
Abstract
The peripheral (PNS) and central nervous system (CNS) are delicate structures, highly sensitive to homeostatic changes—and crucial for basic vital functions. Thus, a selection of barriers ensures the protection of the nervous system from noxious blood-borne or surrounding stimuli. In this chapter, anatomy and functioning of the blood–nerve (BNB), the blood–brain (BBB), and the blood–spinal cord barriers (BSCB) are presented and the key tight junction (TJ) proteins described: claudin-1, claudin-3, claudin-5, claudin-11, claudin-12, claudin-19, occludin, Zona occludens-1 (ZO-1), and tricellulin are by now identified as relevant for nerval barriers. Different diseases can lead to or be accompanied by neural barrier disruption, and impairment of these barriers worsens pathology. Peripheral nerve injury and inflammatory polyneuropathy cause an increased permeability of BNB as well as BSCB, while, e.g., diseases of the CNS such as amyotrophic lateral sclerosis, multiple sclerosis, spinal cord injury, or Alzheimer’s disease can progress and worsen through barrier dysfunction. Moreover, the complex role and regulation of the BBB after ischemic stroke is described. On the other side, PNS and CNS barriers hamper the delivery of drugs in diseases when the barrier is intact, e.g., in certain neurodegenerative diseases or inflammatory pain. Understanding of the barrier - regulating processes has already lead to the discovery of new molecules as drug enhancers. In summary, the knowledge of all of these mechanisms might ultimately lead to the invention of drugs to control barrier function to help ameliorating or curing neurological diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Anatomy and function
Barriers in peripheral nerves: blood–nerve and myelin barriers
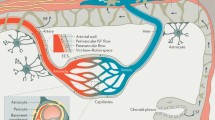
The peripheral nerve has several protective measures to shield itself from external influence. Each axon is surrounded by the endoneurial layer (Fig. 1). A number of axons together form a fascicle, surrounded by a protective layer, the perineurium. A group of fascicles, in turn, is comprised by the rather solid epineurium, an extension of the dura mater. While complexity and thickness vary across species, two structures have been shown to be key to the blood–nerve barrier (BNB)—the perineurium and endoneurial vessels. The perineurium can be divided into two parts, the external pars fibrosa and the internal pars epitheloidea. While the first has mainly mechanic functions, it is the latter that serves as diffusion barrier. It wraps around the fascicle in several lamellar layers, each one cell thick. Every layer is covered by a basal lamina, composed of epithelioid myofibroblasts, which account for stretch compliance properties [11]. The cells, flattened squamous cells of non-polarized architecture, are interconnected by tight junctions (TJs), gap junctions, and adherens junctions to maintain homeostasis. TJ proteins in the perineurium include zona occludens-1 (ZO-1), claudin-1, claudin-3, and occludin [87] as well as claudin-19 and tricellulin [94]. Perineurial cells originate from the central nervous system (CNS), arising as ventral spinal cord glia before migrating into the periphery [61]. Schwann cell-derived desert hedgehog protein (DHH) signals the formation of the connective tissue sheath around peripheral nerves [82].

Barriers in the peripheral nerve. The blood–nerve barrier (BNB) consists of the perineurium (right insert) and endoneurial vessels. The mesaxon, Schmidt–Lantermann incisures, and the perinodal region form the myelin barrier (left insert). Schematic display of main components as well as TJ proteins found in the histological sections. Red dots mark TJs formed in compact myelin
The endoneurium contains endoneurial fluid, similar to the cerebrospinal fluid of the CNS. To maintain homeostasis and protect the nerval microenvironment, blood-borne toxins have to be minimized, yet a controlled blood-nerve exchange must be allowed to nourish nerval and other tissues. This regulation is maintained by endothelial cells of endoneurial vessels, an intrinsic vasculature, occupying about 1% of the endoneurial area. They are sealed by TJs, more permeable than the perineurium. Rather leaky at birth, endoneurial TJs develop and tighten only gradually [57] [87]. Their main constituents are claudin-5, occludin, and ZO-1 [74].
Besides the BNB, myelinating Schwann cells constitute a further feature to protect the peripheral nerve, the myelin barrier. These glia cells wrap around neurons in multiple sheaths to protect and isolate neurons thereby increasing their conductance velocity. The mesaxon is the double-layered membrane of a Schwann cell that envelops a nerve axon. While the outer mesaxon connects to the compact myelin sheath, the inner mesaxon continues to the last sheath opposite the neuronal cell membrane. TJ proteins expressed are claudin-1, claudin-3, claudin-19, occludin, ZO-1, and tricellulin [2].
The process of myelination leaves small clefts referred to as Schmidt–Lantermann incisures, which allow communication between layers by connecting Schwann cell cytoplasm to the inner layer of the myelin sheath. TJ proteins found here are claudin-1, claudin-3, claudin-5, claudin-12, occludin, and ZO-1. Myelination of the nerve is interrupted by node of Ranvier to facilitate salutatory nerve conduction. Close by, in the paranodal region, the barrier is sealed by claudin-1, claudin-19, occludin, ZO-1, and tricellulin [2].
Associated with the myelin barrier is an additional protein, peripheral myelin protein 22 (PMP22). It is a 22-kDa transmembrane glycoprotein made up of 160 amino acids. PMP22 expression is highest in myelinating Schwann cells of peripheral nerves, where it plays an essential role in the formation and maintenance of compact myelin [78]. PMP22-like immunoreactivity is associated with markers of the tight junctional complex, including ZO-1 and occludin. Upon disruption of intercellular contacts, PMP22 is internalized into vesicles [78]. PMP22 deficiency affects nerve conduction not through removal of myelin, but through disruption of myelin junctions [32]. It is the underlying cause for an inherited neuropathy, hereditary neuropathy with liability to pressure palsies (HNPP). Duplication of PMP22 leads to a neuropathy called Charcot–Marie–Tooth disease type 1A (CMT1A) (both reviewed in [102]).
Blood–brain barrier
The existence of the blood–brain barrier (BBB) was first described by Ehrlich more than 100 years ago. The endothelial BBB, composed of the highly specialized CNS microvascular endothelial cells, and the epithelial blood–cerebrospinal fluid barrier (BCSFB), composed of the choroid plexus epithelial cells, protect the CNS from the constantly changing milieu in the blood stream, as well as infections and toxins (reviewed in [108]). In addition, brain capillaries are covered by mature pericytes sharing the basement membrane with the endothelium. Astrocytic end feet form the outer layer of the mature capillaries. Pericytes and astrocytes secret matrix proteins of the basement membrane. The highly coordinated activity of multiple cell types including vascular cells (endothelial cells, pericytes, and smooth muscle cells), glia (astrocytes, oligodendroglia, and microglia), and neurons is summoned as the neurovascular unit.
Endothelial cells seal the barrier via TJ proteins including claudin-3, claudin-5, and claudin-12, summarized in [34, 79]. The expression of claudin-1 is low and probably dependent on the species. Claudin-5 is mainly localized in capillary endothelial cells and less in choroid plexus epithelial cells. Claudin-5 is >100 times higher expressed than any other claudin and it dominates the TJs of the BBB [80]. Besides claudins, occludin and tricellulin are found in the BBB [43]: numerous reports demonstrate cell–cell contact localization of occludin in vivo and in primary brain ECs; however, its function is beyond barrier sealing (reviewed in [34, 79]). Tricellulin was recently identified in tricellular and less in bicellular contacts of human brain endothelium [43]. In addition to TJ proteins, endothelial cells express multiple substrate-specific transport systems that control transport of nutrients, energy metabolites, and other essential molecules from blood into the brain and the transport of metabolic waste products from the brain’s interstitial fluid into the blood. Pericytes share a basement membrane with endothelial cells and form direct synaptic-like peg-socket focal contacts with endothelium through N-cadherin and connexins, allowing exchanges of ions, metabolites, second messengers, and ribonucleic acids between the two cell types. Astrocytes contribute to the dynamic regulations in the neural system, but it remains unclear whether astrocytes are essential for BBB maintenance.
Blood–spinal cord barrier
The spinal cord, part of the CNS, connects the brain to the PNS. Its center contains the gray matter, neuronal perikarya, while the surrounding white matter consists of axons (reviewed in [6]). The spinal cord is, analogous to the brain, surrounded by three meninges, pia, arachnoidea, and dura. As a homeostatic microenvironment is also of utmost importance to the spinal cord function, it is protected from external, blood-borne influence by the blood–spinal cord barrier (BSCB). As it is mainly formed by non-fenestrated capillary endothelial cells, a basal lamina, and pericyte and astrocyte foot processes, one crucial TJ constituent is claudin-5, along with occludin and ZO-1 [109]. Compared with the BBB, it is more permeable for tracers [86] and cytokines [81], partly due to a relative decrease in occludin and ZO-1 [6]. Expression of claudin-1 and claudin-5, however, resembles the BBB. The BSCB and BBB together are sometimes referred to as blood–CNS barrier (BCNSB).
Specific TJ proteins in the nervous system
ZO-1
ZO-1 is not a transmembrane protein but a cytoplasmatic TJ-associated protein, organizing its composition (reviewed in [31]). TJ proteins are connected to cortical actin cytoskeleton via multi-domain scaffolding proteins of the peripheral membrane-associated guanylate kinase (MAGUK) family, i.e., ZO-1, ZO-2, and ZO-3. ZO-1 and ZO-2 independently determine whether and where claudins are polymerized [101]. ZO-1 deficiency disrupts TJs, and reduced ZO-1 levels are associated with barrier breakdown in many neurological disorders [50].
Claudin-1
The first and most prominent member of the claudin family, claudin-1, was described in 1998 by Furuse et al. [25]. It is a 22-kDa protein, consisting of 211 amino acids and present in various tissues. Besides liver and intestines, claudin-1 is expressed both in the PNS as well as in the CNS and plays a pivotal role especially in maintaining the BNB. In the peripheral nerve, it is found mainly in the perineurium, in endoneurial vessels, as well as in the mesaxon, Schmidt–Lantermann incisures, and paranodal loops of myelinating Schwann cells [2, 85]. Claudin-1 binds to the PDZ domain of ZO-1 [42]. Moreover, it forms dimers with other claudins, such as claudin-2 and claudin-3 (reviewed in [7]). Claudin-1 null mice have a lethal phenotype, owing to cutaneous dehydration caused by absence in epidermal stratum granulosum TJs [26]. This suggests such crucial role for claudin-1 in TJs that cannot be compensated for by other TJ proteins.
Claudin-1 is regulated by several factors. Serin threonine kinases such as glycogen synthase kinase 3 (GSK-3) control TJ stability by inducing expression of claudin-1 and occludin, amongst others [95]. Another group of regulators are microRNAs (miR). MiRs are additional post-transcriptional regulators of gene expression that can silence target genes by translation inhibition, messenger RNA (mRNA) decay, or both. In intestinal epithelia, for example, application of miR-29b leads to downregulation of claudin-1 and consequent breakdown of the epithelial intestinal barrier [110]. Perineurial injection of miR-183 mimics reduces both claudin-1 RNA and protein expression [107].
Claudin-3
Claudin-3, a sealing protein of 23 kDa, is ubiquitously expressed in almost all organs with the highest levels of expression in intestine, lung, liver, and testis. Expression in the whole brain is rather low, but it is detectable at the borders of BBB-forming endothelial cells in various species (reviewed in [34]). In the peripheral nerve, claudin-3 is located in the Schmidt–Lantermann incisures and in the mesaxon in humans but not in rodents [2]. As early as 23 weeks, claudin-3 is detectable in the fetus, and Schmidt–Lantermann incisures can be identified by 37 weeks [87].
Claudin-3 is important for the maintenance of the BBB [34]. Claudin-3 KO have an impaired BCSFB and experience a more rapid onset and exacerbated clinical signs of experimental autoimmune encephalomyelitis. This coincides with enhanced levels of infiltrated leukocytes in their CSF [54]. In brain endothelial cells, inactivation of β-catenin causes significant downregulation of claudin-3, up-regulation of plasmalemma vesicle-associated protein, and BBB breakdown [68].
Claudin-5
Claudin-5 has first been described as mutated in congenital velo-cardio-facial syndrome and DiGeorge syndrome, causing cleft palate, heart defects, and facial dysmorphism [97], hence its initial symbol transmembrane protein deleted in VCFS (TMVCF). It is a 31.6-kDa protein, expressed in a multitude of tissues including lung, heart, gut, and skeletal muscle. Like claudin-1, it can form homo-dimers [7]. It was the first claudin to be identified as specifically endothelial by Morita et al. in 1999 [74]. It appears crucial in maintaining the BBB, causing a stronger barrier sealing in the BBB than in endothelia of other tissues. This is supported by the KO phenotype: null mice show BBB permeation for molecules of ∼800 Da. Though macroscopically normal, mice die within 10 days after birth [77]. In the peripheral nerve, it is present in endothelial cells of endoneurial vessels, as well as mesaxons and Schmidt–Lantermann incisures [85].
Claudin-11
When claudin-11 was first described, it was named oligodendrocyte-specific protein (OSP). As a 24-kDa transmembrane protein, it was then assigned to the group of TJ proteins (claudin-11) found in CNS myelin and testis [28]. Tight junctions of the BCSFB are characterized by parallel running particle strands probably induced by claudin-11. In addition to its sealing properties, claudin-11 acts as an autoantigen in the development of autoimmune demyelinating disease [51]. Recent evidence demonstrated that anti-claudin-11 antibodies do not recognize native glial claudin-11 and may therefore rather represent an epiphenomenon in multiple sclerosis [4].
In myelinated nerve fibers, claudin-11 is located in the outer mesaxon and paranodal loops and in Schmidt–Lantermann incisures [15]. Claudin-11 affords rapid nerve conduction principally for small diameter myelinated axon. Claudin-11 KO mice have motor deficiencies, slowed CNS conduction, and males are sterile. Myelin and axonal structures are kept, but the insulating properties are damaged thereby hampering proper signal conduction. In addition, the action potential threshold is increased and potassium channels are activated in claudin-11 KO [16]. Myelin lacking claudin-11 is more permeable to water and small osmolytes but has a preserved structure, stability, and membrane interaction [15]. In summary, claudin-11 creates an electrically tight barrier in myelinated nerves.
Claudin-12
Claudin-12, a 27.1-kDa protein, is one of the few claudins that do not possess a PDZ binding motif. Phylogenetically, it appears to be only distantly related to all other claudins (reviewed in [31]). Claudin-12 is located in the intestine as well as in the BBB and in the BNB. In rat brain capillary cells, expression of claudin-12 mRNA is 751-fold lower than that of claudin-5 [80]. In absence of claudin-5, claudin-12-based TJ in brain–blood vessels appear to function as a molecular sieve: they allow only small molecules (less than 800 Da) to pass across TJs [77]. Claudin-12 seems to be involved in paracellular Ca2+ permeation, since knock-down decreases the effect of vitamin D-induced Ca2+ transfer across epithelial cell cultures [22].
Claudin-19
Claudin-19, of 23 kDa, is the only claudin expressed in the PNS but not in the CNS. It is located not only in Schwann cells paranodal region, mesaxon [2], facilitating TJ assembly [32] but also in the perineurium [94]. Claudin-19 KO mice are vital and fertile but have motor deficit resembling a peripheral neuropathy and lack TJ building in Schwann cells [72]. However, Schwann cell wrapping and forming of node of Ranvier is not impaired, suggesting no role of claudin-19 in cell polarity [72]. Moreover, claudin-19 functions as Cl− blocker. Together with claudin-16 as Na+ channel, it regulates paracellular ion reabsorption in TJs, e.g., in the kidney [38].
Occludin
Occludin was the first integral TJ membrane protein to be discovered by Furuse et al. in 1993 [23]. It is a 56-kDa peptide with four transmembrane segments. It is associated with ZO-1 through its long COOH-terminal cytoplasmic domain that is required for TJ localization [24]. In the peripheral nervous system, it is present in the perineurium, in endothelial cells of endoneurial vessels as well as in Schmidt–Lantermann incisures and mesaxon of myelinating Schwann cells [2]. In the CNS, it constitutes endothelial TJs of both BSCB and BNB. Occludin KO mice are vital but have retarded postnatal growth [91]. Fertility is reduced in males whereas females produce expected litters when mated with wild-type. In occludin KO mice, TJs appear intact, as does the intestinal barrier function. Yet, histological pathologies were observed, such as chronic inflammation and hyperplasia of the gastric epithelium, calcification in the brain, testicular atrophy, loss of cytoplasmic granules in striated duct cells of the salivary gland, and thinning of the compact bone [91]. This is in line with findings that lack of occludin does not necessarily compromise TJ function: Ikenouchi et al. have shown that tricellulin, a protein located in tricellular junctions, can partially take over occludin function in bicellular junctions [41]. Tyrosine phosphorylation in occludin leads to disassembly and dysfunction of the BBB, e.g., through oxidative stress. In this context, it seems that the MAPK-ERK pathway is crucial in maintaining the barrier [92].
Tricellulin
Tricellulin (also known as MARVELD2) is localized mainly in tricellular TJs and sparsely in bicellular TJs [40]. It is considered to play a key role in the organization and function of both tricellular and bicellular TJs [45]. Tricellulin is widely expressed in various epithelial cells, including the intestine, hepatocytes, nasal mucosa, cochlear hair cells and the perineurium [94]. It is also detected in non-epithelial cells, such as nerve fibers, microglia, and Schwann cells [53]. Tricellulin is localized in the same areas as claudin-19: the mesaxon, Schmidt–Lantermann incisures, and paranodal loops. The expression level of tricellulin mRNA is about 10-fold higher in the sciatic nerve than in the spinal cord or cerebrum, pointing towards a critical role of tricellulin in the peripheral nerve [53]. Tricellulin protein and mRNA are also found in choroidal epithelial cells in vitro localized at the tricellular contacts, co-localizing with occludin.
Overexpression of tricellulin in epithelial cells forms a barrier in tricellular TJs that only prevents macromolecule penetration [58]. As tricellular TJs are supposed to be a weak point of the total network, tricellulin plays a regulating and tightening role here to establish the paracellular barrier. Tricellulin KO mice suffer from early-onset rapidly progressive hearing loss associated with the degeneration of hair cells [45]. Mutations in tricellulin lead to nonsyndromic hearing loss (DFNB49) [75]. Downregulation of tricellulin by small interfering RNA (siRNA) increases the blood–cerebrospinal fluid barrier permeability corroborated by decreased transendothelial electrical resistance and an increased FITC-dextran flux [99].
In conclusion, tricellulin and occludin regulate the passage of macromolecules through the TJ. Tricellulin directly limits the passage through tricellular TJs and removes this limit when downregulated [60]. Occludin downregulation seems to increase macromolecule permeability either directly by own action or indirectly by pulling tricellulin out of tricellular TJs or both [1, 59, 13].
Diseases
Nerve injury
Nerve injury denotes the damage of a peripheral nerve, i.e., either damage of the myelin with the axon intact (neuropraxia), the damage of the axon itself, but sparing the epineurium (axonotmesis), or damage of the entire neuron (neurotmesis). In animals, peripheral nerve injury leads to a disruption of the BNB. After crush injury of the rat sciatic nerve, mRNA for claudin-1, claudin-5, and occludin are quickly downregulated in the endoneurium and perineurium, but gradually recover from day 2 on [36]. Also in rats, after chronic constriction injury (CCI; a ligation of the sciatic nerve causing neuropathic pain), BNB leakage is observed as early as 6 h after surgery [73]. BNB opening occurs thus before the development of a neuropathic phenotype which evolves over days. As compared with sham-operated animals, mRNA and protein for both claudin-5 and occludin are decreased even earlier, starting 3 h after CCI. Downregulation peaks at 7 days, i.e., concordant with the phenotype, but is maintained for 2 months. A similar pattern is observed for the corresponding proteins in endothelial cells of endoneurial vessel [73]. Moreover, Echeverry et al. showed that peripheral nerve injury (partial sciatic nerve ligation) leads to BSCB disruption including downregulation ZO-1 and occludin possibly due to spinal inflammatory reaction [18]. The proteins and thereby TJ function can be rescued by application of anti-inflammatory cytokines such as TGF-β1.
Inflammatory polyneuropathy
Inflammatory polyneuropathies in patients as well as preclinical models often exhibit an enhanced BNB permeability. In fact, barrier disruption with consequent edema [100] is often regarded as key component leading to the full phenotype of, e.g., autoimmune polyneuropathies, such as the acute Guillain–Barré syndrome (GBS) or chronic inflammatory demyelinating polyradiculoneuropathy CIDP [49]. In GBS, while demyelination remains the pathognomonic factor, also axonal damage occurs. The BNB appears compromised especially in the endoneurial endothelia, an observation endorsed by in vitro experiments: incubation with GBS sera leads to disruption of the endoneurial barrier [46]. Yet, it is not understood whether antibodies directly cause the barrier breakdown or whether opening via inflammatory factors elicits these changes and ultimately causes demyelination. Recently, an animal model of GBS was established showing extensive BNB leakage [106].
In biopsy samples of CIDP patients, Kanda et al. observed a downregulation of claudin-5 but not of claudin-1 or occludin [47]. The same group later corroborated these findings through an in vitro study: incubation with sera from CIDP patients leads decreased expression of claudin-5, but not of occludin protein, in endothelial BNB cells [96]. Interestingly, application of hydrocortisone leads to recovery of claudin-5 (but does not affect occludin) and barrier resistance [49]. The authors argue that TJ breakdown favors entry of inflammatory cytokines and immunoglobulins into the endoneurium distorting the microenviroment and, ultimately, worsening the neuropathy. In a very small study conducted by Manole et al., an increase of claudin-1 protein was observed in biopsy samples from CIDP patients while the analysis of occludin remained inconclusive [70].
Spinal cord injury
Damage to the vasculature and breakdown of the BSCB is a universal consequence of spinal cord injury (SCI), clinically as well as in animal models [6]. Within minutes, the barrier is open to tracers of various sizes. Occludin, claudin-5, and ZO-1 are rapidly degraded (8 h to 1 day) and pericytes dissociate from endothelial cells. In parallel, metalloproteinases (MMP) are upregulated and endoplasmic reticulum stress is activated. Several other factors are involved in the pathophysiology: oxidative stress, free radicals, as well as endothelin-1 as a vasoconstricting peptide worsen SCI while aquaporin-4 provides protection by facilitating edema clearance [105].
The elucidation of the pathophysiology led to the discovery of several inhibitors preventing the degradation of TJ proteins and thereby improving recovery from SCI. These include MMP-inhibitors, antidepressants, hormones and chemical chaperones. After SCI, MMP-3 is rapidly upregulated via NFκB, degrades occludin, ZO-1, and claudin-5, and activates MMP-9 [66]. The cascade opens the BSCB, which is mimicked by intrathecal MMP-3 injection and lost in MMP-3 KO mice or after treatment with MMP-3 siRNA or a MMP-3 inhibitor. Likewise, fluoxetine, an antidepressant and serotonin reuptake inhibitor, prevents BSCB disruption and occludin/ZO-1 downregulation and improves function recovery from spinal cord injury via inhibition of MMPs [65]. Similarly, estrogen can improve BSCB function after spinal cord injury [67]. Both treatments reduce the extravasation of neutrophils/macrophages and decrease proinflammatory cytokine and chemokine production. The chemical chaperone, phenylbutyric acid, has been recently applied to various diseases that involve a regulatory mechanism of the endoplasmic reticulum stress response. Application of phenylbutyric acid prevented the loss of tight proteins (occludin and claudin-5) in mice with SCI and in vitro in brain microvascular endothelial cells [111]. In summary, inhibition of barrier breakdown improves the outcome after SCI.
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder concerning the first and second motoneurons. It causes progressive motoric paresis including muscles of respiration, the paralysis of which is usually the lethal factor. While the exact pathogenesis is not yet clear, BSCB disruption is established as one crucial early factor. As early as 1984, blood-borne factors have been described in the motor cortex and spinal cord of ALS patients, suggesting BCNSB impairment [17]. Later on, Zhong et al. could show BSCB permeability in SOD1 mutant mice, an ALS rodent model [109]. Protein levels of ZO-1, occludin, and claudin-5 are reduced, leading to microhemorrhage, entry of neurotoxic hemoglobin products, and, ultimately, hypoperfusion. As this endothelial damage occurs prior to motoneuron degeneration and neurovascular inflammation, the authors argue that this barrier dysfunction might be the initial event leading to ALS. This is endorsed by the fact that closing the BSCB at an early disease stage retards the advancement of ALS [104]. Analogous observations have been made in post-mortem tissue of ALS patients: ZO-1 was decreased in medulla, cervical, and lumbar spinal cord samples, in both gray and white matter, whereas occludin and claudin-5 protein were reduced only in the medulla and cervical spinal cord [27]. It has recently been proposed that claudin-5 expression is diminished in ALS via an altered β-catenin/AKT/FoxO1 pathway [71]. In summary, BSCB breakdown facilitates entry of noxious products and stimulates ALS disease progress.
Multiple sclerosis
In CNS diseases, particular tumors, and inflammation or multiple sclerosis, the BBB is disrupted as a primary or secondary process in the pathophysiology of the disease. Hallmarks of multiple sclerosis are perivascular infiltrates of autoreactive, encephalitogenic T cells, demyelination, neuronal loss, and BBB disruption in acute inflammatory lesions. However, the regulation and relevance of TJ proteins in autoimmune neuroinflammation are not completely understood. Claudin-5 [20] and claudin-3 [83] are decreased in the endothelium in experimental autoimmune encephalomyelitis (EAE). Several molecules and pathways have been identified in the past two decades.
Cytokines regulate the BBB in EAE: TNF-α increases the permeability of the BBB by reducing the expression of occludin and claudin-5. Vascular endothelial growth factor affects the BBB integrity by downregulating claudin-5 [3], and interleukin-17 also diminishes the expression of ZO-1 and occludin [52]. Nerve/glial antigen 2+ glia is the major CNS cellular target of IL-17 in EAE [48].
Netrins are laminin-related proteins that regulate cell migration and influence cell–cell and cell–substrate adhesion best known for their axonal guidance functions (reviewed in [88]). Apart from this function, netrin-1 supports BBB integrity by upregulating endothelial junctional protein expression [84]. Netrin-1 treatment reduces diffusion across the BBB in vitro and in vivo in EAE. This is accompanied by prevention of claudin-5, occludin, and JAM-A loss [103] and an improved clinical phenotype in EAE mice. Also, miR-155 is highly elevated in acute multiple sclerosis (MS) lesions in patients and in EAE in mice [69]. It induces delocalization of ZO-1 from the cell–cell contacts and targets claudin-1.
Several approaches have been used to prevent BBB disruption in experimental EAE. Treatment with a small molecule inhibitor of PKC-Cβ presumably stabilizing the cytoskeleton improves the clinical features of EAE in mice [63]. Similarly, overexpression of claudin-1 significantly reduces BBB leakiness and disease burden in the chronic phase of EAE independent of immune cell infiltration [83]. In summary, BBB sealing via claudin-1 overexpression, IFN-γ [76], NG2 KO or microRNA-155 antagomirs, netrin-1, and PKC-Cβ inhibition ameliorate the clinical picture of MS/EAE.
Ischemic stroke
Ischemia is a hypoperfusion of tissue or an organ, mostly caused by stenosis or occlusion of blood vessels. In the brain, it is the leading cause of stroke. Ischemic stroke can be divided into two stages, ischemia and reperfusion, as well as two regions, the core area and the ischemic penumbra. The impaired supply of oxygen and glucose leads to anaerobic stress and starts a cascade of ATP depletion, efflux of excitotoxic glutamate, ion shifts, and metabolic imbalance with acidosis, oxidative stress, and initiation of inflammation. These events directly affect the BBB in the core area and lead to cytotoxic edema: lactic acidosis leads to cell swelling, and protease induction (e.g., tPAs and MMPs) contributes to degradation of the extracellular matrix and detachment of cells from the extracellular matrix [30]. In vitro experiments in bEND.3 cells showed that hypoxia alters both claudin-5 localization and expression in the plasma membrane leading to a decrease in transendothelial electrical resistance and permeability of the barrier for smaller molecules with a phenotype resembling the claudin-5 KO mouse [56]. Moreover, occludin and ZO-1 expression is decreased after experimental induction of cerebral embolism in isolated rat brain capillaries [44]. ZO-1 (and ZO-2) is relocated towards the cell nucleus [21].
The molecular underlying pathways are multiple. Kinases such as tyrosine kinases and myosin light-chain kinase are known to regulate TJ proteins and might be activated here by calcium dysregulation [10]. Furthermore, inflammatory cytokines like TNF-α disrupt the BBB by activating MMPs and NFκB [9, 37]. Last but not least, oxidative stress is crucial in altering the BBB: in Caco-2 cells, it elicits tyrosine phosphorylation of ZO-1 and occludin, amongst others, leading to redistribution, less dimer-formation of the two, and decrease in transepithelial electric resistance [89]. The reperfusion phase is marked by initial paracellular disassembly due to an increased cerebral blood flow and loss of cerebral autoregulation. Early experimental data using mid-cerebral artery occlusion (MCAO) in cats suggest two further permeability peaks [39, 62]. This biphasic model has been endorsed by other studies but may vary due to extent and duration of the ischemia. It is argued that after the initial hyperemia has faded into a latent hypoperfusion, TJs first re-assembly and regenerate before a next phase of permeability is caused by free radicals released after inflammatory and oxidative stress in combination with enzymatic activity and degradation of the extracellular matrix [35]. The last peak in permeability is proposed to be caused by neoangiogenesis which reconstructs the BBB with alternating assembly and disassembly phases [93]. Ultimately, this leads to a vasogenic edema peaking about 2–5 days after onset [35]. It is to be noted that the size of this edema is the main determinant of the clinical outcome. As it has a propensity to white matter, its effect is more pronounced in humans than, e.g., in rodents. To sum up, BBB disruption and reconstruction due to volatility in perfusion, oxidative stress, and neoangiogenesis is a key component in vasogenic edema after ischemic stroke.
Alzheimer’s disease
Alzheimer’s disease (AD) patients develop an early neurovascular dysfunction, progressive neurodegeneration, selective loss of neurons, and amyloid accumulation in the brain. In particular, AD is characterized by the presence of amyloid (Aβ) plaques and neurofibrillary tangles in the brain. The neurovascular hypothesis of AD proposes that cerebrovascular dysfunction and disruption in the neurovascular integrity contribute to the onset and progression of cognitive decline. Primary damage of the cerebrovasculature leads to brain accumulation of blood-derived neurotoxins, and the decrease in brain perfusion causes neuronal injury. Prominent variants of the amyloid precursor protein (APP) Aβ consist of the first 40 (Aβ1–40) and 42 (Aβ1–42) amino acids. Aβ produced in the brain binds to low-density lipoprotein receptor-related protein-1 (LRP-1) at the abluminal side of the BBB, causing its rapid internalization into endothelial cells and clearance through the blood. A recently created brain endothelial cell-specific LRP-1 KO confirmed this pathway [98].
In vitro, amyloid peptide Aβ1–42 enhances permeability, reduces ZO-1, claudin-5 and occludin expression, and increases intracellular calcium and MMP secretion in cultured endothelial cells [55]. Similarly, in vivo, cerebral amyloid angiopathy is accompanied by a dramatic loss of occludin, claudin-5, and ZO-1 in Aβ-laden capillaries surrounded by NADPH oxidase-2-positive activated microglia. Likewise, TJs in 5XFAD mice, a model for AD, appeared to be of significantly shorter lengths than TJs seen in littermate mice. Aβ is toxic to brain endothelial cells via binding to RAGE and induction of reactive oxygen species, which also ultimately leads to disruption of TJs and loss of BBB integrity [12]. In vivo intracerebroventricular injection of Aβ1–42 first decreases ZO-1, claudin-1, and occludin and subsequently claudin-5, increases MMP gene expression in the choroid plexus epithelial cells and opens the BCSFB but not the BBB [8]. In summary, evidence supports the barrier opening properties of Aβ peptide presumably via MMPs. Nevertheless, it should be kept in mind that age per se is associated with decreased clearance of Aβ by LRP-1 and a loss of ZO-1 and occludin in the mouse brain [19].
Therapeutic barrier opening for drug delivery
Although barrier function of the BBB, BNB, and BSCB are crucial to maintain the function of the respective compartment of the nervous system, they also hinder the penetration of drugs. Several approaches have been explored to overcome the barrier, e.g., for the treatment of brain neoplasias, certain neurodegenerative disorders, metabolic diseases, or pain treatment.
Opening of the BBB
Drug delivery across the BBB has been a challenge for decades [5]. So far, explored approaches include receptor-mediated transcytosis and BBB disruption as well as certain carriers. Synthetic carriers involve liposomes, metallic nanoparticles, and polymersomes (reviewed in [64]). Furthermore, certain TJ modulators have therapeutic potentials including claudin-5 and occludin siRNAs, peptides derived from zonula occludens toxin, as well as synthetic peptides targeting the extracellular loops of TJs (reviewed elsewhere in [29]).
Opening of the BNB for analgesics
Current treatment of pain is limited to common pain killers and opioids with known side effects. Analgesic drugs applied near the nerve are restricted to lipophilic local anesthetics such as lidocaine, which block all nerve fibers including motor neurons. Ideally, pharmaceutical agents for regional analgesia should only block pain receptors (nociceptors) and spare motor and sensory neurons. Two classes of hydrophilic opioids and voltage-gated sodium channel blockers are specific for nociceptors but cannot penetrate the perineurial barrier, which is sealed by claudin-1. To target this barrier, several approaches have been developed comprising of non-specific methodologies like hypertonic saline and MMP-9 [33] (Fig. 2) as well as more specific methods like claudin-1 siRNA [33] or claudin-1 peptidomimetics [14, 94, 112]. Barrier opening with hypertonic saline is not simply a mechanical disruption of TJ [90] but rather a receptor-mediated process involving the release of MMP-9 and binding of the non-catalytic domain of MMP-9 (MMP-9-PEX) to low-density lipoprotein receptor-related protein 1 (LRP-1). Other agonists of LRP-1 include recombinant tissue plasminogen activator (rtPA). Therefore, the BNB can transiently be opened for drug delivery via rtPA or microRNA-183, which is upregulated after rtPA [107]. All of these enhancers allow for a transient opening of the barrier between 5 h and 5 days without nerve damage as seen by the absence of histomorphological and functional nerve changes. Via these enhancers, selective analgesics can be applied for pain relief with preserved motor function and sensitivity.

Opening of the perineurial barrier for delivery of opioids or Nav blockers for analgesia. Peripheral neurons including nociceptors (yellow) are surrounded by the perineurium composed of perineurial cells (orange). Pain arising in the periphery is transduced via nociceptors to the dorsal horn of the spinal cord. On nociceptors, μ opioid receptors (green) and voltage-gated sodium channels (red, e.g., NaV1.7) are expressed. Both are specific targets for analgesics like opioids (green triangles) or Nav blockers (red circles) on nociceptors. Perineurial cells express LRP-1. Physiologically, the BNB is sealed between pericytes by TJ proteins like claudin-1. For barrier opening, LRP-1 agonists like tissue plasminogen activator (rtPA) or catalytically inactive rtPA (rtPAi) can be applied locally. This induces Erk phosphorylation and upregulation of microRNA-29b (miR-29b) or miR-183. Both miRs bind to the untranslated region of the claudin-1 gene. microRNA binding leads to transcriptional repression and reduced formation of claudin-1 in the BNB. This process allows sodium channel blockers or opioids to pass paracellularly, bind to their receptors on nociceptors, and inhibit transmission of painful signals from the periphery to the spinal cord [33, 107]
Summary and conclusions
The nervous system is very well protected by several barriers including the BBB, BSCB, and BNB. Distinct tight junction proteins form these barriers including claudin-1, claudin-3, claudin-5, claudin-11, claudin-12, claudin-19, occludin, and tricellulin. Some of these claudins are ubiquitously distributed, while others are only found in certain barriers or compartment of barriers. Inflammatory as well as traumatic and degenerative diseases of the nervous system are frequently accompanied by barrier opening. Frequently, barrier opening is the first hallmark preceding clinical symptoms of the diseases. Known factors regulating barrier tightness include cytokines, growth factors, metalloproteinases, microRNAs, as well as protein kinases amongst others. Understanding the pathophysiology of these barrier regulators might stimulate the discovery of new drugs and novel treatments to facilitate barrier sealing early in the disease process and limit further progression. On the other hand, the design of drug enhancers to transiently overcome these barriers might prove effective for the treatment of pain, neoplasias, or degenerative disorders.
References
Al-Sadi R, Khatib K, Guo S, Ye D, Youssef M, Ma T (2011) Occludin regulates macromolecule flux across the intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol 300:G1054–G1064
Alanne MH, Pummi K, Heape AM, Grenman R, Peltonen J, Peltonen S (2009) Tight junction proteins in human Schwann cell autotypic junctions. J Histochem Cytochem 57:523–529
Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR (2009) VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci U S A 106:1977–1982
Aslam M, Kalluri SR, Cepok S, Kraus V, Buck D, Srivastava R, Hemmer B (2010) The antibody response to oligodendrocyte specific protein in multiple sclerosis. J Neuroimmunol 221:81–86
Banks WA (2016) From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov 15:275–292
Bartanusz V, Jezova D, Alajajian B, Digicaylioglu M (2011) The blood-spinal cord barrier: morphology and clinical implications. Ann Neurol 70:194–206
Blasig IE, Winkler L, Lassowski B, Mueller SL, Zuleger N, Krause E, Krause G, Gast K, Kolbe M, Piontek J (2006) On the self-association potential of transmembrane tight junction proteins. Cell Mol Life Sci 63:505–514
Brkic M, Balusu S, Van Wonterghem E, Gorle N, Benilova I, Kremer A, Van Hove I, Moons L, De Strooper B, Kanazir S, Libert C, Vandenbroucke RE (2015) Amyloid beta oligomers disrupt blood-CSF barrier integrity by activating matrix metalloproteinases. J Neurosci 35:12766–12778
Brown RC, Mark KS, Egleton RD, Huber JD, Burroughs AR, Davis TP (2003) Protection against hypoxia-induced increase in blood-brain barrier permeability: role of tight junction proteins and NFkappaB. J Cell Sci 116:693–700
Brown RC, Mark KS, Egleton RD, Davis TP (2004) Protection against hypoxia-induced blood-brain barrier disruption: changes in intracellular calcium. Am J Physiol Cell Physiol 286:C1045–C1052
Bunge MB, Wood PM, Tynan LB, Bates ML, Sanes JR (1989) Perineurium originates from fibroblasts: demonstration in vitro with a retroviral marker. Science 243:229–231
Carrano A, Hoozemans JJ, van der Vies SM, van Horssen J, de Vries HE, Rozemuller AJ (2012) Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener Dis 10:329–331
Cording J, Gunther R, Vigolo E, Tscheik C, Winkler L, Schlattner I, Lorenz D, Haseloff RF, Schmidt-Ott KM, Wolburg H, Blasig IE (2015) Redox regulation of cell contacts by tricellulin and occludin: redox-sensitive cysteine sites in tricellulin regulate both tri- and bicellular junctions in tissue barriers as shown in hypoxia and ischemia. Antioxid Redox Signal 23:1035–1049
Dabrowski S, Staat C, Zwanziger D, Sauer RS, Bellmann C, Gunther R, Krause E, Haseloff RF, Rittner H, Blasig I (2015) Redox-sensitive structure and function of the first extracellular loop of the cell-cell contact protein claudin-1—lessons from molecular structure to animal. Antioxid Redox Signal 22:1–14
Denninger AR, Breglio A, Maheras KJ, LeDuc G, Cristiglio V, Deme B, Gow A, Kirschner DA (2015) Claudin-11 tight junctions in myelin are a barrier to diffusion and lack strong adhesive properties. Biophys J 109:1387–1397
Devaux J, Gow A (2008) Tight junctions potentiate the insulative properties of small CNS myelinated axons. J Cell Biol 183:909–921
Donnenfeld H, Kascsak RJ, Bartfeld H (1984) Deposits of IgG and C3 in the spinal cord and motor cortex of ALS patients. J Neuroimmunol 6:51–57
Echeverry S, Shi XQ, Rivest S, Zhang J (2011) Peripheral nerve injury alters blood-spinal cord barrier functional and molecular integrity through a selective inflammatory pathway. J Neurosci 31:10819–10828
Elahy M, Jackaman C, Mamo JC, Lam V, Dhaliwal SS, Giles C, Nelson D, Takechi R (2015) Blood-brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun Ageing 12:2
Errede M, Girolamo F, Ferrara G, Strippoli M, Morando S, Boldrin V, Rizzi M, Uccelli A, Perris R, Bendotti C, Salmona M, Roncali L, Virgintino D (2012) Blood-brain barrier alterations in the cerebral cortex in experimental autoimmune encephalomyelitis. J Neuropathol Exp Neurol 71:840–854
Fischer S, Wiesnet M, Marti HH, Renz D, Schaper W (2004) Simultaneous activation of several second messengers in hypoxia-induced hyperpermeability of brain derived endothelial cells. J Cell Physiol 198:359–369
Fujita H, Sugimoto K, Inatomi S, Maeda T, Osanai M, Uchiyama Y, Yamamoto Y, Wada T, Kojima T, Yokozaki H, Yamashita T, Kato S, Sawada N, Chiba H (2008) Tight junction proteins claudin-2 and -12 are critical for vitamin D-dependent Ca2+ absorption between enterocytes. Mol Biol Cell 19:1912–1921
Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S (1993) Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 123:1777–1788
Furuse M, Itoh M, Hirase T, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S (1994) Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J Cell Biol 127:1617–1626
Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S (1998) Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol 141:1539–1550
Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, Noda T, Kubo A, Tsukita S (2002) Claudin-based tight junctions are crucial for the mammalian epidermal barrier; a lesson from claudin-1-deficient mice. J Cell Biol 156:1099–1111
Garbuzova-Davis S, Hernandez-Ontiveros DG, Rodrigues MC, Haller E, Frisina-Deyo A, Mirtyl S, Sallot S, Saporta S, Borlongan CV, Sanberg PR (2012) Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res 1469:114–128
Gow A, Southwood CM, Li JS, Pariali M, Riordan GP, Brodie SE, Danias J, Bronstein JM, Kachar B, Lazzarini RA (1999) CNS myelin and sertoli cell tight junction strands are absent in Osp/claudin-11 null mice. Cell 99:649–659
Greene C, Campbell M (2016) Tight junction modulation of the blood brain barrier: CNS delivery of small molecules. Tissue Barriers 4:e1138017
Grossmann J (2002) Molecular mechanisms of “detachment-induced apoptosis—Anoikis”. Apoptosis 7:247–260
Gunzel D, Yu AS (2013) Claudins and the modulation of tight junction permeability. Physiol Rev 93:525–569
Guo J, Wang L, Zhang Y, Wu J, Arpag S, Hu B, Imhof BA, Tian X, Carter BD, Suter U, Li J (2014) Abnormal junctions and permeability of myelin in PMP22-deficient nerves. Ann Neurol 75:255–265
Hackel D, Brack A, Fromm M, Rittner HL (2012) Modulation of tight junction proteins in the perineurium for regional pain control. Ann N Y Acad Sci 1257:199–206
Haseloff RF, Dithmer S, Winkler L, Wolburg H, Blasig IE (2015) Transmembrane proteins of the tight junctions at the blood-brain barrier: structural and functional aspects. Semin Cell Dev Biol 38:16–25
Heo JH, Han SW, Lee SK (2005) Free radicals as triggers of brain edema formation after stroke. Free Radic Biol Med 39:51–70
Hirakawa H, Okajima S, Nagaoka T, Takamatsu T, Oyamada M (2003) Loss and recovery of the blood-nerve barrier in the rat sciatic nerve after crush injury are associated with expression of intercellular junctional proteins. Exp Cell Res 284:196–210
Hosomi N, Ban CR, Naya T, Takahashi T, Guo P, Song XY, Kohno M (2005) Tumor necrosis factor-alpha neutralization reduced cerebral edema through inhibition of matrix metalloproteinase production after transient focal cerebral ischemia. J Cereb Blood Flow Metab 25:959–967
Hou J, Renigunta A, Konrad M, Gomes AS, Schneeberger EE, Paul DL, Waldegger S, Goodenough DA (2008) Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J Clin Invest 118:619–628
Huang ZG, Xue D, Preston E, Karbalai H, Buchan AM (1999) Biphasic opening of the blood-brain barrier following transient focal ischemia: effects of hypothermia. Can J Neurol Sci 26:298–304
Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S, Tsukita S (2005) Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol 171:939–945
Ikenouchi J, Sasaki H, Tsukita S, Furuse M, Tsukita S (2008) Loss of occludin affects tricellular localization of tricellulin. Mol Biol Cell 19:4687–4693
Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S (1999) Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol 147:1351–1363
Iwamoto N, Higashi T, Furuse M (2014) Localization of angulin-1/LSR and tricellulin at tricellular contacts of brain and retinal endothelial cells in vivo. Cell Struct Funct 39:1–8
Kago T, Takagi N, Date I, Takenaga Y, Takagi K, Takeo S (2006) Cerebral ischemia enhances tyrosine phosphorylation of occludin in brain capillaries. Biochem Biophys Res Commun 339:1197–1203
Kamitani T, Sakaguchi H, Tamura A, Miyashita T, Yamazaki Y, Tokumasu R, Inamoto R, Matsubara A, Mori N, Hisa Y, Tsukita S (2015) Deletion of tricellulin causes progressive hearing loss associated with degeneration of Cochlear hair cells. Sci Rep 5:18402
Kanda T, Yamawaki M, Mizusawa H (2003) Sera from Guillain-Barre patients enhance leakage in blood-nerve barrier model. Neurology 60:301–306
Kanda T, Numata Y, Mizusawa H (2004) Chronic inflammatory demyelinating polyneuropathy: decreased claudin-5 and relocated ZO-1. J Neurol Neurosurg Psychiatry 75:765–769
Kang Z, Wang C, Zepp J, Wu L, Sun K, Zhao J, Chandrasekharan U, DiCorleto PE, Trapp BD, Ransohoff RM, Li X (2013) Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. Nat Neurosci 16:1401–1408
Kashiwamura Y, Sano Y, Abe M, Shimizu F, Haruki H, Maeda T, Kawai M, Kanda T (2011) Hydrocortisone enhances the function of the blood-nerve barrier through the up-regulation of claudin-5. Neurochem Res 36:849–855
Katsuno T, Umeda K, Matsui T, Hata M, Tamura A, Itoh M, Takeuchi K, Fujimori T, Nabeshima Y, Noda T, Tsukita S (2008) Deficiency of zonula occludens-1 causes embryonic lethal phenotype associated with defected yolk sac angiogenesis and apoptosis of embryonic cells. Mol Biol Cell 19:2465–2475
Kaushansky N, Hemo R, Eisenstein M, Ben-Nun A (2007) OSP/claudin-11-induced EAE in mice is mediated by pathogenic T cells primarily governed by OSP192Y residue of major encephalitogenic region OSP179-207. Eur J Immunol 37:2018–2031
Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A (2007) Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med 13:1173–1175
Kikuchi S, Ninomiya T, Tatsumi H, Sawada N, Kojima T (2010) Tricellulin is expressed in autotypic tight junctions of peripheral myelinating Schwann cells. J Histochem Cytochem 58:1067–1073
Kooij G, Kopplin K, Blasig R, Stuiver M, Koning N, Goverse G, van der Pol SM, van Het Hof B, Gollasch M, Drexhage JA, Reijerkerk A, Meij IC, Mebius R, Willnow TE, Muller D, Blasig IE, de Vries HE (2014) Disturbed function of the blood-cerebrospinal fluid barrier aggravates neuro-inflammation. Acta Neuropathol 128:267–277
Kook SY, Hong HS, Moon M, Ha CM, Chang S, Mook-Jung I (2012) Abeta(1)(−)(4)(2)-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca(2)(+)-calcineurin signaling. J Neurosci 32:8845–8854
Koto T, Takubo K, Ishida S, Shinoda H, Inoue M, Tsubota K, Okada Y, Ikeda E (2007) Hypoxia disrupts the barrier function of neural blood vessels through changes in the expression of claudin-5 in endothelial cells. Am J Pathol 170:1389–1397
Kristensson K, Olsson Y (1971) The perineurium as a diffusion barrier to protein tracers. Differences between mature and immature animals. Acta Neuropathol 17:127–138
Krug SM, Amasheh S, Richter JF, Milatz S, Gunzel D, Westphal JK, Huber O, Schulzke JD, Fromm M (2009) Tricellulin forms a barrier to macromolecules in tricellular tight junctions without affecting ion permeability. Mol Biol Cell 20:3713–3724
Krug SM, Amasheh M, Dittmann I, Christoffel I, Fromm M, Amasheh S (2013) Sodium caprate as an enhancer of macromolecule permeation across tricellular tight junctions of intestinal cells. Biomaterials 34:275–282
Krug SM, Schulzke JD, Fromm M (2014) Tight junction, selective permeability, and related diseases. Semin Cell Dev Biol 36:166–176
Kucenas S, Takada N, Park HC, Woodruff E, Broadie K, Appel B (2008) CNS-derived glia ensheath peripheral nerves and mediate motor root development. Nat Neurosci 11:143–151
Kuroiwa T, Ting P, Martinez H, Klatzo I (1985) The biphasic opening of the blood-brain barrier to proteins following temporary middle cerebral artery occlusion. Acta Neuropathol 68:122–129
Lanz TV, Becker S, Osswald M, Bittner S, Schuhmann MK, Opitz CA, Gaikwad S, Wiestler B, Litzenburger UM, Sahm F, Ott M, Iwantscheff S, Grabitz C, Mittelbronn M, von Deimling A, Winkler F, Meuth SG, Wick W, Platten M (2013) Protein kinase Cbeta as a therapeutic target stabilizing blood-brain barrier disruption in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 110:14735–14740
Larsen JM, Martin DR, Byrne ME (2014) Recent advances in delivery through the blood-brain barrier. Curr Top Med Chem 14:1148–1160
Lee JY, Kim HS, Choi HY, Oh TH, Yune TY (2012) Fluoxetine inhibits matrix metalloprotease activation and prevents disruption of blood-spinal cord barrier after spinal cord injury. Brain 135:2375–2389
Lee JY, Choi HY, Ahn HJ, Ju BG, Yune TY (2014) Matrix metalloproteinase-3 promotes early blood-spinal cord barrier disruption and hemorrhage and impairs long-term neurological recovery after spinal cord injury. Am J Pathol 184:2985–3000
Lee JY, Choi HY, Na WH, Ju BG, Yune TY (2015) 17beta-estradiol inhibits MMP-9 and SUR1/TrpM4 expression and activation and thereby attenuates BSCB disruption/hemorrhage after spinal cord injury in male rats. Endocrinology 156:1838–1850
Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, Reis M, Felici A, Wolburg H, Fruttiger M, Taketo MM, von Melchner H, Plate KH, Gerhardt H, Dejana E (2008) Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol 183:409–417
Lopez-Ramirez MA, Wu D, Pryce G, Simpson JE, Reijerkerk A, King-Robson J, Kay O, de Vries HE, Hirst MC, Sharrack B, Baker D, Male DK, Michael GJ, Romero IA (2014) MicroRNA-155 negatively affects blood-brain barrier function during neuroinflammation. FASEB J 28:2551–2565
Manole E, Ceafalan LC, Oproiu AM, Popa-Wagner A, Popescu BO (2015) Claudin-1 and occludin expression in demyelinating peripheral neuropathies. Romanian J Morphol Embryol 56:1097–1102
Meister S, Storck SE, Hameister E, Behl C, Weggen S, Clement AM, Pietrzik CU (2015) Expression of the ALS-causing variant hSOD1(G93A) leads to an impaired integrity and altered regulation of claudin-5 expression in an in vitro blood-spinal cord barrier model. J Cereb Blood Flow Metab 35:1112–1121
Miyamoto T, Morita K, Takemoto D, Takeuchi K, Kitano Y, Miyakawa T, Nakayama K, Okamura Y, Sasaki H, Miyachi Y, Furuse M, Tsukita S (2005) Tight junctions in Schwann cells of peripheral myelinated axons: a lesson from claudin-19-deficient mice. J Cell Biol 169:527–538
Moreau N, Mauborgne A, Bourgoin S, Couraud PO, Romero IA, Weksler BB, Villanueva L, Pohl M, Boucher Y (2016) Early alterations of hedgehog signaling pathway in vascular endothelial cells after peripheral nerve injury elicit blood-nerve barrier disruption, nerve inflammation, and neuropathic pain development. Pain 157:827–839
Morita K, Sasaki H, Furuse M, Tsukita S (1999) Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol 147:185–194
Nayak G, Lee SI, Yousaf R, Edelmann SE, Trincot C, Van Itallie CM, Sinha GP, Rafeeq M, Jones SM, Belyantseva IA, Anderson JM, Forge A, Frolenkov GI, Riazuddin S (2013) Tricellulin deficiency affects tight junction architecture and cochlear hair cells. J Clin Invest 123:4036–4049
Ni C, Wang C, Zhang J, Qu L, Liu X, Lu Y, Yang W, Deng J, Lorenz D, Gao P, Meng Q, Yan X, Blasig IE, Qin Z (2014) Interferon-gamma safeguards blood-brain barrier during experimental autoimmune encephalomyelitis. Am J Pathol 184:3308–3320
Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S (2003) Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol 161:653–660
Notterpek L, Roux KJ, Amici SA, Yazdanpour A, Rahner C, Fletcher BS (2001) Peripheral myelin protein 22 is a constituent of intercellular junctions in epithelia. Proc Natl Acad Sci U S A 98:14404–14409
Obermeier B, Daneman R, Ransohoff RM (2013) Development, maintenance and disruption of the blood-brain barrier. Nat Med 19:1584–1596
Ohtsuki S, Sato S, Yamaguchi H, Kamoi M, Asashima T, Terasaki T (2007) Exogenous expression of claudin-5 induces barrier properties in cultured rat brain capillary endothelial cells. J Cell Physiol 210:81–86
Pan W, Banks WA, Kastin AJ (1997) Permeability of the blood-brain and blood-spinal cord barriers to interferons. J Neuroimmunol 76:105–111
Parmantier E, Lynn B, Lawson D, Turmaine M, Namini SS, Chakrabarti L, McMahon AP, Jessen KR, Mirsky R (1999) Schwann cell-derived desert hedgehog controls the development of peripheral nerve sheaths. Neuron 23:713–724
Pfeiffer F, Schafer J, Lyck R, Makrides V, Brunner S, Schaeren-Wiemers N, Deutsch U, Engelhardt B (2011) Claudin-1 induced sealing of blood-brain barrier tight junctions ameliorates chronic experimental autoimmune encephalomyelitis. Acta Neuropathol 122:601–614
Podjaski C, Alvarez JI, Bourbonniere L, Larouche S, Terouz S, Bin JM, Lecuyer MA, Saint-Laurent O, Larochelle C, Darlington PJ, Arbour N, Antel JP, Kennedy TE, Prat A (2015) Netrin 1 regulates blood-brain barrier function and neuroinflammation. Brain 138:1598–1612
Poliak S, Matlis S, Ullmer C, Scherer SS, Peles E (2002) Distinct claudins and associated PDZ proteins form different autotypic tight junctions in myelinating Schwann cells. J Cell Biol 159:361–372
Prockop LD, Naidu KA, Binard JE, Ransohoff J (1995) Selective permeability of [3H]-D-mannitol and [14C]-carboxyl-inulin across the blood-brain barrier and blood-spinal cord barrier in the rabbit. J Spinal Cord Med 18:221–226
Pummi KP, Heape AM, Grenman RA, Peltonen JT, Peltonen SA (2004) Tight junction proteins ZO-1, occludin, and claudins in developing and adult human perineurium. J Histochem Cytochem 52:1037–1046
Rajasekharan S, Kennedy TE (2009) The netrin protein family. Genome Biol 10:239
Rao RK, Basuroy S, Rao VU, Karnaky KJ Jr, Gupta A (2002) Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J 368:471–481
Rittner HL, Hackel D, Yamdeu RS, Mousa SA, Stein C, Schafer M, Brack A (2009) Antinociception by neutrophil-derived opioid peptides in noninflamed tissue—role of hypertonicity and the perineurium. Brain Behav Immun 23:548–557
Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T, and Tsukita S. complex Phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 11: 4131–4142, 2000.
Samak G, Aggarwal S, Rao RK (2011) ERK is involved in EGF-mediated protection of tight junctions, but not adherens junctions, in acetaldehyde-treated Caco-2 cell monolayers. Am J Physiol Gastrointest Liver Physiol 301:G50–G59
Sandoval KE, Witt KA (2008) Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 32:200–219
Sauer RS, Krug SM, Hackel D, Staat C, Konasin N, Yang S, Niedermirtl B, Bosten J, Gunther R, Dabrowski S, Doppler K, Sommer C, Blasig IE, Brack A, Rittner HL (2014) Safety, efficacy, and molecular mechanism of claudin-1-specific peptides to enhance blood-nerve-barrier permeability. J Control Release 185:88–98
Severson EA, Kwon M, Hilgarth RS, Parkos CA, Nusrat A (2010) Glycogen synthase kinase 3 (GSK-3) influences epithelial barrier function by regulating occludin, claudin-1 and E-cadherin expression. Biochem Biophys Res Commun 397:592–597
Shimizu F, Sawai S, Sano Y, Beppu M, Misawa S, Nishihara H, Koga M, Kuwabara S, Kanda T (2014) Severity and patterns of blood-nerve barrier breakdown in patients with chronic inflammatory demyelinating polyradiculoneuropathy: correlations with clinical subtypes. PLoS One 9:e104205
Sirotkin H, Morrow B, Saint-Jore B, Puech A, Das Gupta R, Patanjali SR, Skoultchi A, Weissman SM, Kucherlapati R (1997) Identification, characterization, and precise mapping of a human gene encoding a novel membrane-spanning protein from the 22q11 region deleted in velo-cardio-facial syndrome. Genomics 42:245–251
Storck SE, Meister S, Nahrath J, Meissner JN, Schubert N, Di Spiezio A, Baches S, Vandenbroucke RE, Bouter Y, Prikulis I, Korth C, Weggen S, Heimann A, Schwaninger M, Bayer TA, Pietrzik CU (2016) Endothelial LRP1 transports amyloid-beta(1-42) across the blood-brain barrier. J Clin Invest 126:123–136
Su P, Zhao F, Cao Z, Zhang J, Aschner M, Luo W (2015) Mir-203-mediated tricellulin mediates lead-induced in vitro loss of blood-cerebrospinal fluid barrier (BCB) function. Toxicol in Vitro 29:1185–1194
Uceyler N, Necula G, Wagemann E, Toyka KV, Sommer C (2016) Endoneurial edema in sural nerve may indicate recent onset inflammatory neuropathy. Muscle Nerve 53:705–710
Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, Matsui T, Tsukita S, Furuse M (2006) ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell 126:741–754
van Paassen BW, van der Kooi AJ, van Spaendonck-Zwarts KY, Verhamme C, Baas F, de Visser M (2014) PMP22 related neuropathies: Charcot-Marie-tooth disease type 1A and hereditary neuropathy with liability to pressure palsies. Orphanet J Rare Dis 9:38
Wen J, Qian S, Yang Q, Deng L, Mo Y, Yu Y (2014) Overexpression of netrin-1 increases the expression of tight junction-associated proteins, claudin-5, occludin, and ZO-1, following traumatic brain injury in rats. Exp Ther Med 8:881–886
Winkler EA, Sengillo JD, Sagare AP, Zhao Z, Ma Q, Zuniga E, Wang Y, Zhong Z, Sullivan JS, Griffin JH, Cleveland DW, Zlokovic BV (2014) Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc Natl Acad Sci U S A 111:E1035–E1042
Wu Q, Zhang YJ, Gao JY, Li XM, Kong H, Zhang YP, Xiao M, Shields CB, Hu G (2014) Aquaporin-4 mitigates retrograde degeneration of rubrospinal neurons by facilitating edema clearance and glial scar formation after spinal cord injury in mice. Mol Neurobiol 49:1327–1337
Yang M, Rainone A, Shi XQ, Fournier S, Zhang J (2014) A new animal model of spontaneous autoimmune peripheral polyneuropathy: implications for Guillain-Barre syndrome. Acta Neuropathol Commun 2:5
Yang S, Krug SM, Heitmann J, Hu L, Reinhold AK, Sauer S, Bosten J, Sommer C, Fromm M, Brack A, Rittner HL (2016) Analgesic drug delivery via recombinant tissue plasminogen activator and microRNA-183-triggered opening of the blood-nerve barrier. Biomaterials 82:20–33
Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV (2015) Establishment and dysfunction of the blood-brain barrier. Cell 163:1064–1078
Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, O’Banion MK, Stojanovic K, Sagare A, Boillee S, Cleveland DW, Zlokovic BV (2008) ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci 11:420–422
Zhou Q, Costinean S, Croce CM, Brasier AR, Merwat S, Larson SA, Basra S, Verne GN (2015) MicroRNA 29 targets nuclear factor-kappaB-repressing factor and claudin 1 to increase intestinal permeability. Gastroenterology 148:158–169
Zhou Y, Ye L, Zheng B, Zhu S, Shi H, Zhang H, Wang Z, Wei X, Chen D, Li X, Xu H, Xiao J (2016) Phenylbutyrate prevents disruption of blood-spinal cord barrier by inhibiting endoplasmic reticulum stress after spinal cord injury. Am J Transl Res 8:1864–1875
Zwanziger D, Hackel D, Staat C, Bocker A, Brack A, Beyermann M, Rittner H, Blasig IE (2012) A peptidomimetic tight junction modulator to improve regional analgesia. Mol Pharm 9:1785–1794
Acknowledgement
The Interdisciplinary Centre for Clinical Research (IZKF) of the Medical Faculty of the University of Wuerzburg supported the research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Reinhold, A., Rittner, H. Barrier function in the peripheral and central nervous system—a review. Pflugers Arch - Eur J Physiol 469, 123–134 (2017). https://doi.org/10.1007/s00424-016-1920-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-016-1920-8