
Abstract
Trigeminal neuralgia is a rare and debilitating disorder that affects one or more branches of the trigeminal nerve, leading to severe pain attacks and a poor quality of life. It has been reported that the CaV3.1 T-type calcium channel may play an important role in trigeminal pain and a recent study identified a new missense mutation in the CACNA1G gene that encodes the pore forming α1 subunit of the CaV3.1 calcium channel. The mutation leads to a substitution of an Arginine (R) by a Glutamine (Q) at position 706 in the I-II linker region of the channel. Here, we used whole-cell voltage-clamp recordings to evaluate the biophysical properties of CaV3.1 wild-type and R706Q mutant channels expressed in tsA-201 cells. Our data indicate an increase in current density in the R706Q mutant, leading to a gain-of-function effect, without changes in the voltage for half activation. Moreover, voltage clamp using an action potential waveform protocol revealed an increase in the tail current at the repolarization phase in the R706Q mutant. No changes were observed in the voltage-dependence of inactivation. However, the R706Q mutant displayed a faster recovery from inactivation. Hence, the gain-of-function effects in the R706Q CaV3.1 mutant have the propensity to impact pain transmission in the trigeminal system, consistent with a contribution to trigeminal neuralgia pathophysiology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Trigeminal neuralgia (TN) is the most common type of craniofacial neuropathic pain, and it is defined as a sudden unilateral severe shock-like pain in one or more branches of the trigeminal nerve [4, 14]. TN is a rare condition, with a prevalence of 4 to 8 cases per 100,000 patients/year with some reported familial occurrences, suggesting a genetic component for TN [13, 19]. TN is classified as “classical,” when a neurovascular compression of the trigeminal root leads to morphological changes; “idiopathic,” when the cause is unknown; and “secondary,” which is usually associated with other neurological disease such as multiple sclerosis [9, 14, 19].
Recent studies have identified several new missense variants in ion channels in TN patients, including some that could be involved in regulating neuronal excitability in trigeminal ganglion neurons [10,11,12, 25]. This includes voltage-gated calcium channels (VGCCs) which allow depolarization-induced calcium influx that in turn triggers different calcium-dependent processes such as neurotransmitter release, and the regulation of neuronal excitability [18, 27]. T-type calcium channels contribute to both of these processes, and their dysregulation has been associated with various chronic pain states [17]. Moreover, mutations in the CACNA1H gene that encodes the CaV3.2 calcium channels have been associated with TN and shown to cause a gain-of-function of channel activity [16].
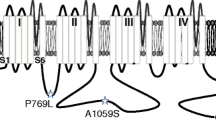
Among the mutations previously described, a new missense variant in the CACNA1G gene that encodes the CaV3.1 channel was reported in a single patient [11]. This variant contains a substitution of an arginine (R) at position 706 to glutamine (Q), with a possibly damaging prediction by PolyPhen2 [1]. This substitution is located in the domain I-II linker region of the CaV3.1 channel, a region involved in channel gating and trafficking (Fig. 1) [3, 29]. Here, we investigated the electrophysiological properties of this R706Q variant, demonstrating that this mutation causes a gain-of-function in channel activity.

Schematic representation of the CaV3.1 channel with approximate location of the R706Q mutation. The CaV3.1 channel is composed of 4 transmembrane domains (DI-IV), and each domain is composed of 6 transmembrane segments. The fourth segment (S4) is positively charged and is responsible for sensing changes in voltage. The fifth and sixth segments (S5 and S6) form the pore region. The R706Q mutation is located in the intracellular loop that links DI and DII
Methods
Molecular cloning
Site-directed mutagenesis using a wild-type (WT) human CaV3.1 subunit clone in pcDNA3.1 (kindly provided by Dr. Terrance Snutch) was performed using PCR (Pfu polymerase) along with forward and reverse mutagenesis primers for insertion of the R706Q missense mutation. The constructs were analyzed by DNA sequencing after the mutagenesis protocol.
Cell culture and cDNA transfection
Human embryonic kidney tsA-201 cells were cultured in standard DMEM medium supplemented with 10% fetal bovine serum (heat inactivated) and 50 U/ml penicillin and 50 μg/ml streptomycin. Cells were kept at 37 °C in a humidified incubator with 5% CO2. Cells were transiently transfected with 3 μg of hCaV3.1 WT or R706Q cDNA, and 0.5 μg of eGFP cDNA using the calcium phosphate method [30]. To avoid batch to batch and day to day variability, CaV3.1 WT or R706Q mutant were transfected on the same day, with a total of 8 transfections, with both WT and mutant channels tested on the same day.
Electrophysiology recordings
Whole-cell voltage-clamp recordings in tsA-201 cells were performed 72 h post transfection at room temperature (22–24 °C). Currents were recorded using an Axopatch 200B amplifier linked to a computer equipped with pClamp 11.2 software. Patch pipettes were filled with an internal solution containing (in mM): 130 CsCl, 2.5 MgCl2, 10 HEPES, 5 EGTA, 0.5 Mg-GTP 3, and Na-ATP (pH 7.4 adjusted with CsOH). The external solution contained (in mM): 10 BaCl2, 125 CsCl, 1 MgCl2, 10 HEPES, and 10 glucose (pH 7.4 adjusted with CsOH). The linear leak component of the current was corrected using a P/4 subtraction protocol and current traces were digitized at 10 kHz and filtered at 2 kHz. Junction potentials were left uncorrected because they were very small (~ 3 mV in tsA-201 cells).
Current density-voltage (IV) relation curves were obtained from the peak current of 140 ms depolarizing pulses between − 80 and + 20 mV in 5 mV increments from a holding potential of – 110 mV. The current density–voltage relationship was obtained by dividing the peak current at each voltage by the cell capacitance. Data from individual cells were fitted using the following Boltzmann equation: I = ((Vm – Vrev) × Gmax)/(1 + exp((Vm – Va)/ka) where I is the peak current, Vm and Vrev is the membrane potential and the reversal potential, respectively, Gmax is the maximum conductance, Va is the voltage for half activation, and ka is the slope factor. Conductance curves were obtained from the IV curves by dividing the peak current at each depolarizing step by the driving force according to the equation: G = I/(Vm − Vrev) and normalized against the maximum conductance (Gmax). Time constants of activation and inactivation (τact and τinact) were obtained by mono-exponential fits to the rising phase and the decaying of the current.
Action potential waveforms were obtained from mouse trigeminal ganglion recordings, and cells were clamped at a holding potential of – 60 mV which was close to the measured resting membrane potential of TG neurons. The total charge movement was calculated as the integral of the area under the current evoked by the protocol. Two charge movements were calculated, from 0 to 12 ms and 12.5 to 20 ms (ramp and tail current, respectively).
Steady-state inactivation curves were obtained by applying 1-s conditioning pre-pulses from − 100 to − 15 mV in 5 mV increments followed by a 50-ms test pulse to – 30 mV. Curves were fitted with the equation: Inormalized = 1/(1 + exp(−(Vm − Vh)/kh)) where Vh is the potential for half inactivatio and kh is the slope factor. Recovery from inactivation was evaluated by applying two test pulses (P1 and P2) from a holding potential of – 110 mV; a 2-s P1 pulse and a 150-ms P2 pulse, at – 20 mV that were separated by an interval ranging from 1 ms to 8 s. P2/P1 was plotted as a function of time and fitted using the following equation: Fractional recovery = A1x(1-exp(-t/τfast) + A2x (1-exp(-t/τslow), where τfast and τslow are the recovery time constants and A1 and A2 are the relative normalized fractional amplitudes of the fast and slow recovery components.
Statistical analysis
Electrophysiology data were analyzed using Clampfit 11.2 software (Molecular Devices) and fitted using GraphPad Prism version 9 (GraphPad Software). D'Agostino-Pearson normality test was performed. Statistical analysis was performed using two-tailed Student’s t test. All averaged data are plotted as mean ± SEM, and the results were considered statistically significant if p < 0.05.
Results
The R706Q missense variant increases CaV3.1 channel current density
tsA-201 cells were transfected with human CaV3.1 WT channels or the R706Q mutant (Fig. 1) and then subjected to patch clamp analysis. Representative traces for each condition are shown in Fig. 2(A). Current density-voltage curves for wild type and mutant revealed that the R706Q mutant exhibited an increase in current density (Fig. 2(B)). Corroborating the data in the IV curve, an increase in the peak current density was observed with the mutant (Fig. 2(D)). When we analyzed the maximum conductance, we observed an increase in channel conductance in the R706Q mutant compared to WT (Table 1). No significant changes were observed in the voltage for half activation and the time constant for activation (Fig. 2(C, E) and Table 1).

The R706Q mutation increases channel current density. (A) Representative whole cell current traces recorded in response to depolarizing steps from − 80 to + 20 mV from a holding potential of – 110 mV from tsA-201 cells expressing CaV3.1 WT or R706Q channels. (B) Average current density–voltage (IV) curves for cells expressing CaV3.1 WT or R706Q channels. (C) Voltage dependence of activation. Activation curves were constructed based on the parameters obtained from the IV curves. (D) Average peak current density from cells expressing CaV3.1 WT or R706Q channels. (E) Time constant of current activation as a function of voltage. Numbers in parentheses represent the number of cell recordings per variant. Data are expressed as mean ± SEM, two-tailed Student’s t test **p < 0.01 vs. CaV3.1 WT
R706Q increases ion flux in response to action potential-like stimuli
In step protocols, we observed that the R706Q mutant increased the channel current density (Fig. 2(C and D)). To investigate of the effect of the mutation in a more physiological context, we examined the movement of Ba2+ using an action potential-voltage-clamp protocol, using an action potential waveform (Fig. 3(A)) recorded from a trigeminal ganglion neuron [16]. Overall, we observed an increase in Ba2+ influx in the R706Q mutant compared to CaV3.1 WT (Fig. 3(B)). An increase in barium current was observed at the ramp phase (from 0 to 12 ms) of the action potential (Fig. 3(C)) and at the repolarization phase (from 12.5 to 20 ms) (Fig. 3(D)). Moreover, the tail current in the R706Q mutant was significantly larger when compared to CaV3.1 WT channels (Fig. 3(E)).

The R706Q mutation increases Ba2+ influx in response to an action potential waveform. (A) Action potential waveform from mouse trigeminal ganglion neuron. (B) Current density in response to action potential waveform for each condition. (C) Ba2+ flux during the ramp phase (from 0 to 12 ms). (D) Ba2+ movement during the repolarization phase (from 12.5 to 20 ms). (E) Peak tail current density. Numbers in parentheses represent the number of cell recordings per variant. Data are expressed as mean ± SEM, two-tailed Student’s t test *p < 0.05, **p < 0.01 vs. CaV3.1 WT
The R706Q mutation increases the recovery from inactivation
Steady-state inactivation curves were recorded to assess channel availability. No significant difference was observed in the voltage for half inactivation or slope between the conditions (Fig. 4(A) Table 1). On the other hand, analysis of recovery from inactivation demonstrated a gain-of-function in the R706Q mutant, with the mutant channel exhibiting a faster recovery from inactivation (Fig. 4(B), Table 1). No major difference was observed in the inactivation kinetics (Fig. 4(C)).

R706Q displays a faster recovery from inactivation. (A) Steady-state inactivation curves for CaV3.1 WT and R706Q channels. The data were fitted with the Boltzmann equation. (B) Time course of recovery from inactivation for CaV3.1 WT and R706Q channels. The solid lines are bi-exponential fits. (C) Time constant of current inactivation kinetics as a function of voltage. Numbers in parentheses represent the number of cell recordings per variant. Data are expressed as mean ± SEM., asterisks denote statistical significance (t test)
Discussion
Some mutations in the CACNA1G gene have been associated with cerebellar ataxia and epilepsy [7, 22, 32]. A previous study reported a new missense variant in the CACNA1G gene in a single patient with trigeminal neuralgia [11]. It is important to highlight that the same patient also harbors a gain-of-function mutation in the CACNA1A gene that encodes CaV2.1 channels [15] and hence it is unclear to what extent the two channels contribute to the clinical phenotype, and whether there is synergism in the effects of the two mutations. Not all individuals with CaV3.1 gain-of-function mutations present with trigeminal neuralgia. This may be due to splice isoform specific functional effects of such mutations, or alternatively the specific biophysical properties that are altered by the mutations that may or may not manifest themselves strongly within in the specific firing properties of trigeminal neurons.
The R706Q missense variant is located in the I-II linker region, an important region in T-type calcium channels that is involved in channel trafficking, and this may explain the increase in current density [2, 3, 26, 29]. Indeed, previous findings showed that single-nucleotide polymorphisms in the I-II linker region of T-type channels increase the channel's cell surface expression levels [29]. The increase in current density observed in the R706Q CaV3.1 channel could thus be due to an increase in channel trafficking, but alternatively through changes in single channel conductance and/or maximum open probability. The latter possibility would not be without precedent, since a previous study from our group demonstrated that a single point mutation in the I-II linker region of the CaV3.2 channel increases current density without changing channel trafficking [16].
The major contribution of T-type channels during the action potential is at the repolarizing phase, where the strong driving force creates a tail current that supports substantial ion influx [5, 21]. This increase in ion influx in the R706Q mutant could potentially trigger the hyper-activation of various calcium-dependent signaling pathways as well as alterations in low threshold neurotransmitter release [31]. The recovery from inactivation is a critical property of T-type calcium channels, as recovery from inactivation is closely linked to rebound bursting [23]. The faster recovery from inactivation observed here would thus be consistent with an increase in trigeminal neuron excitability and thus the clinical pain phenotype. Moreover, during rapid trains of action potentials, a more rapid recovery from inactivation would be expected to lead to less accumulation of inactivated channels, thus leading to maintenance of T-type channel mediated calcium entry [6]. That said, we note that there is heterogeneity in the firing properties of trigeminal ganglion neurons, including burst firing, and the physiological impact of the observed biophysical alterations may therefore vary with neuronal subtype [20, 24, 28]. Also, in line with a potential role of CaV3.1 in trigeminal neuralgia is evidence from CaV3.1 null mice. In a model of trigeminal neuropathic pain, CaV3.1 null mice still develop mechanical hyperalgesia, however, to a lesser degree when compared to wild type littermates [8], which then may suggest that a gain-of-function in these channels may have the opposite effect.
In summary, the R706Q variant is the first CaV3.1 channel mutation to be associated with a chronic pain condition. Our functional analysis of the CaV3.1 variant revealed an overall gain-of-function that could contribute to trigeminal pain. It is important to highlight that the analysis was conducted in a heterologous system, providing only a snapshot of the phenotype of the mutation. Therefore, additional analysis in native tissues will be necessary to further validate these findings. Nonetheless, the CaV3.1 calcium channel may emerge as a potential target for treating trigeminal neuralgia.
Data availability
The data used in our study are available from the authors on reasonable request.
Abbreviations
- TN:
-
Trigeminal neuralgia
- WT:
-
Wild type
- VGCCs:
-
Voltage-gated calcium channels
References
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249. https://doi.org/10.1038/nmeth0410-248
Arias O II, Vitko I, Fortuna M, Baumgart JP, Sokolova S, Shumilin IA, Van Deusen A, Soriano-Garcia M, Gomora JC, Perez-Reyes E (2008) Characterization of the gating brake in the I-II loop of Ca(v)3.2 T-type Ca(2+) channels. J Biol Chem 283:8136–8144. https://doi.org/10.1074/jbc.M708761200
Baumgart JP, Vitko I, Bidaud I, Kondratskyi A, Lory P, Perez-Reyes E (2008) I-II loop structural determinants in the gating and surface expression of low voltage-activated calcium channels. PLoS One 3:e2976. https://doi.org/10.1371/journal.pone.0002976
Bendtsen L, Zakrzewska JM, Heinskou TB, Hodaie M, Leal PRL, Nurmikko T, Obermann M, Cruccu G, Maarbjerg S (2020) Advances in diagnosis, classification, pathophysiology, and management of trigeminal neuralgia. Lancet Neurol 19:784–796. https://doi.org/10.1016/S1474-4422(20)30233-7
Cain SM, Snutch TP (2010) Contributions of T-type calcium channel isoforms to neuronal firing. Channels (Austin) 4:475–482. https://doi.org/10.4161/chan.4.6.14106
Chemin J, Monteil A, Bourinet E, Nargeot J, Lory P (2001) Alternatively spliced alpha(1G) (Ca(V)3.1) intracellular loops promote specific T-type Ca(2+) channel gating properties. Biophys J 80:1238–1250. https://doi.org/10.1016/S0006-3495(01)76100-0
Chemin J, Siquier-Pernet K, Nicouleau M, Barcia G, Ahmad A, Medina-Cano D, Hanein S, Altin N, Hubert L, Bole-Feysot C, Fourage C, Nitschke P, Thevenon J, Rio M, Blanc P, Vidal C, Bahi-Buisson N, Desguerre I, Munnich A et al (2018) De novo mutation screening in childhood-onset cerebellar atrophy identifies gain-of-function mutations in the CACNA1G calcium channel gene. Brain 141:1998–2013. https://doi.org/10.1093/brain/awy145
Choi S, Yu E, Hwang E, Llinas RR (2016) Pathophysiological implication of CaV3.1 T-type Ca2+ channels in trigeminal neuropathic pain. Proc Natl Acad Sci U S A 113:2270–2275. https://doi.org/10.1073/pnas.1600418113
Cruccu G, Finnerup NB, Jensen TS, Scholz J, Sindou M, Svensson P, Treede RD, Zakrzewska JM, Nurmikko T (2016) Trigeminal neuralgia: new classification and diagnostic grading for practice and research. Neurology 87:220–228. https://doi.org/10.1212/WNL.0000000000002840
Devor M, Amir R, Rappaport ZH (2002) Pathophysiology of trigeminal neuralgia: the ignition hypothesis. Clin J Pain 18:4–13. https://doi.org/10.1097/00002508-200201000-00002
Di Stefano G, Yuan JH, Cruccu G, Waxman SG, Dib-Hajj SD, Truini A (2020) Familial trigeminal neuralgia—a systematic clinical study with a genomic screen of the neuronal electrogenisome. Cephalalgia 40:767–777. https://doi.org/10.1177/0333102419897623
Dong W, Jin SC, Allocco A, Zeng X, Sheth AH, Panchagnula S, Castonguay A, Lorenzo LE, Islam B, Brindle G, Bachand K, Hu J, Sularz A, Gaillard J, Choi J, Dunbar A, Nelson-Williams C, Kiziltug E, Furey CG et al (2020) Exome sequencing implicates impaired GABA signaling and neuronal ion transport in trigeminal neuralgia. iScience 23:101552. https://doi.org/10.1016/j.isci.2020.101552
Eide PK (2022) Familial occurrence of classical and idiopathic trigeminal neuralgia. J Neurol Sci 434:120101. https://doi.org/10.1016/j.jns.2021.120101
Gambeta E, Chichorro JG, Zamponi GW (2020) Trigeminal neuralgia: an overview from pathophysiology to pharmacological treatments. Mol Pain 16:1744806920901890. https://doi.org/10.1177/1744806920901890
Gambeta E, Gandini MA, Souza IA, Ferron L, Zamponi GW (2021) A CACNA1A variant associated with trigeminal neuralgia alters the gating of Cav2.1 channels. Mol. Brain 14:4. https://doi.org/10.1186/s13041-020-00725-y
Gambeta E, Gandini MA, Souza IA, Zamponi GW (2022) Ca V 3.2 calcium channels contribute to trigeminal neuralgia. Pain 163:2315–2325. https://doi.org/10.1097/j.pain.0000000000002651
Harding EK, Zamponi GW (2022) Central and peripheral contributions of T-type calcium channels in pain. Mol Brain 15:39. https://doi.org/10.1186/s13041-022-00923-w
Iftinca MC, Zamponi GW (2009) Regulation of neuronal T-type calcium channels. Trends Pharmacol Sci 30:32–40. https://doi.org/10.1016/j.tips.2008.10.004
Maarbjerg S, Benoliel R (2021) The changing face of trigeminal neuralgia—a narrative review. Headache 61:817–837. https://doi.org/10.1111/head.14144
Marchenkova A, van den Maagdenberg AM, Nistri A (2016) Loss of inhibition by brain natriuretic peptide over P2X3 receptors contributes to enhanced spike firing of trigeminal ganglion neurons in a mouse model of familial hemiplegic migraine type-1. Neuroscience 331:197–205. https://doi.org/10.1016/j.neuroscience.2016.06.034
McCobb DP, Beam KG (1991) Action potential waveform voltage-clamp commands reveal striking differences in calcium entry via low and high voltage-activated calcium channels. Neuron 7:119–127. https://doi.org/10.1016/0896-6273(91)90080-j
Morino H, Matsuda Y, Muguruma K, Miyamoto R, Ohsawa R, Ohtake T, Otobe R, Watanabe M, Maruyama H, Hashimoto K, Kawakami H (2015) A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia. Mol Brain 8:89. https://doi.org/10.1186/s13041-015-0180-4
Perez-Reyes E (2003) Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev 83:117–161. https://doi.org/10.1152/physrev.00018.2002
Puil E, Miura RM, Spigelman I (1989) Consequences of 4-aminopyridine applications to trigeminal root ganglion neurons. J Neurophysiol 62:810–820. https://doi.org/10.1152/jn.1989.62.3.810
Rappaport ZH, Devor M (1994) Trigeminal neuralgia: the role of self-sustaining discharge in the trigeminal ganglion. Pain 56:127–138. https://doi.org/10.1016/0304-3959(94)90086-8
Shcheglovitov A, Vitko I, Bidaud I, Baumgart JP, Navarro-Gonzalez MF, Grayson TH, Lory P, Hill CE, Perez-Reyes E (2008) Alternative splicing within the I-II loop controls surface expression of T-type Ca(v)3.1 calcium channels. FEBS Lett 582:3765–3770. https://doi.org/10.1016/j.febslet.2008.10.013
Simms BA, Zamponi GW (2014) Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron 82:24–45. https://doi.org/10.1016/j.neuron.2014.03.016
Tseng WT, Tsai ML, Iwata K, Yen CT (2012) Long-term changes in trigeminal ganglionic and thalamic neuronal activities following inferior alveolar nerve transection in behaving rats. J Neurosci 32:16051–16063. https://doi.org/10.1523/JNEUROSCI.1828-12.2012
Vitko I, Bidaud I, Arias JM, Mezghrani A, Lory P, Perez-Reyes E (2007) The I-II loop controls plasma membrane expression and gating of Ca(v)3.2 T-type Ca2+ channels: a paradigm for childhood absence epilepsy mutations. J Neurosci 27:322–330. https://doi.org/10.1523/JNEUROSCI.1817-06.2007
Weiss N, Black SA, Bladen C, Chen L, Zamponi GW (2013) Surface expression and function of Cav3.2 T-type calcium channels are controlled by asparagine-linked glycosylation. Pflugers Arch 465:1159–1170. https://doi.org/10.1007/s00424-013-1259-3
Weiss N, Hameed S, Fernandez-Fernandez JM, Fablet K, Karmazinova M, Poillot C, Proft J, Chen L, Bidaud I, Monteil A, Huc-Brandt S, Lacinova L, Lory P, Zamponi GW, De Waard M (2012) A Ca(v)3.2/syntaxin-1A signaling complex controls T-type channel activity and low-threshold exocytosis. J Biol Chem 287:2810–2818. https://doi.org/10.1074/jbc.M111.290882
Weiss N, Zamponi GW (2020) Genetic T-type calcium channelopathies. J Med Genet 57:1–10. https://doi.org/10.1136/jmedgenet-2019-106163
Acknowledgements
We are grateful to Lina Chen for her technical support.
Code availability
Not applicable.
Funding
This work was supported by a Project Grant from the Canadian Institutes of Health Research (CIHR) to GWZ. EG held a Alberta Innovates Scholarship. GWZ is a Canada Research Chair.
Author information
Authors and Affiliations
Contributions
AMA and EG performed experiments and data analysis. EG designed the study and drafted the manuscript. GWZ supervised the study and co-wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Alaklabi, A.M., Gambeta, E. & Zamponi, G.W. Electrophysiological characterization of a CaV3.1 calcium channel mutation linked to trigeminal neuralgia. Pflugers Arch - Eur J Physiol 475, 711–718 (2023). https://doi.org/10.1007/s00424-023-02808-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-023-02808-w