
Abstract
Bulbospinal catecholaminergic neurons located in the rostral aspect of the ventrolateral medulla (C1 neurons) or within the ventrolateral pons (A5 neurons) are involved in the regulation of blood pressure and sympathetic outflow. A stimulus that commonly activates the C1 or A5 neurons is hypoxia, which is also involved in breathing activation. Although pharmacological and optogenetic evidence suggests that catecholaminergic neurons also regulate breathing, a specific contribution of the bulbospinal neurons to respiratory control has not been demonstrated. Therefore, in the present study, we evaluated whether the loss of bulbospinal catecholaminergic C1 and A5 cells affects cardiorespiratory control during resting, hypoxic (8% O2), and hypercapnic (7% CO2) conditions in unanesthetized rats. Thoracic spinal cord (T4-T8) injections of the immunotoxin anti-dopamine β-hydroxylase-saporin (anti-DβH-SAP—2.4 ng/100 nl) and the retrograde tracer Fluor-Gold or ventrolateral pontine injections of 6-OHDA were performed in adult male Wistar rats (250–280 g, N = 7–9/group). Anti-DβH-SAP or 6-OHDA eliminated most bulbospinal C1 and A5 neurons or A5 neurons, respectively. Serotonergic neurons and astrocytes were spared. Depletion of the bulbospinal catecholaminergic cells did not change cardiorespiratory variables under resting condition, but it did affect the response to hypoxia and hypercapnia. Specifically, the increase in the ventilation, the number of sighs, and the tachycardia were reduced, but the MAP increased during hypoxia in anti-DβH-SAP-treated rats. Our data reveal that the bulbospinal catecholaminergic neurons (A5 and C1) facilitate the ventilatory reflex to hypoxia and hypercapnia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The rostral aspect of the ventrolateral medulla (RVLM) is composed of chemically distinct populations of cells that project to the intermediolateral column (IML), C1 neurons and non-C1 neurons. Approximately 70% of spinally projecting RVLM neurons contain the enzymes phenylethanolamine-N-methyltransferase (PNMT), tyrosine hydroxilase (TH), and dopamine-β-hydroxylase (DβH), as well as other enzymes involved in catecholamine biosynthesis [22, 64], and are part of the C1 neurons. Although the role of the bulbospinal C1 neurons in cardiovascular regulation is well established, the specific contributions of these neurons in breathing regulation are still unclear. Recently, a subset of studies from Dr. Guyenet’s laboratory showed that C1 cell photostimulation increased breathing in mammals [1, 10, 25, 26]. The respiratory changes produced by C1 cell stimulation mimic the response to hypoxia and differ from those produced by hypercapnia. Specifically, the selective activation of C1 neurons located in the RVLM causes tachypnea, sighs, and blood pressure increases, suggesting that bulbospinal C1 cells can contribute to cardiorespiratory adjustments induced by hypoxia [10]. Besides the direct evidence to support a role of the C1 cell population in breathing regulation, no one has shown the effect of selective elimination of the bulbospinal C1 cells in the control of breathing under resting and hypoxic or hypercapnic condition.
Using the standard immunotoxin that consists of the ribosome-inactivating toxin saporin bound to an antibody to DβH (anti-DβH-SAP), which can selectively destroy the catecholaminergic neurons, several studies have directly assessed the role of the C1 cell population in sympathetic outflow and cardiovascular function [36, 48, 61, 66]. Injection of anti-DβH-SAP directly into the RVLM appears to effectively lesion neurons of the C1 cell group [6, 35,34,37]. However, with this approach, the elimination of C1 cells is not selective for the bulbospinal C1 neurons. In addition, injection of the toxin directly into the RVLM produces an unavoidable measure of nonselective local damage to the area of interest. The anti-DβH-SAP is retrogradely transported; therefore, we injected the toxin directly into spinal cord in order to destroy catecholaminergic neurons projecting to IML [28, 48, 49]. We then used this model to determine the role of bulbospinal C1 neurons for the control of breathing under hypoxia (8% O2) and hypercapnia (7% CO2) condition in unanesthetized rats.
Material and methods
Animals
Experiments were performed in 41 adult male Wistar rats weighing 250–280 g. The animals were housed individually in cages in a room with controlled temperature (24 ± 2 °C) and humidity (55 ± 10%). Lights were on from 7:00 a.m. to 7:00 p.m. Standard Bio Base rat chow (Águas Frias, SC, Brazil), and tap water was available ad libitum. Animals were used in accordance with the guidelines approved by the Animal Experimentation Ethics Committee of the Institute of Biomedical Science at the University of São Paulo (protocol number 07/2014).
Immunotoxin lesions
All surgical procedures were performed under anesthesia with a mixture of ketamine (100 mg kg−1 of body weight) and xylazine (7 mg kg−1 of body weight) intraperitoneal (i.p.), and, after surgical procedure, the rats received antibiotic and anti-inflammatory protection (160,000 U kg−1 benzylpenicillin and 33.3 mg kg−1 dihydrostreptomycin, intramuscular).
For selective lesion of bulbospinal catecholaminergic neurons, the rats (N = 9/group) were fixed to a stereotaxic frame, and the bilateral injections of the toxin anti-dopamine beta-hydroxylase-saporin (anti-DβH-SAP, two injections/site—100 nl/injection: total volume was 400 nl) or saline (control), combined with the bilateral injection of the retrograde tracer Fluor-Gold (FG—2%, one injection/site—50 nl/side: total volume of 100 nl) (Fluorochrome, Denver, CO, USA), were made bilaterally into the thoracic portion of spinal cord (T4-T8) [48, 49] using the following coordinates: 0.5 mm lateral to the midline and 0.5–0.8 mm below the dorsal surface of the spinal cord. The rats were allowed to recover for 3 weeks prior to the physiological experiments. Based on a previous publication from our laboratory, we did not notice any differences in neuroanatomical or physiological experiments in animals that received IgG-saporin or saline into the C1 region [37, 61]. Thus, in the present study, the sham-operated rats were injected with saline (0.15 M; N = 9).
In order to discriminate the specific contribution of the ventrolateral pontine region (the catecholaminergic A5 region) on the breathing responses, normoxia, hypoxia, or hypercapnia, in a separate group of rats (N = 7/group), bilateral injection of 6-OHDA (12 μg/μl in saline solutions with 0.3% ascorbic acid; 200 nl/site) or vehicle (0.3% ascorbic acid—control) was performed into the ventrolateral pons, as previously described [35, 61]. The stereotaxic coordinates (relative to bregma level) were 9.8 mm caudal to bregma and ± 1.8 mm lateral to the midline and 8.3 mm ventral to the dorsal surface of the brain. After surgery, the animals were kept in recovery for 2 weeks before they were used in physiological experiments.
The dose of anti-DβH-SAP and 6-OHDA used in the present study was determined based on previous experiments using anti-DβH-SAP and 6-OHDA to reduce catecholaminergic neurons [6, 35,34,37, 48, 49, 61].
Determination of pulmonary ventilation
The ventilatory response was assessed using barometric, unrestrained whole body plethysmography (EMKA Technologies). Rats were acclimatized to the plethysmography chambers (Bonther, Ribeirão Preto, SP, Brazil) 2–3 days prior to the experiments. On the experiment day, freely moving rats were kept in a 5-L plethysmography chamber with room air for 45–60 min before the ventilatory parameters were recorded. The plethysmography chamber was continuously flushed with 1.5 L min−1 and regulated by computer-driven mass flow controllers for O2, N2, and CO2 (Alicat Scientific, Inc., Tucson, AZ, USA). The flow controllers were adjusted to 21% O2 balanced with N2 in the normoxia condition, 8% O2 balanced with N2 in the hypoxia condition and 7% CO2; 21% O2 and 72% N2 in the hypercapnia condition. The ambient temperature (23–26 °C) and humidity (50–60%) were continuously recorded inside the plethysmography chamber and used to calculate the tidal volume. Rectal temperature was used as a core body temperature index and was measured twice: before and at the end of the experiments. The values were averaged. The ventilatory parameters measured by the plethysmography system included respiratory frequency (fR, bpm), inspiratory time (TI, ms), expiratory time (TE, ms), tidal volume (VT, mL/kg), and minute volume (VE, ml/min/kg).
Arterial blood pressure recording
In a second set of experiments (N = 4/5 groups of rats), we recorded blood pressure in unanesthetized freely moving rats through a chronically indwelling catheter. To instrument the rats, they were anesthetized as discussed in section “Immunotoxin lesions” above, and a catheter made from polyethylene tubing (PE-10 connected to PE-50; Clay Adams, Parsippany, NJ, USA) was inserted into the descending aorta through a femoral artery. The other end of the catheter was tunneled subcutaneously and emerged on the rat’s back for access. The rats were given a 24-h recovery period at which time the rats resumed normal food and water consumption with no impairment of motor activity or locomotion and no apparent differences in reactivity during handling. Mean arterial pressures (MAP; mmHg) and heart rate (HR; bpm) were measured from the AP recording using LabChart 8.0 (model Powerlab 8SP ADInstruments).
Chemoreflex analysis
Unanesthetized rats were allowed at least 30–40 min to acclimatize to the chamber environment at normoxia/normocapnia (21% O2, 79% N2 and < 0.5% CO2) before measurements of breathing parameters were taken. Hypoxia was induced by lowering the O2 concentration in the inspired air down to a level of 8% for 10 min. Hypercapnia was induced by titrating CO2 into the respiratory mixture up to a level of 7% (21% O2, 69% N2) for 10 min.
Histology
The rats were deeply anesthetized with pentobarbital (60 mg kg−1, i.p.), then injected with heparin (500 units, intracardially) and perfused through the ascending aorta first with 250 ml of phosphate-buffered saline (PB—pH 7.4) and then with 500 ml of 4% phosphate-buffered paraformaldehyde (0.1 M, pH 7.4). The brains and the thoracic spinal cords (T4-T8) were extracted, cryoprotected by overnight immersion in a 20% sucrose solution in phosphate-buffered saline at 4 °C, sectioned in the coronal plane at 40 μm on a sliding microtome, and stored in cryoprotectant solution (20% glycerol plus 30% ethylene glycol in 50 mM phosphate buffer, pH 7.4) at − 20 °C. All histochemical procedures were completed using free-floating sections according to previously described protocols [5, 54, 60].
All of the primary antibodies were diluted in PB containing 1% normal donkey serum (017-000-121, Jackson Immuno Research Laboratories) and 0.3% Triton X-100 and incubated for 24 h. Tyrosine hydroxylase (TH) immunoreactivity was detected using mouse anti-TH (lot#2716631, 1:1000; Chemicon, Temecula, CA, USA), followed by goat anti-mouse Cy3 (lot#100686) or Alexa488 IgG (lot#7107586) (1:200; Jackson Laboratories; West Grove, PA, USA). Tryptophan hydroxylase (TrpOH) immunoreactivity was detected using TrpOH anti-mouse antibody (lot#2492095, 1:2000; Sigma, St. Louis, MO, USA), followed by goat anti-mouse Cy3 IgG (1:200; Jackson Laboratories, West Grove, PA, USA). GFAP immunoreactivity was detected using mouse anti-GFAP (lot#5804, 1:1000; Chemicon, Temecula, CA, USA), followed by goat anti-mouse Cy3 IgG (1:200; Jackson Laboratories, West Grove, PA, USA). The sections were mounted on gelatin-coated slides, coverslipped with DPX (Aldrich, Milwaukee, WI, USA).
Cell counting, imaging, and data analysis
A multifunction microscope Zeiss Axioimager A1 microscope (Zeiss, Muenchen, Germany) was used to image the sections and perform subsequent analysis. Immunofluorescence was examined under epifluorescence illumination.
The locations of TH and/or FG (TH-ir and/or FG) into the rostral ventrolateral medulla were plotted 1:6 sections from 11.12 to 14.00 mm caudal to the bregma (13 sections/animal) or into the ventrolateral pons were plotted in 1:6 sections from 9.00 to 10.68 mm caudal to the bregma (8 sections/animal). The profile counts of the animals that received anti-DβH-SAP or 6-OHDA bilateral microinjections reflected the average of both sides of the medulla and were compared with the control rats.
Digital color photomicrographs were acquired using a Zeiss Axiocam HRc camera. Images of double immunofluorescence-stained sections were acquired and analyzed with the Axiovision software (Zeiss), which permits the acquisition of images from several separate fluorescence channels. Image J (version 1.41; National Institutes of Health, Bethesda, MD) was used for cell counting, and Canvas software (ACD Systems, Victoria, Canada, v. 9.0) was used for line drawings. The neuroanatomical nomenclature employed during experimentation was described relative to the caudal pole of the facial motor nucleus (bregma = − 11.6 mm) and was also confirmed by Paxinos and Watson [42].
Statistics
Data normality was assessed using the Shapiro-Wilk test, and all the normally-distributed data were expressed as the mean ± SEM. The data regarding number of neurons (TH+, TH+/FG+) and breathing activity (i.e., VT, fR, VE, TI, and TE) were compared between groups and across time points using two-way ANOVA, with repeated measures for only the time factor (Figs. 1 and 2, 4, 6 and 7, and 8). When applicable, the Student-Newman-Keuls post hoc test was used. Inter-group differences in sighs were evaluated with one-way ANOVA (Fig. 10). When applicable, the Student-Newman-Keuls post hoc test was used. Inter-group differences in VT, fR, VE, TI, and TE during basal conditions were evaluated with unpaired Student’s t tests (Tables 1 and 2). Two-tailed Pearson’s correlation was used to test for a relationship between the number of catecholaminergic neurons and the hypoxia or hypercapnia-induced change in breathing (Figs. 5 and 9). Sigma Stat version 3.0 package (Jandel Corporation, Point Richmond, CA, USA) was used for all analysis. Significance level used was p < 0.05.

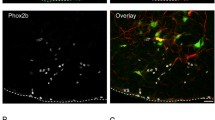
Injection of anti DβH-SAP into the spinal cord reduces the bulbospinal C1 neurons. a Schematic draw of the saporin toxin and the retrograde-labeling technique used to identify spinally projecting neurons in the RVLM by microinjections of anti-DβH-SAP and Fluor-Gold (FG, 2%) into the intermediolateral (IML) cell column at the T4-T8 level of the spinal cord. b Photomicrograph and composite photomicrograph of FG injection into the thoracic spinal cord. c, d, e and g, h, i Photomicrographs at the level of the RVLM (− 11.84 mm from Bregma) from one c, d, e control rat and one g, h, i anti-DβH-SAP rat. f Average number of TH-ir neurons per section from control (N = 9) or anti-DβH-SAP (N = 9) rats. j Average number of TH-ir and FG neurons per section from control or anti-DβH-SAP. Counts were made in one in six series of 40 μm coronal sections. Catecholaminergic C1 neurons were identified immunohistochemically as TH-positive neurons. Scale bar in I represents 100 μm and applies to c, d, e and g, h, i. IO inferior olive, FG Fluor-Gold, FN facial motor nucleus, TH tyrosine hydroxylase. *Different from control, two-way repeated measure ANOVA, p < 0.05

Injection of anti DβH-SAP into the spinal cord reduces the bulbospinal A5 neurons a, b, c and e, f, g photomicrographs at the level of the ventrolateral pons (− 9.96 mm from Bregma) from one a, b, c control rat and one e, f, g anti-DβH-SAP rat. d Average number of TH-ir neurons per section from control (N = 9) or anti-DβH-SAP (N = 9) rats. h Average number of TH-ir and FG neurons per section from control or anti- DβH-SAP. Counts were made in one in six series of 40 μm coronal sections. Catecholaminergic C1 neurons were identified immunohistochemically as TH-positive neurons. Scale bar in g represents 100 μm and applies to a, b, c and e, f, g. FG Fluor-Gold, LSO lateral superior olive, TH tyrosine hydroxylase. *Different from control, two-way repeated measures ANOVA, p < 0.05
Results
-
1)
Depletion of bulbospinal catecholaminergic neurons by anti-DβH-SAP
In order to evaluate the contribution of bulbospinal catecholaminergic neurons in the respiratory response to hypoxia or hypercapnia, the following experiments were performed by injecting saline or anti-DβH-SAP in the thoracic spinal cord (T4-T8). We also injected the retrograde tracer Fluor-Gold (FG) to estimate the number of the bulbospinal neurons eliminated by the saporin toxin (spread of the injections, 480 ± 0.4 μm). Examination of the thoracic spinal cord revealed that the injections were centered approximately in the region of the intermediolateral cell column in the upper thoracic spinal cord, but the injectate had clearly spread beyond this region into the levels of the dorsal and ventral horns (Fig. 1a and b). According to the spinal cord histologies, the compounds diffused predominantly along the length of the spinal cord, but did not affect the midline or lateral edges of the spinal cord (Fig. 1b). It is important to notice that the animals that received injections into the spinal cord did not show deficits in locomotion (data not shown).
The appearance of FG in TH-ir neurons demonstrated that the bulbospinal C1 cells were located at the rostral aspect of the C1 cell group, with 68% of the TH+/FG+ neurons located between − 11.60 and − 12.80 mm caudal to bregma (Fig. 1c–f, j). Anti-DβH-SAP-lesioned rats (N = 9), co-injected with FG, showed an average depletion of 72 ± 3% of TH-ir neurons (range 61–87%; bregma level: − 11.60 to − 12.80 mm caudal) (Fig. 1c–j). In the caudal aspect of the VLM (− 13.04 to − 14.00 mm caudal to bregma), the TH-ir neurons did not contain FG (Fig. 1j), and the number of TH-ir neurons at these levels was unaffected by treatment with anti-DβH-SAP (Fig. 1f). Control rats (saline-treated rats) did not show changes in the number of C1 neurons at any medullary levels (Fig. 1c–j).
Considering the fact that intraspinal injection of the anti-DβH-SAP is able to eliminate the bulbospinal catecholaminergic neurons in the brainstem [48], the next step of our work is to better investigate if our toxin injections affected the catecholaminergic A5 cell group located in the ventrolateral pons (Fig. 2). We noticed that intraspinal injection of the toxin anti-DβH-SAP produced a significant reduction of the A5 noradrenergic neurons (Fig. 2). Quantification of the TH-ir neurons in lesioned rats revealed a 67 ± 3% reduction (range, 56–82%) compared with control rats (Fig. 2a–g). We also noticed an 81 ± 3% depletion of TH-ir bulbospinal A5 neurons (TH+/FG+) (range, 66–90%) compared with saline-treated rats (Fig. 2h). No changes in the TH-ir neurons were observed in the A1, A6 (locus coeruleus), and A7 regions after treatment with anti-DβH-SAP (data not shown).
In the present experimental protocol, we also examined two populations of noncatecholaminergic cells (serotonergic neurons and astrocytes) to determine whether the toxicity of anti-DβH-SAP was selective for catecholaminergic neurons. Serotonergic neurons revealed by immunohistochemical detection of TrpOH in animals that had been injected with anti-DβH-SAP (N = 9) or saline (N = 9) into the spinal cord were analyzed in the medullary sections from − 11.60 to − 12.80 mm (Fig. 3a and b). We also analyzed the astrocyte profile, identified by GFAP-ir between − 11.60 and − 12.80 mm. Similarly, the GFAP-ir profiles in control rats were comparable with rats treated with anti-DβH-SAP (Fig. 3c and d).
-
2)
Impairment of the chemosensory control of breathing after anti-DβH-SAP injections into the spinal cord

Selectivity of the anti-DβH-SAP injection into the spinal cord representative photomicrographs of a, c one control rats and b, d one rat treated with anti-DβH-SAP showing tryptophan hydroxylase-immunoreactive (TrpOH-ir) serotonergic neurons (a, b) and GFAP-immunoreactive (GFAP-ir) glial cells (c, d). Scale bar in d represents 100 μm and applies to all panels. IO inferior olive
Depletion of bulbospinal catecholaminergic neurons did not change the ventilatory parameters during normoxia (21% O2, balanced with N2) (Table 1). Hypoxia (8% O2, balanced with N2) or hypercapnia (7% CO2, 21% O2, balanced with N2) induced an increase in VE in all groups (p < 0.05, ANOVA), which resulted from increases of fR and VT in saline treated-rats (Fig. 4a–d). Hypoxia or hypercapnia also produced a reduction in TI and TE (Fig. 4e and f).

Depletion of bulbospinal catecholaminergic neurons impairs the ventilatory responses to hypoxia and hypercapnia A) Whole-body plethysmography was used to measure tidal volume (VT, ml/kg), respiratory frequency (fR, bpm) and minute ventilation (VE, ml/kg/min) in unanesthetized control or in anti-DβH-SAP treated rats during exposure to normoxia (N), hypoxia (8% O2) or hypercapnia (7% CO2). Summary data plotted as B) VT, C) fR, D) VE, E) inspiratory time (TI, ms) and F) expiratory time (TE, ms) show that anti-DβH-SAP animals have a decreased hypoxia and hypercapnia-induced responses in breathing. *Different from control, two-way repeated measures ANOVA, p < 0.05. N = 9/group of rats
Depletion of bulbospinal catecholaminergic neurons blunted the increase in VT (p = 0.015), fR (p = 0.031), and VE (p = 0.001) produced by hypoxia (Fig. 4a–d). The attenuation of fR in rats subjected to depletion of bulbospinal neurons was accompanied by an increase in TI (p = 0.036), without changing TE (p > 0.05) (Fig. 4e and f). To better analyze the association between the depletion of bulbospinal catecholaminergic neurons and the attenuation of the ventilatory response induced by hypoxia, we analyzed the correlation between the changes in VT (r 2 = 0.273; p = 0.026), fR (r 2 = 0.292; p = 0.021), and VE (r 2 = 0.406; p = 0.005) elicited by hypoxia and the total number of TH+-ir/FG+ cells in RVLM and A5 region. Our results indicate that there is a strong and significant correlation between these factors (Fig. 5a–c).

Correlations between the number of bulbospinal catecholaminergic neurons and the respiratory physiological response elicited by hypoxia or hypercapnia a, b, c, d, e, f X-Y plots of the increase in a, d tidal volume (ΔVT), b, e respiratory frequency (ΔfR) and c, f minute ventilation (ΔVE) elicited by hypoxia or hypercapnia relative to the number of TH+/FG+ bulbospinal neurons. Pearson’s r 2 and significance (p) are indicated
Lesions on the bulbospinal neurons did not affect fR (p = 0.404), TI (p = 0.460), and TE (p = 0.529) produced by hypercapnia (Fig. 4c; e and f). However, depletion of the bulbospinal catecholaminergic neurons attenuated the increase in VT (p = 0.044), and VE (p = 0.012) produced hypercapnia (Fig. 4a, b and d). We found a significant correlation between the changes in VT (r 2 = 0.319; p = 0.015) and VE (r 2 = 0.275; p = 0.025) elicited by hypercapnia and the total number of TH+-ir/FG+ cells in the RVLM and A5 region (Fig. 5d and f). No significant changes were observed between changes in fR and the total number of TH+-ir/FG+ cells in RVLM and A5 region (Fig. 5e).
-
3)
Changes in the mean arterial pressure and heart rate produced by anti-DβH-SAP injections into the spinal cord
Depletion of bulbospinal catecholaminergic neurons reduced the baseline values of mean arterial pressure (MAP) (normoxia—N) (p = 0.041) (Fig. 6a). No changes in heart rate (HR) were noticed between the saline and anti-DβH-SAP groups under the baseline condition (Fig. 6b). In control rats (N = 5), exposure to acute hypoxia (8% O2, balanced with N2) did not change MAP (Fig. 6a) but did increase HR; p = 0.024) (Fig. 6b). In anti-DβH-SAP-treated rats (N = 4), hypoxia elicited an increase in MAP (p = 0.004) (Fig. 6a). The tachycardia response induced by hypoxia was blunted in rats that received anti-DβH-SAP (Fig. 6b).

Depletion of bulbospinal catecholaminergic neurons affects the cardiovascular adjustments induced by hypoxia and hypercapnia a Mean arterial pressure (MAP, mmHg), and b heart rate (HR, bpm) of control (saline) and anti-DβH-SAP-treated rats under resting, hypoxia (8% O2) or hypercapnia (7% CO2) conditions. *Different from control (p < 0.05) and # different from the same group in normoxia. One-way ANOVA, p < 0.05. N = 4–5/group of rats
Hypercapnia (7% CO2, 21% O2, balanced with N2) did not change MAP, but it decreased HR in control rats (Fig. 6a and b). Interestingly, the rats subjected to anti-DβH-SAP injection into the spinal cord and exposed to the hypercapnia challenge showed an increase in MAP and HR (Fig. 6a and b).
-
4)
Depletion of ventrolateral pontine catecholaminergic neurons by 6-OHDA
Considering that we had a massive reduction in the number of TH-ir neurons in the ventrolateral pontine region, i.e., A5 region, after anti-DβH-SAP into the spinal cord (Fig. 2), in the next series of experiment, we performed selective lesion of the TH-ir neurons of the ventrolateral pons by injecting 6-OHDA toxin directly into the A5 region. This protocol allowed us to evaluate the breathing effects caused by A5 cell depletion without the interference of the C1 cells elimination (Fig. 7).

Injection of 6-OHDA into the ventrolateral pons reduces only the A5 neurons
a, b Photomicrographs at the level of the ventrolateral pons (− 9.96 mm from Bregma) from one a control rat and one b 6-OHDA rat. c Average number of TH-ir neurons per section from control (N = 7) or 6-OHDA (N = 7) rats. d, e Photomicrographs at the level of the RVLM (− 11.84 mm from Bregma) from d one control rat and e one 6-OHDA rat. f Average number of TH-ir neurons per section from control or 6-OHDA rats. Counts were made in one in six series of 40 μm coronal sections. Catecholaminergic C1 neurons were identified immunohistochemically as TH-positive neurons. Scale bar in e represents 100 μm and applies to a, b, d, e. VIIn facial nerve, FN facial motor nucleus, LSO lateral superior olive, IO inferior olive, TH tyrosine hydroxylase. *Different from control, two-way repeated measures ANOVA, p < 0.05.
The injection of 6-OHDA into the A5 region produced a massive reduction in the number of TH-expressing neurons (Fig. 7a–c). In the seven rats treated with 6-OHDA, counts of the TH-ir between − 9.00 and − 10.68 mm caudal to bregma revealed an average depletion of 61 ± 4% (range between 47 and 70%) (Fig. 7a–c). At the VLM region (− 11.36 to − 13.28 mm caudal to bregma), the number of TH-ir neurons was unaffected by treatment with 6-OHDA (Fig. 7d–f). No changes in the TH-ir neurons in the A6 (locus coeruleus) and A7 regions were observed after treatment with 6-OHDA (data not shown).
-
5)
Impairment of the chemosensory control of breathing after 6-OHDA injections into the ventrolateral pons
Depletion of catecholaminergic neurons in ventrolateral pons (A5 region) did not change the ventilatory parameters during normoxia (Table 2). Depletion of the A5 neurons reduced the increase in VT (p = 0.003) and VE (p = 0.004) elicited by hypoxia (Fig. 8a, b and d). Lesion of the A5 neurons did not change the increase in fR produced by hypoxia (p = 0.9) (Fig. 8c). Loss of the A5 neurons did not attenuate the reduction in TI (p = 0.648) and TE (p = 0.651) produced by hypoxia (Fig. 8e and f). We also showed a strong and significant correlation between the changes in VT (r 2 = 0.640; p < 0.001) and VE (r 2 = 0.641; p < 0.001) elicited by hypoxia and the total number of TH+-ir cells in the A5 region (Fig. 9a and c). No correlation was observed between fR and the total number of TH+-ir cells in the A5 region (Fig. 9b).

Depletion of A5 catecholaminergic neurons impairs the ventilatory responses to hypoxia and hypercapnia a Whole-body plethysmography was used to measure tidal volume (VT, ml/kg), respiratory frequency (fR, bpm) and minute ventilation (VE, ml/kg/min) in unanesthetized control or 6-OHDA treated rats during exposure to normoxia (N), hypoxia (8% O2) or hypercapnia (7% CO2). Summary data plotted as b VT, c fR, d VE, e inspiratory time (TI, ms) and f expiratory time (TE, ms) show that 6-OHDA animals have a decreased hypoxia and hypercapnia-induced responses in breathing. *Different from contro, two-way repeated measures ANOVA; p < 0.05. N = 7/group of rats

Correlations between the number of catecholaminergic A5 neurons and the respiratory physiological response elicited by hypoxia or hypercapnia a, b, c, d, e, f X-Y plots of the increase in a, d Tidal volume (ΔVT), b, e Respiratory frequency (ΔfR) and c, f Minute ventilation (ΔVE) elicited by hypoxia or hypercapnia relative to the number of TH+ neurons in the A5 region. Pearson’s r 2 and significance (p) are indicated
Lesion of A5 neurons also reduced VT (p = 0.032) and VE (p = 0.05) and responses induced by hypercapnia (Fig. 8a, b and d). However, the changes in fR (p = 0.944), TI (p = 0.892), and TE (p = 0.891) produced by hypercapnia were not affected by depletion of the A5 neurons (Fig. 8c, e and f). No significant correlation was observed between changes in VT, fR, and VE elicited by hypercapnia and the total number of TH+-ir cells in the A5 region (Fig. 9d–f).
-
6)
Depletion of bulbospinal catecholaminergic neurons in the brainstem reduces the number of sighs during hypoxia
As expected, during the hypoxia condition, we observed a large number of sighs (22.6 ± 0.8 vs. normoxia 1.3 ± 0.5 sighs/5 min; p < 0.001), which are characterized by a large-amplitude event that is triggered by a smaller eupneic event and are followed by a respiratory pause. In the anti-DβH-SAP experimental group, we noticed a significant reduction in the sigh frequency induced by hypoxia (p < 0.001) compared to control, without affecting the number of sighs observed during normoxia (p = 0.302) (Fig. 10a). In the 6-OHDA rats, we also demonstrated an attenuation of number of sighs frequency during hypoxia (p < 0.001), without changing the number of sighs during normoxia (p = 0.678) (Fig. 10b).

Depletion of catecholaminergic neurons reduces the number of sighs generated by hypoxia a Hypoxia-induced sigh rates after saline (control) or spinal cord injections of anti-DβH-SAP to ablate TH-expressing neurons in RVLM. b Hypoxia-induced sigh rates after vehicle (control) or A5 injections of 6-OHDA to ablate TH-expressing neurons in ventrolateral pons. *Different from control, one-way ANOVA, p < 0.05. N = 7–9/group of rats
Discussion
The rostral ventrolateral medulla (RVLM) that harbors the bulbospinal catecholaminergic neurons is considered a heterogeneous neuronal population critical for the autonomic control and much remains unknown about the role of different subclasses of RVLM neurons and the related control of homeostasis that include the breathing activity. The present study provides new findings regarding the contribution of bulbospinal catecholaminergic neurons for breathing control. Firstly, anti-DβH-SAP treatment was effective to deplete bulbospinal catecholaminergic neurons (C1 and A5 neurons) but spare other cell populations such as the serotonergic neurons and astrocytes. Secondly, besides the well-described role of catecholaminergic bulbospinal neurons for the control of sympathetic vasomotor tone, our data demonstrate that selective depletion of catecholaminergic neurons blunted the increase of ventilation induced by hypoxia and hypercapnia. After depletion of the bulbospinal C1 and A5 cells, the hypoxia ventilatory response was reduced due to a reduction in the tachypneic response to hypoxia, while the selective depletion of A5 neurons elicited a reduction in the hypoxia ventilatory response mainly due to a reduction in tidal volume. Finally, lesion of bulbospinal catecholaminergic neurons reduced the number of sighs elicited by hypoxia. Our findings suggest that the bulbospinal catecholaminergic neurons modulate breathing during hypoxic and hypercapnic challenge.
Selective depletion of bulbospinal catecholaminergic neurons by anti-DβH-SAP
The anti-DβH-SAP toxin is considered a very potent tool to selectively destroy neurons that express DβH on their plasma membrane [48]. Previous studies have shown that anti-DβH-SAP was able to effectively eliminate neurons when deposited in the region of the axon terminals or even the cell bodies (C1 cells in the RVLM) [5, 35, 36, 48, 61].
Anti-DβH-SAP into the IML produced a significant reduction of bulbospinal C1 neurons. We believe that the reduction is a reflection of the cell depletion and not a reduction in the immunolabeling of the TH immunohistochemistry for three reasons. First, the quality of the TH stain was evaluated in each rat by counting the number of the TH immunoreactive profiles in the caudal aspects of the VLM (A1 group) or dorsal aspect of the brainstem (A2 group) that do not project to the spinal cord [16, 43]. Our counts in these areas were unaffected by the toxin, then we believe that the TH immunoreactivity was the same in control and lesioned rats. Second, our results of the ablation of cells were also based on counts of retrogradely labeled neurons. If anti-DβH-SAP had merely reduced the level of expression of TH without killing the bulbospinal C1 cells, the number of FG-positive neurons of the rostral aspect of the VLM should have remained unchanged. Third, anti-DβH-SAP also eliminated bulbospinal A5 neurons rather than reduced the TH immunoreactivity.
In addition, the numbers of serotonergic (TrpOH+) and non-catecholaminergic neurons of the VLM bulbospinal projection (FG+/TH−) in rats treated with anti-DβH-SAP were similar to those in control rats. We also found that the number of astrocytes in the VLM was comparable with the control rats. It is important to report that rats treated with anti-DβH-SAP displayed no changes in locomotor activity or behavior, suggesting that other descending spinal projections were intact.
The rostral ventrolateral medulla catecholaminergic neurons regulate the ventilatory response to hypoxia
After elimination of most of the bulbospinal catecholaminergic neurons, rats breathe normally in room air [present results; 48], suggesting that the bulbospinal catecholaminergic neurons (C1 and A5 neurons) are not essential to maintain breathing under resting condition. In fact, we did not expect resting breathing impairment with depletion of the bulbospinal neurons. We should consider that resting breathing is largely maintained by a core of a heterogeneous population of neurons located in the preBötzinger Complex [51]. We can also speculate that brainstem connection of the bulbospinal neurons could be reinnervated by the surviving cells or compensate by some postsynaptic adaptive response.
The fact that C1 bulbospinal neurons do not make an essential contribution to resting physiological function is well discussed in the literature [22]. Based on the various effects attributed to the bulbospinal neurons, it is reasonable to believe that these cells appear to be differentially recruited by pain, hypoxia, inflammation, hemorrhage, and hypoglycemia to produce a repertoire of stereotyped autonomic, metabolic, and neuroendocrine responses that help the organism to survive.
There are some evidences that the increase in breathing elicited by C1 stimulation is mediated via respiratory column activation [10, 26, 55]. The catecholaminergic C1 cells (bulbospinal and non-bulbospinal) are highly collateralized and projected to several pontomedullary structures in addition to the sympathetic preganglionic neurons [3, 19, 22]. A direct connection between the bulbospinal C1 cells and the ventral respiratory column is plausible. Burke and colleagues [10] showed that the selective activation of the bulbospinal C1 neurons increases the activity of neurons in the respiratory column, providing functional evidence that bulbospinal C1 cells can contribute to breathing control. Here, we demonstrated that selective ablation of the bulbospinal neurons reduced the hypoxic ventilatory response in unanesthetized rats. The reduction in the ventilatory response to hypoxia cannot be explained by the existence of a central nervous system hypoxic impairment of the respiratory network [10] because the respiratory rate and amplitude can still be greatly increased by hypercapnia (7% CO2). It is possible that the reduction occurs directly at the level of the bulbospinal C1 or A5 neurons because these cells are strongly activated by hypoxia [15, 26, 40, 56, 63]. The activation of C1 or A5 neurons by hypoxia can produce a breathing stimulation through a connection with the respiratory column, including the inspiratory premotor neurons in the rVRG [10, 11, 21, 22, 26, 27, 55] or other brainstem regions involved in breathing regulation such as the parabrachial/Kolliker-Fuse complex [12, 50, 52], the nucleus of the solitary tract (NTS) [18, 53], or the respiratory rhythm-generating neurons located in the preBötzinger complex [27, 44]. It is important to consider that chemoreceptor neurons located in the retrotrapezoid nucleus (RTN) contribute to the acute ventilatory response to hypoxia [17, 58]; however, a recent study indicates that RTN neurons may temporarily reduce their activity after several minutes of hypoxia due to respiratory alkalosis [7]. Considering this information, we do not believe that bulbospinal neurons in the C1 region will activate breathing through RTN chemoreceptor activation. Ablation of the C1 cells did not affect the ventilatory response to hypercapnia [37; present results]. Evidences also indicate that C1 neurons are not activated by hypercapnia [30, 40]. As previously shown, the ventilatory response to hypercapnia is normally attributed to other neuronal groups such as the classic chemoreceptor neurons within the RTN, the locus coeruleus, or serotonergic and orexinergic neurons [8, 21, 24, 25, 41, 60].
Role of noradrenergic A5 region in breathing control
The catecholaminergic neurons located in the ventrolateral pons, i.e., A5 region, are described to be involved in cardiorespiratory response to hypoxia and hypercapnia [23, 61]. In the present study, our lesion represents approximately 60% of reduction in noradrenergic neurons in the A5 region. This reduction is associated with a decrease in the ventilatory response to hypoxia and hypercapnia, indicating that this nucleus exerts an influence on the chemoreceptor-drive to breathe.
A5 neurons are activated in animals exposed to hypoxia [23, 63]. Inhibition of neural activity in the A5 area attenuated the carotid sympathetic chemoreflex without changing the activity of the phrenic nerve in anesthetized rats or reduced preparations [29, 61, 62]. There is an important issue that must be taken into consideration to interpret these results with those presented in this manuscript. Here, we performed experiments in unanesthetized rats with bilateral lesion of the A5 region, while the previous study performed the experiments in urethane-anesthetized rats [61]. Urethane-anesthesia could certainly interfere with the activity of neurons in the brainstem as demonstrated before [2, 34, 39] leading to a blockade of the role of A5 region in the hypoxia ventilatory response.
The fact that catecholaminergic pontine neurons are activated by hypercapnia stimulation is already known [8, 26, 61, 63]. For example, the result is consistent with the fact that A5 neurons express Fos in animals exposed to hypercapnia [63]. According to the literature, the increase in the A5 neuron activity to hypercapnia was very similar of the A6 activity and the effect of hypoxia on A5 neurons was incomparably larger than the effect of hypercapnia [26].
Our data clearly showed that A5 neurons have a contribution in the chemosensory control of breathing by regulating ventilation mainly due to controlling tidal volume in response to hypoxia and hypercapnia, with no changes in respiratory rate. Under these conditions, i.e., unrestrained awake animals and chemoreceptor activation, it seems that the control of breathing amplitude involves pontine regions, while the control of respiratory rate may recruit medullary areas.
The role of C1 and A5 catecholaminergic neurons in the sigh generation during hypoxia
Hypoxia can trigger sighs in mammals in order to reinflate alveoli [26, 31, 32, 45, 65]. PreBötzinger complex is considered a key area to be involved in the sigh rhythmic activity [31, 32]. Here, we show that hypoxia-triggered sighing is considerably reduced after ablation of the bulbospinal C1 or A5 cells. This effect is consistent with the fact that C1 or A5 neurons can influence the activity of a subset of preBötzinger complex neurons responsible for generating sighs [27]. This increase is presumably mediated by β-adrenergic receptor stimulation [46, 65]. Therefore, C1 and A5 neurons could conceivably be sources of major catecholaminergic inputs to the pre-Bötzinger neurons.
Bulbospinal catecholaminergic neurons and cardiovascular control
The role of bulbospinal catecholaminergic cells in the regulation of sympathetic outflow and blood pressure under resting and hypoxic conditions is well described in the literature [19, 22, 36, 38, 48, 67]. In the present study, MAP was reduced 21 days after selective depletion of the bulbospinal catecholaminergic neurons in conscious unrestrained rats. The reduction in the MAP in our sample is due to the extensive destruction of the major core of the neurons that control sympathetic outflow, suggesting that C1 and A5 neurons may be contributed to the maintenance of resting blood pressure.
The acute hypoxia (10 min of low levels of O2 exposure) used in the present study did not change MAP. This observation is consistent with data from our laboratory showing that hypoxia did not change the LF component of systolic pressure [37]. It seems that our hypoxia protocol produces a biphasic blood pressure response, i.e., hypertension (in the first 20–30 s) followed by hypotension (1–2 min), a return to normal levels, and then stabilization. After depletion of bulbospinal catecholaminergic cells, we expected an opposite result, i.e., a large drop in MAP under hypoxia in bulbospinal-lesioned rats. The MAP increase in lesioned rats is unexpected and should be taken into consideration for at least five reasons. First, our lesions were not large enough to impair the ability of the C1 and A5 cells to buffer blood pressure. Second, other neuronal groups including glutamatergic, sympathoexcitatory, and projections to the spinal cord (e.g., C3 or A6 neurons) could contribute to the maintenance of blood pressure during hypoxia. Third, the exposure of animals to 8% O2 is severe and the reflex could be predominantly mediated by a direct effect of hypoxia in the brainstem and elsewhere rather than by activation of the carotid bodies [4]. Fourth, we should also consider that chronic brain lesions cause circuit reorganization and do not necessarily reproduce the effect of an acute silencing of the targeted neurons [35, 48]. Finally, breathing stimulation during hypoxia was considerably reduced in the lesioned rats. We speculate that this phenomenon could be related with arterial desaturation, and brainstem hypoxia must have been far more severe in the lesioned rats than in the control rats and had impacts on sympathetic activity. However, future experiments are necessary to confirm all of the possibilities described above to better understand the further increase in blood pressure in bulbospinal-lesioned rats.
Our hypoxia challenge produced an increase in HR suggesting the recruitment of sympathetic drive to the heart. Ablation of the C1 and A5 neurons prevented the increase in HR due to a presumably reduction of sympathetic outflow to the heart. Therefore, in this scenario, the C1 and A5 neurons may be considered as the main source of sympathetic input to the heart, and other presympathetic neurons (paraventricular nucleus of the hypothalamus) could compensate for the loss of these cells in order to control sympathetic activity to the blood vessels.
Technical consideration
The toxin anti DβH-SAP is very likely to have an effect on blood pressure at rest and during hypoxia stimulation which can indirectly influence respiratory responses rather being due to a direct input of C1 neurons on the respiratory network. The role of bulbospinal C1 cells in the regulation of sympathetic outflow and blood pressure under resting and hypoxia condition is a well-recognized function of these neurons [13, 19, 22, 25, 33, 35, 36, 38]. Here, we found a small decrease in resting MAP after selective depletion of the bulbospinal C1 neurons in conscious unrestrained rats, suggesting that bulbospinal C1 neurons may be important for the maintenance of resting blood pressure. On the other hand, our lesion did not affect breathing at rest. The depletion of bulbospinal C1 cells only affects breathing under hypoxia condition. This effect is in accordance with the literature, suggesting that the bulbospinal C1 cells are enrolled in emergency situation, such as hypoxia [67]. In addition, a direct connection between the C1 cells and elements of the central respiratory generator is plausible because these cells have axonal collaterals within the VLM that contact respiratory cells [3, 33, 55]. Alternatively, indirect connections between the C1 cells and the respiratory neurons may occur via C1 projections to the RTN, the lateral parabrachial region, the periqueductal gray matter, or even via hypothalamic nuclei such as the dorsomedial nucleus/perifornical region. Accordingly, several evidences indicate that the increase in breathing elicited by C1 stimulation can also be mediated via respiratory column activation [10, 27, 55]. In the present study, we performed experiments in order to better evaluate the role of A5 ablation on breathing control in conscious animals. These experiments indicated that the A5 lesion did not mimic the combined effect of bulbospinal lesion on breathing control. However, we did not properly investigate the effects of A6 and A7 lesions. Therefore, it remains possible that some changes attributed to bulbospinal C1/A5 or A5 neurons could be also due the loss of the A6 and/or A7 neurons. It is important to highlight that the depletion of A6 neurons affects only the breathing response to hypercapnia [8, 9]. Future studies should approach how bulbospinal C1 neurons might contribute to breathing control without interfering with change in blood pressure. In addition, the next set of experiments should also clarify the role of A7 neurons to breathing control during hypoxia and hypercapnia.
Perspectives and conclusions
In the absence of the majority of bulbospinal catecholaminergic neurons, animals continued to have a normal breathing and sighs. Although changes in resting breathing are difficult to accurately determine, rats treated with anti-DβH-SAP or 6-OHDA clearly maintained a measurable baseline ventilatory activity. We believe that the toxins may have caused only minor changes, because the remaining bulbospinal C1 and/or A5 cells combined with other adaptive changes in the ventral respiratory column or in spinal cord can compensate for the loss of a majority of the bulbospinal catecholaminergic cells (Fig. 11). A second interpretation is that seemingly normal breathing function is observed after lesions of the C1 or A5 neurons, because the preBötzinger complex is the main responsible to maintain breathing automaticity (Fig. 11). In fact, our work showed a role of bulbospinal catecholaminergic cells to control breathing under hypoxic and hypercapnic conditions, suggesting that these cells are not just involved in the regulation of sympathetic outflow, but also to maintain homeostasis in a chemosensory challenge.

Contribution of the C1 cells to the autonomic and respiratory responses to hypoxia and hypercapnia Schematic model showing that hypoxia activates the C1 or A5 cells via a disynaptic pathway that consists of carotid body afferents activated by a reduction in PO2 or increase in PCO2/H+ and a projection from the caudal aspect of the NTS to the ventrolateral medulla or the ventrolateral pontine region. Central hypoxia may also directly or indirectly (via the glia) activate the C1 cells (Angelova et al. [4]). C1 cells regulate sympathetic nerve activity through the preganglionic sympathetic neurons (classic pathway described by C1 cells in autonomic control; Guyenet et al. [22]). Activation of the C1 cells by hypoxia may also increase breathing via stimulation of the ventral respiratory column (VRC). Hypoxia also probably activates the cardiovagal outflow via direct inputs from C1 neurons to cardiovagal neurons located in the dorso motor vagus nucleus (Depuy et al. [14]). In addition, hypoxia activates C1 neurons that project to the hypothalamus in order to increase CRH and ACTH release (Schiltz and Sawchenko [47]; Silva et al. [50])
Signals from central chemoreceptors may directly or indirectly affect the activity of several medullary areas, including the RTN/pFRG, which affect sympathetic nerve activity and breathing (Guyenet [20]; Takakura et al. [59]). An essential step for hypercania-induced breathing is activation of RTN/pFRG neurons by increased levels of PCO2/H+, which, in turn, send excitatory signals to activate the VRC, either directly or through activation of excitatory receptors in the C1 region (Takakura and Moreira [57]; Takakura et al. [59]). Signals from the RTN that activate excitatory receptors in the C1 region may also increase sympathetic activity.
References
Abbott SB, DePuy SD, Nguyen T, Coates MB, Stornetta RL, Guyenet PG (2013) Selective optogenetic activation of rostral ventrolateral medullary catecholaminergic neurons produces cardiorespiratory stimulation in conscious mice. J Neurosci 33:3164–3177
Accorsi-mendonça D, Leao RM, Aguiar JF, Varanda WA, Machado BH (2007) Urethane inhibits the GABAergic neurotransmission in the nucleus of solitary tract (NTS) of rat brainstem slices. Am J Physiol Regul Integr Comp Physiol 292:R396–R402
Agassandian K, Shan Z, Raizada M, Sved AF, Card JP (2012) C1 catecholamine neurons form local circuit synaptic connections within the rostroventrolateral medulla of rat. Neuroscience 227:247–259
Angelova PR, Kasymov V, Christie I, Sheikhbahaei S, Turovsky E, Marina N, Korsak A, Zwicker J, Teschemacher AG, Ackland GL, Funk GD, Kasparov S, Abramov AY, Gourine AV (2015) Functional oxygen sensitivity of astrocytes. J Neurosci 35:10460–10473
Barna BF, Takakura AC, Moreira TS (2014) Acute exercise-induced activation of Phox2b-expressing neurons of the retrotrapezoid nucleus in rats may involve the hypothalamus. Neuroscience 258:355–363
Barna BF, Takakura AC, Mulkey DK, Moreira TS (2016) Purinergic receptor blockade in the retrotrapezoid nucleus attenuates the respiratory chemoreflexes in awake rats. Acta Physiol 217:80–93
Basting TM, Burke PG, Kanbar R, Viar KE, Stornetta DS, Stornetta RL, Guyenet PG (2015) Hypoxia silences retrotrapezoid nucleus respiratory chemoreceptors via alkalosis. J Neurosci 35:527–543
Biancardi V, Bícego KC, Almeida MC, Gargaglioni LH (2008) Locus coeruleus noradrenergic neurons and CO2 drive to breathing. Pflugers Arch 455:1119–1128
Biancardi V, da Silva LT, Bícego KC, Gargaglioni LH (2010) Role of locus coeruleus noradrenergic neurons in cardiorespiratory and thermal control during hypoxia. Respir Physiol Neurobiol 28:150–156
Burke PG, Abbott SB, Coates MB, Viar KE, Stornetta RL, Guyenet PG (2014) Optogenetic stimulation of adrenergic C1 neurons causes sleep state-dependent cardiorespiratory stimulation and arousal with sighs in rats. Am J Respir Crit Care Med 190:1301–1310
Card JP, Sved JC, Craig B, Raizada M, Vazquez J, Sved AF (2006) Efferent projections of rat rostroventrolateral medulla C1 catecholamine neurons: implications for the central control of cardiovascular regulation. J Comp Neurol 499:840–859
Damasceno RS, Takakura AC, Moreira TS (2014) Regulation of the chemosensory control of breathing by Kölliker-Fuse neurons. Am J Physiol Regul Integr Comp Physiol 307:R57–R67
Dampney RA (1994) Functional organization of central pathways regulating the cardiovascular system. Physiol Rev 74:323–364
DePuy SD, Stornetta RL, Bochorishvili G, Deisseroth K, Witten I, Coates M, Guyenet PG (2013) Glutamatergic neurotransmission between the C1 neurons and the parasympathetic preganglionic neurons of the dorsal motor nucleus of the vagus. J Neurosci 33(4):1486–1497
Erickson JT, Millhorn DE (1994) Hypoxia and electrical stimulation of the carotid sinus nerve induce c-Fos-like immunoreactivity within catecholaminergic and serotoninergic neurons of the rat brainstem. J Comp Neurol 348:161–182
Freiria-Oliveira AH, Blanch GT, Pedrino GR, Cravo SL, Murphy D, Menani JV, Colombari DS (2015) Catecholaminergic neurons in the comissural region of the nucleus of the solitary tract modulate hyperosmolality-induced responses. Am J Physiol Regul Integr Comp Physiol 309:R1082–R1091
Gourine AV, Llaudet E, Dale N, Spyer KM (2005) Release of ATP in the ventral medulla during hypoxia in rats: role in hypoxic ventilatory response. J Neurosci 25:1211–1218
Gozal D, Xue YD, Simakajornboon N (1999) Hypoxia induces c-Fos protein expression in NMDA but not AMPA glutamate receptor labeled neurons within the nucleus tractussolitarii of the conscious rat. Neurosci Lett 262:93–96
Guyenet PG (2006) The sympathetic control of blood pressure. Nat Rev Neurosci 7:335–346
Guyenet PG (2014) Regulation of breathing and autonomic outflows by chemoreceptors. Compr Physiol 4(4):1511–1562
Guyenet PG, Bayliss DA (2015) Neural control of breathing and CO2 homeostasis. Neuron 87:946–961
Guyenet PG, Stornetta RL, Bochorishvili G, DePuy SD, Burke PGR, Abbott SBG (2013) C1 neurons: the body’s EMTs. Am J Physiol Regul Integr Comp Physiol 305:187–204
Hirooka Y, Polson JW, Potts PD, Dampney RAL (1997) Hypoxia-induced Fos expression in neurons projecting to the pressor region in the rostral ventrolateral medulla. Neuroscience 80:1209–1224
Hodges MR, Tattersall GJ, Harris MB, McEvoy SD, Richerson DN, Deneris ES, Johnson RL, Chen ZF, Richerson GB (2008) Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. J Neurosci 28:2495–2505
Kanbar R, Stornetta RL, Cash DR, Lewis SJ, Guyenet PG (2010) Photostimulation of Phox2b medullary neurons activates cardiorespiratory function in conscious rats. Am J Respir Crit Care Med 182:1184–1194
Kanbar R, Depuy SD, West GH, Stornetta RL, Guyenet PG (2011) Regulation of visceral sympathetic tone by A5 noradrenergic neurons in rodents. J Physiol 589:903–917
Kang JJ, Liang WH, Lam CS, Huang XF, Yang SJ, Wong-Riley MT, Fung ML, Liu YY (2016) Catecholaminergic neurons in synaptic connections with pre-Bötzinger complex neurons in the rostral ventrolateral medulla in normoxic and daily acute intermittent hypoxic rats. Exp Neurol 287:165–175
King TL, Ruyle BC, Kline DD, Heesch CM, Hasser EM (2015) Catecholaminergic neurons projecting to the paraventricular nucleus of the hypothalamus are essential for cardiorespiratory adjustments to hypoxia. Am J Physiol Regul Integr Comp Physiol 309:R721–R731
Koshiya N, Guyenet PG (1994) A5 noradrenergic neurons and the carotid sympathetic chemoreflex. Am J Phys 267:R519–R526
Lazarenko RM, Milner TA, Depuy SD, Stornetta RL, West GH, Kievits JA, Bayliss DA, Guyenet PG (2009) Acid sensitivity and ultrastructure of the retrotrapezoid nucleus in Phox2b-EGFP transgenic mice. J Comp Neurol 517:69–86
Li AJ, Wang Q, Davis H, Wang R, Ritter S (2015) Orexin-a enhances feeding in male rats by activating hindbrain catecholamine neurons. Am J Physiol Regul Integr Comp Physiol 309:R358–R367
Lieske SP, Thoby-Brisson M, Telgkamp P, Ramirez JM (2000) Reconfiguration of the neural network controlling multiple breathing patterns: eupnea, sighs and gasps. Nat Neurosci 6:600–607
Lipski J, Kanjhan R, Kruszewska B, Smith M (1995) Barosensitive neurons in the rostral ventrolateral medulla of the rat in vivo: morphological properties and relationship to C1 adrenergic neurons. Neuroscience 69:601–618
Machado BH, Bonagamba LG (1992) Microinjection of S-nitrosocysteine into the nucleus tractus solitarii of conscious rats decreases arterial pressure but L-glutamate does not. Eur J Pharmacol 221:179–182
Madden CJ, Sved AF (2003) Cardiovascular regulation after destruction of the C1 cell group of the rostral ventrolateral medulla in rats. Am J Physiol Heart Circ Physiol 285:H2734–H2748
Madden CJ, Ito S, Rinaman L, Wiley RG, Sved AF (1999) Lesions of the C1 catecholaminergic neurons of the ventrolateral medulla in rats using anti-DbetaH-saporin. Am J Phys 277:R1063–R1075
Malheiros-Lima MR, Takakura AC, Moreira TS (2017) Depletion of rostral ventrolateral medullary catecholaminergic neurons impairs the hypoxic ventilatory response in conscious rats. Neuroscience 351:1–14
Menuet C, Le S, Dempsey B, Connelly AA, Kamar JL, Jancovski N, Bassi JK, Walters K, Simms AE, Hammond A, Fong AY, Goodchild AK, McMullan S, Allen AM (2017) Excessive respiratory modulation of blood pressure triggers hypertension. Cell Metab 25:739–748
Moreira TS, Takakura AC (2011) Initiating inspiration outside the medulla does produce eupneic breathing. What is the role of brain stem neurons in eupneic breathing. J Appl Physiol 110:857–859
Moreira TS, Takakura AC, Colombari E, Guyenet PG (2006) Central chemoreceptors and sympathetic vasomotor outflow. J Physiol 577:369–386
Nattie E, Li A (2010) Central chemoreception in wakefulness and sleep: evidence for a distributed network and a role for orexin. J Appl Physiol 108:1417–1424
Paxinos G & Watson C (1998). The rat brain in stereotaxic coordinates
Pedrino GR, Maurino I, de Almeida Colombari DS, Cravo SL (2006) Role of catecholaminergic neurones of the caudal ventrolateral medulla in cardiovascular responses induced by acute changes in circulating volume in rats. Exp Physiol 91:995–1005
Pilowsky PM, Jiang C, Lipski J (1990) An intracellular study of respiratory neurons in the rostral ventrolateral medulla of the rat and their relationship to catecholamine-containing neurons. J Comp Neurol 301:604–617
Qureshi M, Khalil M, Kwiatkowski K, Alvaro RE (2009) Morphology of sighs and their role in the control of breathing in preterm infants, term infants and adults. Neonatology 96:43–49
Ramirez JM, Viemari JC (2005) Determinants of inspiratory activity. Respir Physiol Neurobiol 147:145–157
Schiltz JC, Sawchenko PE (2007) Specificity and generality of the involvement of catecholaminergic afferents in hypothalamic responses to immune insults. J Comp Neurol 502(3):455–467
Schreihofer AM, Guyenet PG (2000) Sympathetic reflexes after depletion of bulbospinal catecholaminergic neurons with anti-DH-saporin. Am J Physiol Regul Integr Comp Physiol 279:R729–R742
Schreihofer AM, Guyenet PG (2003) Baro-activated neurons with pulse-modulated activity in the rat caudal ventrolateral medulla express GAD67 mRNA. J Neurophysiol 89:1265–1277
Silva JN, Lucena EV, Silva TM, Damasceno RS, Takakura AC, Moreira TS (2016) Inhibition of the pontine Kölliker-Fuse nucleus reduces genioglossal activity elicited by stimulation of the retrotrapezoid chemoreceptor neurons. Neuroscience 22:328–9-21
Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL (1991) Pre-Bötzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science 254:726–729
Song G, Poon CS (2009) Lateral parabrachial nucleus mediates shortening of expiration during hypoxia. Respir Physiol Neurobiol 165:1–8
Song G, Xu H, Wang H, Macdonald SM, Poon CS (2011) Hypoxia-excited neurons in NTS send axonal projections to Kölliker-Fuse/parabrachial complex in dorsolateral pons. Neuroscience 175:145–153
Stornetta RL, Sevigny CP, Guyenet PG (2003) Inspiratory augmenting bulbospinal neurons express both glutamatergic and enkephalinergic phenotypes. J Comp Neurol 455:113–124
Stornetta RL, Inglis MA, Viar KE, Guyenet PG (2015) Afferent and efferent connections of C1 cells with spinal cord or hypothalamic projections in mice. Brain Struct Funct 221:4027–4044
Sun MK, Reis DJ (1996) Excitatory amino acid-mediated chemoreflex excitation of respiratory neurones in rostral ventrolateral medulla in rats. J Physiol 492:559–571
Takakura AC, Moreira TS (2011) Contribution of excitatory amino acid receptors of the retrotrapezoid nucleus to the sympathetic chemoreflex in rats. Exp Physiol 96(10):989–999
Takakura AC, Moreira TS, Colombari E, West GH, Stornetta RL, Guyenet PG (2006) Peripheral chemoreceptor inputs to retrotrapezoid nucleus (RTN) CO2-sensitive neurons in rats. J Physiol 572:503–523
Takakura AC, Colombari E, Menani JV, Moreira TS (2011) Ventrolateral medulla mechanisms involved in cardiorespiratory responses to central chemoreceptor activation in rats. Am J Physiol Regul Integr Comp Physiol 300(2):R501–510
Takakura AC, Barna BF, Cruz JC, Colombari E, Moreira TS (2014) Phox2b-expressing retrotrapezoid neurons and the integration of central and peripheral chemosensory control of breathing in conscious rats. Exp Physiol 99:571–585
Taxini CL, Takakura AC, Gargaglioni LH, Moreira TS (2011) Control of the central chemoreflex by A5 noradrenergic neurons in rats. Neuroscience 199:177–186
Taxini CL, Moreira TS, Takakura AC, Bícego KC, Gargaglioni LH, Zoccal DB (2017) Role of A5 noradrenergic neurons in the chemoreflex control of respiratory and sympathetic activities in unanesthetized conditions. Neuroscience 354:146–157
Teppema LJ, Veening JG, Kranenburg A, Dahan A, Berkenbosch A, Olievier C (1997) Expression of c-fos in the rat brainstem after exposure to hypoxia and to normoxic and hyperoxic hypercapnia. J Comp Neurol 388:169–190
Tucker DC, Saper CB, Ruggiero DA, Reis DJ (1987) Organization of central adrenergic pathways: I. Relationships of ventrolateral medullary projections to the hypothalamus and spinal cord. J Comp Neurol 259:591–603
Viemari JC, Garcia AJ III, Doi A, Elsen G, Ramirez JM (2013) β-Noradrenergic receptor activation specifically modulates the generation of sighs in vivo and in vitro. Front Neural Circuits 7:1–14
Wenker IC, Sobrinho CR, Takakura AC, Mulkey DK, Moreira TS (2013) P2Y1 receptors expressed by C1 neurons determine peripheral chemoreceptor modulation of breathing, sympathetic activity, and blood pressure. Hypertension 62:263–273
Wenker IC, Abe C, Viar KE, Stornetta DS, Stornetta RL, Guyenet PG (2017) Blood pressure regulation by the rostral Ventrolateral medulla in conscious rats: effects of hypoxia, Hypercapnia, baroreceptor denervation, and anesthesia. J Neurosci 37:4565–4583
Acknowledgements
This work was supported by the São Paulo Research Foundation (FAPESP; grants: 2014/22406-1 to ACT; 2015/23376-1 to TSM) and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; grant: 471263/2013-3 to ACT and 471283/2012-6 to TSM). FAPESP fellowship (2014/07698-6 to MRML) and CNPq fellowship (301219/2016-8 to ACT and 301904/2015-4 to TSM).
Author information
Authors and Affiliations
Contributions
MRML, ACT, and TSM designed the research; MRML and LTT performed the research; MRML, LTT, and TSM analyzed the data; and MRML, ACT, and TSM wrote the paper.
Corresponding author
Rights and permissions
About this article

Cite this article
Malheiros-Lima, M.R., Totola, L.T., Takakura, A.C. et al. Impaired chemosensory control of breathing after depletion of bulbospinal catecholaminergic neurons in rats. Pflugers Arch - Eur J Physiol 470, 277–293 (2018). https://doi.org/10.1007/s00424-017-2078-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-017-2078-8