern of MITE-related sequences
After filtering out the MITE-related sequences with low reads per kilobase of exon model per million mapped reads (RPKM < 1), there left 19,012 MITE-related sequences (3.88 %) which were detected to transcribe in at least two tissues. Among all the seven super-families, micron-like MITEs showed the highest expressed ratio (the total of expressed MITE-related sequences over the total of MITE-related sequences in corresponding superfamily) (Fig. 4). While among the 12 tissues, shoot with 20 cm height demonstrated the highest expressed ratio for MITE-related sequences (Fig. 4). The relative locations of the expressed MITE-related sequences impact the expressed ratio. MITE-related sequences that close to genes, such as in exon, intron and 3` UTR regions, showed a significant higher expressed ratio than the sequences far from genes (>5000 bp, P < 0.001, Chi-square test, Fig. 5). The expressed ratios tend to diminish with increasing distance of the expressed MITE-related sequences to the closest genes.

Statistical analysis of expressed MITE-related sequences of every super-family. The ratio of the total number of expressed MITE-related sequences of every super-family over the total number of corresponding super-family MITE-related sequences is shown at the Y-axis. The seven tissues, the tip of a 20-cm-long shoot (S20), the tip of a 50-cm-long shoot (S50), the rhizome (RH), the root (RT), the panicle at the early stage (P1), the panicle at the flowering stage (P2), and the leaf (LF), are labeled with different colors

Statistical analysis of expressed MITE-related sequences located at different regions of genes. The ratio of the total number of expressed MITE-related sequences located at different regions of genes over the total number of MITE-related sequences located at corresponding regions is shown at the Y-axis. Seven tissues, the tip of a 20-cm-long shoot (S20), the tip of a 50-cm-long shoot (S50), the rhizome (RH), the root (RT), the panicle at the early stage (P1), the panicle at the flowering stage (P2), and the leaf (LF), are marked by different colors
The small RNAs derived from MITE sequences in moso bamboo
We have identified a total of 23,154 siRNAs in the rapid grow internodes of moso bamboo. 3,868 of them belong to 21-nt class and 19,286 to 24-nt class. Among them, 28.5 % of the 21-nt siRNAs (1102) and 32.2 % of the 24-nt siRNAs (6205) are derived from the MITE sequences. To analyze the preference of siRNA derivation, ratio of the total siRNAs derived from the individual MITE super-family over the total of MITE derived siRNAs was calculated. To limit the bias caused by small sample size, the ratio of small RNAs derived from the CACTA-like and micron-like MITE-related sequences were not analyzed as there are few families in both of them. As can be seen from Fig. 6, Tourist and hAT-like MITEs generate most siRNAs.

21-nt siRNA (blue), 24-nt siRNA (red) and miRNA (gray) derived from individual MITEs superfamily. The content in percentage of the number of small RNAs derived from every MITE superfamily in the total number of MITE derived corresponding small RNAs is shown
Previously studies identified 2297 Uni-miRNA in the rapid elongation culm (He et al. 2013), in leaf (Gao et al. 2014; Xu et al. 2014; Zhao et al. 2014b), and in root (Xu et al. 2014). 29.1 % of them (668) are derived from the MITE-related sequences, with Tourist and hAT-like MITEs contributing the most (Fig. 6).
Derivation position polymorphism of MITE-related small RNAs in different MITE super-families
We further investigated the distribution of small RNAs in MITE sequences. Based on the structure, MITEs were divided five regions, in turn including 5′ TIR region, 5′ TIR blanking region (overlap of 5′ TIR region), internal region (no overlap of 5′ and 3′ TIR region), 3′ TIR blanking region (overlap of 3′ TIR region), and 3′ TIR region. Strikingly, the mapping positions of small RNAs in different MITE super-families vary dramatically. For the 21-nt siRNAs, they mainly position in the 5′ and 3′ TIR regions of Stowaway and hAT-like MITEs, while predominantly in the internal region of Mutator-like, Tourist-like and the Unknown MITEs (Fig. 7a). For the 24-nt siRNAs and miRNAs, they mainly position in the 5′ and 3′ TIR regions of Stowaway and Mutator-like MITEs, while predominantly in the internal region of hAT-like, Tourist-like and the Unknown MITEs (Fig. 7b, c).

21-nt siRNA (a), 24-nt siRNA (b) and miRNA (c) derived from the relative position of MITEs. The ratio of the number of small RNA derived from the different position of MITEs over the total number of corresponding small RNA derived from corresponding MITE superfamily is shown. Individual superfamily is marked by different color
Insertion polymorphism of a MITE family
The predicted 30 insertion sites of PhTo25 were selected for verification by PCR. For 24 moso bamboo half sibling seedlings, the insertion sites that contain the intact MITEs or transposition footprints were detected. Sequencing of the corresponding PCR products confirmed the presence or absence of PhTo25, attesting the predictions by the program MITE Digger.
Of the detected 30 insertion sites, only one locus (PhTo25-3) showed polymorphism in all the 24 seedlings. 15 seedlings contained one insertion site and one absence site, 4 seedlings had only insertion sites with the remaining 5 seedlings only the absence sites (Fig. 8).

PCR results of PhTo25 in genomes of 24 Ph. edulis half-sib seedlings. The PCR products close to 600 bp show the deletion of PhTo25. The PCR products close to 1000 bp show the presence of PhTo25 in the genomic location. Numbers represent individual seedlings. M, DNA molecular marker
Discussion
Discovery and characterization of moso bamboo MITEs
In this study, we carried out a systematic and genome-wide analysis to search for MITEs in moso bamboo using MITE Digger (Yang 2013). 362 moso bamboo MITE families were identified and classed into 7 super-families. Moso bamboo MITEs show a huge diversification in TSD patterns, TIR sequences and full length sequences (Table 1). Most families exhibit MITE-specific features: high number of copies and high AT content. This also validated the use of MITE Digger in the prediction of moso bamboo MITEs. Existence of MITEs in the bamboo was also verified via PCR and sequencing.
Previous studies have shown that number of MITEs varies dramatically with species, but still significantly correlates with the genome assembly size (Chen et al. 2013). Papaya has a genome size of 342.68 Mb, and contains only one MITE family with 538 MITE-related sequences. Apple is larger in genome (881.28 Mb) and contains more MITE families (180 with 237,302 MITE-related sequences, Chen et al. 2013). In moso bamboo genome, 362 MITE families with 489,592 MITE-related sequences are identified (Table 1). The number may be reasonable considering bamboo’s genome size of 2.075 Gb.
It should be pointed out that some MITEs may be missed due to the incompleteness of moso bamboo genome. Moso bamboo genome assembly is highly fragmented with over 1 million contigs across the 2.075 Gb genome. Although we combined the structure-based and homology-based approaches in the identification of MITEs, the fragmentation of moso bamboo genome could still cause an underestimation of MITE contents. Furthermore, the existence of a large proportion of repetitive sequences in moso bamboo may impair the identification of MITEs. Thereby, the 489,592 MITE-related sequences identified here may be a conservative figure and more would be found in the future. In fact, our reported percentage of MITE elements in bamboo is obviously lower (~4.7 %) than rice (10 %), this could be due to the low assembly quality of bamboo and the true MITE proportion could be equal to or higher than rice (Chen et al. 2013).
Expansion and diversity pattern of moso bamboo MITEs
The ages of the seven MITE super-families vary greatly (Fig. 1), ranging from 0 to 43 mya ago. Three families might undergo two major expansion events during 8–11 mya ago and 22–28 mya ago, and another two super-families might experience a major expansion event during 6–13 mya (Fig. 1).
For intra MITE families, the diversity analysis indicated that MITE family members are similar in both sequences and sequence lengths. As pointed by Zerjal et al. (2009), this corresponds to a population that experienced several successive steps of amplifications from ancestors. The network topology analysis confirmed that many MITE families have experienced several expansions recently. It is possible that MITE amplifications in moso bamboo genome remained active sporadically and inactive most of the time (Figs. 2, 3). Activation of MITEs may be trigged by the “genome shock” or temporal activation of the cognate transposase (McClintock 1984). Indeed in rice, irradiation, cell culture, or recent domestication, all can activate mPing (Jiang et al. 2003; Nakazaki et al. 2003; Naito et al. 2006).
Distribution of MITEs in moso bamboo genome
Our analysis suggested that moso bamboo MITEs are widely distributed in the genome and preferentially inserted into gene regions, similar to MITEs in other higher organisms such as O. sativa (Jiang et al. 2004). We noted that more moso bamboo MITEs distribute on both upstream and downstream of the closest genes (<2000 bp, 6.317, 5.160 %, respectively) than on the regions distant from the genes (Table 2). It is possible that MITE insertions in intergenic regions are rapidly purged out from a population because they are deleterious (Oki et al. 2008; Hollister and Gaut 2009). Many MITEs have been found to contain poly (A) signal (Bureau and Wessler 1994b), they are likely to be maintained in the 3′ flanking regions to act in regulation. Existence of a large number of moso bamboo MITE insertions in the upstream of the closest genes implies that MITEs play important roles in gene expression by altering regulatory motifs. Due to their high copy numbers, there is a good chance that more MITEs in gene regions would be verified to be functional such as providing regulatory sequences or recruiting epigenetic modifications.
Abundant MITE-derived small RNAs
The siRNAs generated in plants mainly include the 21-nt class and the 24-nt class. The former is known for its regulation of post-transcriptionally related mRNAs while the latter suppresses gene expression at transcriptional level via RNA-dependent DNA methylation and heterochromatin maintenance (Baulcombe 2004).
Our analysis indicates that MITE sequences generate almost 30 % of all small RNAs in moso bamboo, and 60.7 % of all siRNAs, similar to those from O. sativa (Lu et al. 2012; Cantu et al. 2010).
The positions of small RNAs on the MITE sequences from different MITE superfamilies vary dramatically. Some MITE superfamilies produce MITE from their terminals, with relative few small RNAs from the central regions. Others produce small RNAs mainly from their central regions (Fig. 7), with relative few from the terminals. The similar phenomenon is also observed in rice (Lu et al. 2012).
As they are potentially highly mutagenic, the activity of MITEs is usually controlled by the host genome through the siRNA machinery. The specificity of this response is achieved by a surveillance system that detects aberrant RNA (Liu et al. 2004). The proliferative nature of transposable elements makes them prone to insert in the genome in such a way that both sense and anti-sense transcripts are produced, generating dsRNA, and activating the siRNA system (Hollister et al. 2011).
Given the high copy number of MITEs, many siRNAs and miRNAs derived from MITEs and their preferential insertion into gene regions, it will be important to systematically investigate the formation mechanisms of different MITE families and their potential functions in the transcriptional regulation of genes. It is possible that there exist a set of genes behind each genotype and they are regulated by certain MITEs families, whose diversity may in turn contribute to phenotypic diversity in species.
Conclusions
Although MITEs in many higher plant organisms have been extensively investigated, little is known in moso bamboo. Here we identified 362 moso bamboo MITE families by using the available bamboo genome sequences and the recently developed algorism. Analysis of the nucleotide compositions of TSD and TIR indicated that they can be classified into six known and one unknown super-families. Further comparison among the families revealed an evolutionary pathway for MITEs. Importantly, not only did we reveal that MITEs are preferentially inserted into or near genes but also found that close to 1/3 small RNAs might be derived from the MITE-related sequences. On the basis of having shown the roles of MITEs in the transcriptional regulation of genes, further studies will advance our understanding of their mechanism of action and interactions with host genomes.
Author contribution statement
M. B. Zhou designated the experiments, identified and classified the MITEs, and wrote paper; G. Y. Tao and P. Y. Pi estimated MITE insertion sites; Y. H. Zhu and Y. H. Bai analyzed MITE-related sequence expression and performed PCR; X. W. Meng identified siRNA; all authors read and approved the manuscript.
Abbreviations
- MITEs:
-
Miniature inverted-repeat transposable elements
- TIRs:
-
Terminal inverted repeats
- TSDs:
-
Target site duplications
- Mya:
-
Million years ago
References
Akagi H, Yokozeki Y, Inagaki A, Mori K, Fujimura T (2001) Micron, a microsatellite-targeting transposable element in the rice genome. Mol Genet Genomics 266:471–480
Bandelt HJ, Forster P, Rohl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Baulcombe D (2004) RNA silencing in plants. Nature 431:356–363
Bureau TE, Wessler SR (1992) Tourist: a large family of small inverted repeat elements frequently associated with maize genes. Plant Cell 4:1283–1294
Bureau TE, Wessler SR (1994a) Mobile inverted-repeat elements of the Tourist family are associated with the genes of many cereal grasses. Proc Natl Acad Sci USA 91:1411–1415
Bureau TE, Wessler SR (1994b) Stowaway: a new family of inverted repeat elements associated with the genes of both monocotyledonous and dicotyledonous plants. Plant Cell 6:907–916
Cai Y, Zhou Q, Yu C, Wang X, Hu S, Yu J, Yu X (2012) Transposable-element associated small RNAs in Bombyx mori genome. PLoS One 7:e36599
Cantu D, Vanzetti LS, Sumner A, Dubcovsky M, Matvienko M, Distelfeld A, Michelmore RW, Dubcovsky J (2010) Small RNAs, DNA methylation and transposable elements in wheat. BMC Genom 11:408
Chen J, Hu Q, Zhang Y, Lu C, Kuang H (2013) P-MITE: a database for plant miniature inverted-repeat transposable elements. Nucleic Acids Res 42:1176–1181
Depra M, Ludwig A, Valente VL, Loreto EL (2012) Mar, a MITE family of hAT transposons in Drosophila. Mob DNA 3:13
Dong HT, Zhang L, Zheng KL, Yao HG, Chen J, Yu FC, Yu XX, Mao BZ, Zhao D, Yao J, Li DB (2012) A Gaijin-like miniature inverted repeat transposable element is mobilized in rice during cell differentiation. BMC Genom 13:135
Doyle JJ, Doyle JL (1987) A rapid isolation procedure for small quantities of fresh leaf materials. Phytochem Bull 19:11–15
Dufresne M, Hua-Van A, El Wahab HA, Ben M’Barek S, Vasnier C, Teysset L, Kema GH, Daboussi MJ (2007) Transposition of a fungal miniature inverted-repeat transposable element through the action of a Tc1-like transposase. Genetics 175:441–452
El Amrani A, Marie L, Ainouche A, Nicolas J, Couee I (2002) Genome-wide distribution and potential regulatory functions of AtATE, a novel family of miniature inverted-repeat transposable elements in Arabidopsis thaliana. Mol Genet Genomics 267:459–471
Fattash I, Bhardwaj P, Hui C, Yang G (2013a) A rice Stowaway MITE for gene transfer in yeast. PLoS One 8:e64135
Fattash I, Rooke R, Wong A, Hui C, Luu T, Bhardwaj P, Yang G (2013b) Miniature inverted-repeat transposable elements: discovery, distribution, and activity. Genome 56:475–486
Feschotte C, Mouches C (2000) Evidence that a family of miniature inverted-repeat transposable elements (MITEs) from the Arabidopsis thaliana genome has arisen from a pogo-like DNA transposon. Mol Biol Evol 17:730–737
Feschotte C, Swamy L, Wessler SR (2003) Genome-wide analysis of mariner-like transposable elements reveals complex relationships with Stowaway MITEs. Genetics 143:747–758
Gao J, Zhang Y, Zhang C, Qi F, Li X, Mu S, Peng Z (2014) Characterization of the floral transcriptome of Moso bamboo (Phyllostachys edulis) at different flowering developmental stages by transcriptome sequencing and RNA-seq analysis. PLoS One 9:e98910
Han MJ, Shen YH, Gao YH, Chen LY, Xiang ZH, Zhang Z (2010) Burst expansion, distribution and diversification of MITEs in the silkworm genome. BMC Genom 11:520
He CY, Cui K, Zhang JG, Duan AG, Zeng YF (2013) Next-generation sequencing-based mRNA and microRNA expression profiling analysis revealed pathways involved in the rapid growth of developing culms in Moso bamboo. BMC Plant Biol 13:119
Hollister JD, Gaut BS (2009) Epigenetic silencing of transposable elements: a trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res 19:1419–1428
Hollister JD, Smith LM, Guo YL, Ott F, Weigel D, Gaut BS (2011) Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc Natl Acad Sci USA 108:2322–2327
Jiang N, Bao Z, Zhang X, Hirochika H, Eddy SR, McCouch SR, Wessler SR (2003) An active DNA transposon family in rice. Nature 421:163–167
Jiang N, Feschotte C, Zhang X, Wessler SR (2004) Using rice to understand the origin and amplification of miniature inverted repeat transposable elements (MITEs). Curr Opin Plant Biol 7:115–119
Jobling MA, Hurles ME, Tyler-Smith C (2004) Human evolutionary genetics. Garland, Abingdon and New York
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110:462–467
Kawaoka S, Hayashi N, Katsuma S, Kishino H, Kohara Y, Mita K, Shimada T (2008) Bombyx small RNAs: genomic defense system against transposons in the silkworm, Bombyx mori. Insect Biochem Mol Biol 38:1058–1065
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2011) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. doi:10.1186/gb-2013-14-4-r36
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kuang H, Padmanabhan C, Li F, Kamei A, Bhaskar PB, Ouyang S, Jiang J, Buell CR, Baker B (2009) Identification of miniature inverted- repeat transposable elements (MITEs) and biogenesis of their siRNAs in the Solanaceae: new functional implications for MITEs. Genome Res 19:42–56
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:25
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettiga PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Liu J, He Y, Amasino R, Chen X (2004) siRNAs targeting an intronic transposon in the regulation of natural flowering behavior in Arabidopsis. Genes Dev 18:2873–2878
Lu C, Chen J, Zhang Y, Hu Q, Su W, Kuang H (2012) Miniature inverted-repeat transposable elements (MITEs) have been accumulated through amplification bursts and play important roles in gene expression and species diversity in Oryza sativa. Mol Biol Evol 29:1005–1017
Ma J, Jackson SA (2006) Retrotransposon accumulation and satellite amplification mediated by segmental duplication facilitate centromere expansion in rice. Genome Res 16:251–259
McClintock B (1984) The significance of responses of the genome to challenge. Science 226:792–801
McCue AD, Slotkin RK (2012) Transposable element small RNAs as regulators of gene expression. Trends Genet 28:616–623
Miskey C, Papp B, Mates L, Sinzelle L, Keller H, Izsvak Z, Ivics Z (2007) The ancient mariner sails again: transposition of the human Hsmar1 element by a reconstructed transposase and activities of the SETMAR protein on transposon ends. Mol Cell Biol 27:4589–4600
Momose M, Abe Y, Ozeki Y (2010) Miniature inverted-repeat transposable elements of Stowaway are active in potato. Genetics 186:59–66
Moreno-Vazquez S, Ning J, Meyers BC (2005) hATpin, a family of MITE-like hAT mobile elements conserved in diverse plant species that forms highly stable secondary structures. Plant Mol Biol 58:869–886
Naito K, Cho E, Yang G, Campbell MA, Yano K, Okumoto Y, Tanisaka T, Wessler SR (2006) Dramatic amplification of a rice transposable element during recent domestication. Proc Natl Acad Sci USA 103:17620–17625
Naito K, Zhang F, Tsukiyama T, Saito H, Hancock CN, Richardson AO, Okumoto Y, Tanisaka T, Wessler SR (2009) Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 461:1130–1134
Nakazaki T, Okumoto Y, Horibata A, Yamahira S, Teraishi M, Nishida H, Inoue H, Tanisaka T (2003) Mobilization of a transposon in the rice genome. Nature 421:170–172
Oki N, Yano K, Okumoto Y, Tsukiyama T, Teraishi M, Tanisaka T (2008) A genome-wide view of miniature inverted-repeat transposable elements (MITEs) in rice, Oryza sativa ssp. japonica. Genes Genet Syst 83:321–329
Peng Z, Lu Y, Li L, Zhao Q, Feng Q, Gao Z, Lu H, Hu T, Yao N, Liu K, Li Y, Fan D, Guo Y, Li W, Lu Y, Weng Q, Zhou C, Zhang L, Huang T, Zhao Y, Zhu C, Liu X, Yang X, Wang T, Miao K, Zhuang C, Cao X, Tang W, Liu G, Liu Y, Chen J, Liu Z, Yuan L, Liu Z, Huang X, Lu T, Fei B, Ning Z, Han B, Jiang Z (2013) The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat Genet 45:456–461
Piriyapongsa J, Jordan IK (2007) A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One 2:e203
Saito K, Nishida KM, Mori T, Kawamura Y, Miyoshi K, Nagami T, Siomi H, Siomi MC (2006) Specific association of Piwi with rasiRNAs derived from retrotransposon and heterochromatic regions in the Drosophila genome. Genes Dev 20:2214–2222
Sampath P, Lee SC, Lee J, Izzah NK, Choi BS, Jin M, Park BS, Yang TJ (2013) Characterization of a new high copy Stowaway family MITE, BRAMI-1 in Brassica genome. BMC Plant Biol 13:56. doi:10.1186/1471-2229-13-56
Santiago N, Herráiz C, Goñi JR, Messeguer X, Casacuberta JM (2002) Genome-wide analysis of the Emigrant family of MITEs of Arabidopsis thaliana. Mol Biol Evol 19:2285–2293
Wessler SR, Bureau TE, White SE (1995) LTR-retrotransposons and MITEs: important players in the evolution of plant genomes. Curr Opin Genetics 5:814–821
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8:973–982
Xia X, Xie Z (2001) DAMBE: software package for data analysis in molecular biology and evolution. J Hered 92:371–373
Xu P, Mohorianu I, Yang L, Zhao H, Gao Z, Dalmay T (2014) Small RNA profile in moso bamboo root and leaf obtained by high definition adapters. PLoS One 9:e103590
Yang G (2013) MITE Digger, an efficient and accurate algorithm for genome wide discovery of miniature inverted repeat transposable elements. BMC Bioinform 14:186
Yang G, Hall TC (2003) MDM-1 and MDM-2: two mutator-derived MITE families in rice. J Mol Evol 56:255–264
Yang G, Lee YH, Jiang Y, Shi X, Kertbundit S, Hall TC (2005) A two-edged role for the transposable element Kiddo in the rice ubiquitin2 promoter. Plant Cell 17:1559–1568
Zerjal T, Joets J, Alix K, Grandbastien MA, Tenaillon MI (2009) Contrasting evolutionary patterns and target specificities among three Tourist-like MITE families in the maize genome. Plant Mol Biol 71:99–114
Zhang X, Jiang N, Feschotte C, Wessler SR (2004) PIF- and Pong-like transposable elements: distribution, evolution and relationship with Tourist-like miniature inverted-repeat transposable elements. Genetics 166:971–986
Zhao H, Peng Z, Fei B, Li L, Hu T, Gao Z, Jiang Z (2014) BambooGDB: a bamboo genome database with functional annotation and an analysis platform. Database (Oxford) 2014: bau006
Zhao H, Wang L, Dong L, Sun H, Ga Z (2014b) Discovery and comparative profiling of microRNAs in representative monopodial bamboo (Phyllostachys edulis) and sympodial bamboo (Dendrocalamus latiflorus). PLoS One 9:e102375
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Funding
This work was supported by the grant from the National Natural Science Foundation of China (Grant nos. 31270645 and 31470615), and through Talents Program of Natural Science Foundation of Zhejiang Province (Grant no. LR12C16001).
Electronic supplementary material
Below is the link to the electronic supplementary material.
425_2016_2544_MOESM2_ESM.xls
Supplementary Table S2 The structural characteristics, sequences and copy number of the representative sequences of 362 MITE families (XLS 242 kb)
Rights and permissions
About this article

Cite this article
Zhou, M., Tao, G., Pi, P. et al. Genome-wide characterization and evolution analysis of miniature inverted-repeat transposable elements (MITEs) in moso bamboo (Phyllostachys heterocycla). Planta 244, 775–787 (2016). https://doi.org/10.1007/s00425-016-2544-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00425-016-2544-0