
Abstract
The volume-regulated anion channel (VRAC) is a ubiquitously expressed yet highly enigmatic member of the superfamily of chloride/anion channels. It is activated by cellular swelling and mediates regulatory cell volume decrease in a majority of vertebrate cells, including those in the central nervous system (CNS). In the brain, besides its crucial role in cellular volume regulation, VRAC is thought to play a part in cell proliferation, apoptosis, migration, and release of physiologically active molecules. Although these roles are not exclusive to the CNS, the relative significance of VRAC in the brain is amplified by several unique aspects of its physiology. One important example is the contribution of VRAC to the release of the excitatory amino acid neurotransmitters glutamate and aspartate. This latter process is thought to have impact on both normal brain functioning (such as astrocyte-neuron signaling) and neuropathology (via promoting the excitotoxic death of neuronal cells in stroke and traumatic brain injury). In spite of much work in the field, the molecular nature of VRAC remained unknown until less than 2 years ago. Two pioneer publications identified VRAC as the heterohexamer formed by the leucine-rich repeat-containing 8 (LRRC8) proteins. These findings galvanized the field and are likely to result in dramatic revisions to our understanding of the place and role of VRAC in the brain, as well as other organs and tissues. The present review briefly recapitulates critical findings in the CNS and focuses on anticipated impact on the LRRC8 discovery on further progress in neuroscience research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction: general properties and physiological significance of VRAC
The volume-regulated anion channel (VRAC) is a ubiquitously expressed ion channel. It is activated in response to cellular swelling and plays a key role in cell volume homeostasis. This physiological process is essential because in the majority of animal cells, the plasma membrane is highly permeable to water, and they, therefore, either swell or shrink in response to changes in the extracellular and intracellular content of osmotically active molecules. Alterations in cell volume are associated with several “dangers.” Severe osmotic swelling has negative impact on cellular and tissue morphology and integrity and may culminate in osmotic lysis. Conversely, excessive osmotic shrinkage dehydrates cells to such a degree that it causes misfolding of intracellular proteins and dysfunction of organelles, altogether triggering apoptotic cell death. In higher organisms, dramatic changes in osmolarity are largely restricted to the kidneys and gastrointestinal tract, while the rest of the body is protected by the multilevel homeostatic control of systemic osmolarity [28, 156]. Nevertheless, even in highly regulated extracellular milieu, cells experience frequent fluctuations in their volume due to unbalanced transmembrane fluxes of ions and nutrients or due to osmotically significant synthesis or degradation of macromolecules within the cell. The associated moderate swelling and shrinkage do not jeopardize cell integrity but are harmful because they tend to disrupt the precise equilibrium in concentrations of intracellular ions, signaling molecules, enzymes, and substrates and products of biochemical reactions. These latter changes and their impact on cell function are amplified due to the fact that up to 20–30 % of the cellular interior is occupied by macromolecules and the amount of free, unbound water in the cytosol and organellar matrix is rather limited [55]. As a result, changes in concentrations and activities of the intracellular constituents exceed those which would be expected based on degree of cell swelling or shrinkage. For the purposes of protection, swollen and shrunken cells engage a number of compensatory mechanisms leading to regulatory volume decrease (RVD) or regulatory volume increase (RVI), respectively. A comprehensive discussion of cell volume regulation can be found in numerous reviews on this topic (see for example [87, 110, 136, 209]).
In the majority of cell types, VRAC is the first line of defense against abrupt cell swelling, because the activity of this channel sustains the driving force for RVD. Efflux of Cl− and other anions via VRAC offers accompanying charge for the movement of the most abundant cytosolic ion and osmolyte, K+ [87, 110]. Typically, and this is certainly the case for the central nervous system (CNS) cells, the plasma membrane has high background permeability for K+ and much lower permeability for Cl−. In swollen cells, the outwardly directed flux of K+ is severely restricted by the negative membrane potential, unless opening of VRAC allows for functionally coupled release of anions and in such a way facilitates the net loss of KCl and osmotically obligated water (see diagram in Fig. 1). This principle was recognized very early and experimentally tested in several pioneer studies, which measured electrogenic movement of radiolabeled cations and anions in relationship to cell volume in Ehrlich ascites tumor cells and human lymphocytes [70, 86, 88, 181, 182]. Several years later, two groups independently recorded the first volume-sensitive Cl− currents in the intestinal epithelial 407 cells and human lymphocytes using an electrophysiology approach, effectively launching the extensive biophysical studies of VRAC [34, 81]. Within the next few years, swelling-activated Cl− currents were detected in numerous, highly diverse cell types. What emerged from this subsequent work was that regardless of cell type, swelling-activated Cl− currents possessed strong similarities in their properties and were, therefore, likely mediated by a distinct Cl− channel (or group of very similar Cl− channels). This channel was dubbed either VRAC [142], or volume-sensitive outwardly rectifying (VSOR) Cl− channel [148], or volume-sensitive organic osmolyte-anion channel (VSOAC) [193]. Besides the undisputed role of VRAC in cell volume regulation, this channel was putatively linked to transepithelial ion transport, regulation of membrane potential, cell proliferation, apoptosis, migration and invasion, and release of biologically active molecules (reviewed in [87, 110, 136, 209], with additional focus on VRAC [7, 142, 148, 149, 193, 194]). Some of these VRAC functions will be discussed in the context of brain physiology in the next section.

Membrane mechanisms responsible for regulatory volume decrease (RVD) in osmotically swollen cells. Left: Under steady-state conditions, membrane permeability is dominated by K+ channels (outlined in box) and transmembrane water fluxes are equilibrated. Right: In the response to osmotic swelling, cells dramatically increase membrane permeability to Cl− and other anions due to activation of volume-regulated anion channels (VRAC, outlined in box). This facilitates cooperative loss of K+ and Cl−, which is coupled with release of osmotically obligated water. Many cell types additionally activate KCl loss via electroneutral K+,Cl− transporters (KCC, also outlined in box). Another electroneutral transporter, NKCC1, drives the net uptake of Na+/K+/Cl− under basal conditions and is additionally strongly activated by cellular shrinkage. However, in at least one brain cell type, astrocytes, this transporter is paradoxically stimulated by cell swelling and contributes to sustained astrocytic edema in CNS pathologies (asterisk)
The molecular identity of VRAC was unknown until very recently. Therefore, all early functional studies were based on correlating densities of the VRAC-attributed anion currents with various biological phenomena and the use of putative VRAC blockers—such as 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB), tamoxifen, phloretin, and 4-[(2-butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid (DCPIB)—for probing its roles. The “fingerprint” biophysical properties of VRAC, several of which are reflected in its alternative names, include (i) activation by cell swelling, (ii) moderate outward rectification, (iii) intermediate conductance in the range of 50–80 pS at positive potentials and 10–20 pS at negative potentials, (iv) time-dependent inactivation at high positive potentials, (v) so called low-field strength anion selectivity resulting in the Eisenman type I selectivity sequence for anionic molecules (SCN− > I− > NO3 − > Br − > Cl− > F− > HCO3 − > gluconate), (vi) permeability to small negatively charged and uncharged organic molecules, and (vii) requirement of a non-hydrolytic binding of intercellular ATP for channel activation. Although taken individually these properties are not unique, their combination was used for VRAC identification and to discriminate it from other Cl− channels in various cells and tissues (see representative VRAC current traces in brain cells in the following sections). The comprehensive overview of the biophysical properties of VRAC, covering numerous cell types including brain cells, can be found in several outstanding publications [7, 142, 148, 193]; they are also thoroughly discussed in this issue of Pflugers Archiv by Stine Pedersen et al. I have briefly recapitulated VRAC characteristics in the present manuscript because they are essential for understanding the multiple roles of this channel under physiological and pathological conditions.
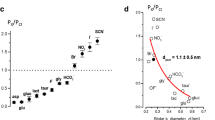
One biophysical feature, which deserves singular attention for our understanding of VRAC functions in the brain, is its permeability for small negatively charged or uncharged organic molecules. The first evidence that Cl− channels (which we now know as VRAC) can constitute a route for transmembrane amino acid fluxes was collected in Madin-Darby canine kidney (MDCK) cells using single-channel electrophysiological recordings. Umberto Banderali and Guy Roy measured transmembrane anion currents using solutions containing aspartic acid, glutamic acid, or the amino sulfonic acid taurine as the main carrier of charge and demonstrated substantial membrane permeability for these molecules [17]. These observations matched numerous independent findings linking cell swelling to a “leak-type” loss of cytosolic amino acids from MDCK and other cell types, including several classes of brain cells [106, 153, 166, 173, 174, 189]. Ironically, the above cited first recordings of the amino acid currents [17] might not have been mediated by VRAC, because they had been performed in the excised membrane patches with ATP-free solutions, which are poorly suitable for VRAC recordings. Nevertheless, the subsequent whole-cell electrophysiological studies—many of which were performed in primary and transformed brain glial cells—supplied strong experimental proof that VRAC is permeable to a variety of amino acids [97, 98, 124, 150]. Such permeability is consistent with the VRAC pore diameter of ~1.1–1.2 nm (11–12 Å) or perhaps slightly larger. The size of the pore was estimated based on the fit of the relative permeabilities of inorganic anions with known radii or derived from electrophysiological recordings in the presence of pore-permeating or pore-blocking sulfonic-calix arenes and polyethylene glycol molecules with various diameters [51, 52, 200]. The VRAC pore may be suitable for conducting molecules as large as ATP [24, 85] and the tripeptide glutathione [178]. This channel poorly discriminates between various anions and neutral molecules due to the weak charge interactions of transported molecules with the amino acid moieties lining its vestibule.
The biggest unsolved question of VRAC biology is the nature of cell volume signal and downstream biophysical and biochemical mechanisms which link channel activity to changes in cell volume. Despite much work in the field, this issue remains unsolved. Numerous intracellular signaling enzymes and cascades have been implicated in the regulation or modulation of VRAC, but none of them appear to play the dominant and universal role across many cell types. Because this particular problem is outside of the scope of the present manuscript, the reader can be referred to several prior publications, which discuss the topic at length [87, 136, 142], and to review by Stine Pedersen et al. in this issue of the journal.
Finally, a few additional aspects of cell volume regulation need mentioning and will be helpful for discussing VRAC functions in the brain. First is the nature of the permeant anion in cell volume regulation during RVD. Certain cell types, including certain types of neurons in the brain, effectively regulate their volume in spite of the very low intracellular Cl− (10 mM or lower) [101]. Therefore, to sustain loss of large quantities of cytosolic K+, these cells likely release appreciable quantities of alternative cytosolic anions, such as bicarbonate and negatively charged amino acids and monocarboxylates, which have been already mentioned above. For this reason, VRAC is frequently called an anion channel rather than a Cl− channel.
The second important point is that many vertebrate cells express electroneutral K+,Cl− cotransporters (KCCs), which are also activated by cell swelling and aid in the process of cell volume regulation (see Fig. 1). Contribution of KCCs to cell volume control was first discovered in red blood cells, where they are entirely responsible for RVD [111]. In the brain, KCCs are highly expressed in neurons. The KCC2 isoform is active under isotonic conditions and helps neurons to maintain intracellular Cl− levels below the Nernst equilibrium potential, allowing for inhibitory actions of GABAA receptor channels [126]. Other KCCs, and particularly KCC3, appear to be important for neural volume regulation [101]. Mentioning swelling-activated KCCs is important in two respects: (i) VRAC and KCCs may cooperate in cell volume regulation since mutations in KCCs lead to brain disorders, which may be associated with cell swelling (see discussion in [101]), and (ii) opening of VRAC can modulate neuronal excitability by changing Cl− permeability in a manner independent of the GABAA receptor function. In contrast to KCCs, the distantly related electroneutral Na+,K+,2Cl− cotransporters (expression of NKCC1 is widespread, while NKCC2 is restricted to the kidney) are activated by cellular shrinkage and mediate RVI (reviewed in [176]). Paradoxically, in one type of brain cells—astrocytes—NKCC1 is activated by cell swelling and also under various pathological conditions. This unique feature of the astrocytic NKCC1 drives uncontrolled changes in cell volume and activation of VRAC in CNS disorders (see Fig. 1 and “Astrocytic VRAC and its contribution to pathological amino acid release and physiological signaling” for references and discussion).
Finally, it is important to mention that VRAC is thought to be a “wet” channel allowing water permeation through its pore [140]. This function will be important for discussion of VRAC activity in certain populations of neuronal cells, which have very low water permeability due to the lack of aquaporin water channels on their membranes.
Common and unique roles of VRAC within the brain
Brain tissue is composed of several diverse classes of cells which greatly differ in terms of their origins and function. Excitable neurons are a major cell type in the CNS; they mediate higher brain functions via synaptic communication in the extensive multicellular networks. The remaining non-excitable cells are collectively called glia or neuroglia. This term neuroglia encompasses three types of cells: astrocytes, oligodendrocytes, and microglia. Astrocytes are actually the most numerous and functionally complex cells within the brain, which create a supportive milieu for neuronal communication. This is accomplished via (i) removal of excessive neurotransmitters from the extrasynaptic space, (ii) buffering fluctuations in the extracellular levels of K+ and other ions, (iii) processing and redistributing energetic substrates delivered with blood flow, and (iv) regulation of blood flow to match the perfusion rates to changing activities in neuronal networks. More recent studies established that besides trophic and homeostatic support, astrocytes communicate with neurons via release and sensing of signaling molecules (gliotransmitters) and, in such a way, actively modulate neuronal communication [80, 152]. The main role of oligodendrocytes is to produce myelin, an insulating structure that covers axonal processes of neuronal cells. Myelin accelerates propagation of action potentials along axonal fibers and also supports their integrity and health. Microglial cells are the “macrophages of the brain”—the resident immune cells in the CNS. They phagocytose apoptotic neurons during development, participate in the pruning of dysfunctional synapses in mature brain, and mediate the inflammatory responses in situations of infection or physical damage to the brain. Finally, it is important to remember that brain tissue is supplied with oxygen and nutrients via an extensive network of blood vessels. Although vascular smooth muscle cells, endothelial cells, and pericytes do not technically belong to the CNS, they actively communicate with the brain’s resident cells, particularly astrocytes, altogether integrating in the so-called neurovascular unit [77]. Many of the functions listed in this paragraph may be directly or indirectly modulated by VRAC.
Electrophysiological studies identified VRAC-like anionic conductance in all types of brain cells with the exception of oligodendrocytes, which have not been extensively studied in this regard. In the present manuscript, I mention only key previous findings and focus on the current views on functional significance of VRAC in the neural tissue. Many helpful additional details about VRAC/VSOR in the CNS can be found in a recent comprehensive review [7]. Historically, astrocytes were studied to a greater extent as compared to other cell classes of brain cells. Therefore, this review is by necessity highly “astrocentric.”
Astrocytic VRAC and its contribution to pathological amino acid release and physiological signaling
The first recordings of swelling-activated Cl− currents in the CNS cells were performed in astroglial cells by the group of Kevin Strange, using the rat glioma C6 cell line in their electrophysiology experiments [97, 98]. Subsequently, many laboratories explored VRAC characteristics and VRAC functions in primary astrocytes isolated from rats and mice [2, 39, 119, 150]. For illustration purposes, I have included here our own recordings of VRAC currents in primary rat astrocytes (Fig. 2a). Independent of the electrophysiological work, several groups started to utilize radiolabeled amino acids and/or HPLC assays to find that swelling of astrocytes triggers release of cytosolic amino acids, including glutamate, aspartate, and taurine [106, 125, 154]. Such release occurs via the permeability pathway, which is sensitive to known VRAC blockers. Representative data demonstrating swelling-activated release of the non-metabolizable glutamate analog d-[3H]aspartate in astrocytes are included in Fig. 2c. The compounds, which inhibit VRAC currents and release of organic osmolytes, also blocked hypoosmotic RVD in the substrate-attached and suspended astrocytes [180]. Overall, the totality of studies on astrocytic VRAC convincingly demonstrated its critical role in cell volume control.

Representative electrophysiology recordings of VRAC currents and swelling-activated glutamate release in brain cells in vitro and in brain cortex in vivo. a Whole-cell recordings of Cl− currents in primary rat astrocytes exposed to hypoosmotic medium (−60 mOsm) with symmetrical concentrations of Cl− in bath and pipette solutions. In the main panel, the time course Cl− current development was recorded in response to brief alterative voltage steps to ±40 mV from the holding potential of 0 mV. In the inset, Cl− currents in response to voltage steps from −100 to +100 mV in 20-mV increments. Note outward rectification and time-dependent current inactivation at high positive potentials. Reproduced with permission from I.F. Abdullaev et al. [2]. b Whole-cell recordings of VRAC Cl− currents performed in rat primary microglial cells. The conditions and composition of experimental solutions were similar to those used in a. Reproduced with permission from T.J. Harrigan et al. [74]. c Swelling-activated glutamate release from primary rat astrocytes traced with its non-metabolizable analog d-[3H]aspartate. Astrocytes were superfused with isoosmotic or hypoosmotic (−100 mOsm) media in the presence or absence of the VRAC blocker DCPIB as indicated. ***p < 0.001, effect of DCPIB on glutamate release in swollen cells. Modified with permission from I.F. Abdullaev et al. [2]. d Swelling-activated glutamate release from primary rat microglial cells measured with the non-metabolizable glutamate analog d-[3H]aspartate. The experimental conditions were identical to those shown in c. ***p < 0.001, effect of DCPIB on glutamate release in swollen cells. Modified with permission from T.J. Harrigan et al. [74]. e Hypoosmotic stimulation of glutamate release from the rat cortical tissue measured using a microdialysis approach. The cortex was perfused with isoosmotic or hypoosmotic artificial cerebrospinal fluid additionally containing the low-affinity VRAC blocker DNDS, which is well tolerated in vivo. Collected microdialysis samples were analyzed off-line for the presence of l-glutamate and other amino acids using HPLC. *p < 0.05, effect of DNDS on glutamate release under hypoosmotic conditions. Reproduced from R.E. Haskew-Layton et al. [75] under the Creative Commons Attribution (CC BY) license
VRAC functions in astrocytes receive a lot of attention because these cells are known to be profoundly swollen in various brain pathologies. Via incompletely defined mechanisms, traumatic brain injury, stroke, hyponatremia, and epilepsy all cause astrocytes to undergo dramatic and sustained swelling, particularly in the areas where the astrocytic processes contact brain vessels (see for example [18, 56, 123] and reviews [103, 104, 129, 134]). Initially, VRAC activation was considered to be a solely protective mechanism assisting in resolution of cellular edema. The release of organic osmolytes from swollen cells was also deemed to be a good thing because it was thought to aid cell volume regulation even in the face of disrupted transmembrane gradients for K+, Cl−, and Na+. To the best of my knowledge, Harold Kimelberg was the first to propose a counterintuitive idea that opening of VRAC may also harm the brain via release of the excitotoxic amino acid neurotransmitters glutamate and aspartate [106].
Ischemia and traumatic brain injury are known to produce buildup of glutamate and aspartate in the extracellular space via a variety of mechanisms [22, 158, 169, 190]. Since these transmitters activate numerous types of glutamate receptors, particularly the Ca2+-permeable N-methyl-D-aspartate receptor (NMDA) subtype of glutamate receptors, they persistently depolarize neurons. Prolonged depolarization causes pathological increases in the intracellular Ca2+ levels and kills neuronal cells via activation of numerous Ca2+-dependent degradative processes. The glutamate receptor-dependent cell damage and death are known under the umbrella term “excitotoxicity” [16, 37, 117]. Perhaps, glutamate toxicity is the price to pay for use of this molecule as both the brain’s main excitatory neurotransmitter and the major cytosolic osmolyte (it is the most abundant amino acid in the CNS). Sustained glutamate release from pathologically swollen cells is incompatible with brain physiology.
The hypothesis of VRAC toxicity was extensively tested in vivo, in animal models of stroke. Systemic administration of the blood-brain barrier-permeable VRAC inhibitor tamoxifen potently protected the brain against injury in rodent models of transient and permanent cerebral ischemia (up to >80 % reduction of brain infarction [105, 107], also see Fig. 3a). These findings were reproduced with the more selective VRAC blocker DCPIB [216] and further verified in larger animal species, in a canine model of stroke [27]. Equally important, a variety of VRAC inhibitors including tamoxifen and DCPIB were found to reduce the intraischemic glutamate release, which was quantified using a microdialysis approach or cortical cup perfusion technique [60, 159, 160, 216] (also shown in Fig. 3b).

The VRAC inhibitor tamoxifen potently protects rat brain against damage in an experimental stroke model and reduces the intraischemic glutamate release in rat cortical tissue. a Representative images of brain sections from rats subjected to 2-h experimental stroke and evaluated 3 days after ischemia. Animals were treated with vehicle (A) or the VRAC blocker tamoxifen (5 mg/kg) given i.v. either 10 min prior to (B) or 3 h after initiation of stroke (C). At the end of the experiment, rats were euthanized and their brains were sliced and stained with triphenyltetrazolium chloride to visualize tissue damage (infarcted tissue is unstained). Reproduced with permission from H.K. Kimelberg et al. [105]. b Dynamics of the microdialysate glutamate levels during and after focal cerebral ischemia. Vehicle control (5 % DMSO), the VRAC blocker tamoxifen (50 μM), or the inhibitor of the glial glutamate transporter GLT-1 dihydrokainate (DHK, 1 mM) was delivered through the microdialysis probe placed in the ischemic penumbra. *p < 0.05, tamoxifen vs. DMSO and DHK during ischemia; #p < 0.05, DHK vs. tamoxifen and control after ischemia (repeated measures ANOVA). Modified with permission from P.J. Feustel et al. [60]. c Hypothetical representation of the processes in the ischemic penumbra, based on the results of microdialysis experiments. Pathological swelling of astrocytes (and perhaps other cells) triggers glutamate release via the VRAC. Increased glutamate levels cause Ca2+-dependent damage and death of neuronal cells due to excessive activation of neuronal NMDA receptors. In the penumbra, glial transporter GLT-1 continues to take up extracellular glutamate and, therefore, addition of DHK leads to high sustained glutamate levels during and after ischemia
The effects of VRAC inhibitors on glutamate release in ischemia vary depending on the location of the probe in the ischemic tissue. It is important to remember that VRAC activity requires non-hydrolytic binding of ATP on its cytosolic side, and the channel is potently blocked by arachidonic acid and other polyunsaturated fatty acids [142, 148, 193]. Therefore, in the ischemic core, VRAC is likely inactive due to depletion of ATP and the phospholipase-driven accumulation of fatty acids. Instead, in this ischemic area, glutamate is unloaded to the extracellular space via reversal of glial and neuronal Na+-dependent glutamate transporters and perhaps other mechanisms [129, 169, 190, 198]. In contrast, on the periphery of ischemic tissue, in the area called the ischemic penumbra, collateral blood flow partially sustains metabolism in brain cells, allowing for VRAC activation. There, tamoxifen and DCPIB suppress the intraischemic glutamate release by up to 70 % [60, 216] (Fig. 3b). Finally, one should ask what cell type, or cell types, contributes to VRAC-mediated glutamate release. There is no definitive answer to this question but two facts point to a dominant role of astrocytes: (i) in the ischemic tissue, astrocytes, but not neurons, show prominent swelling; (ii) neuronal VRAC is paradoxically insensitive to tamoxifen and therefore unlikely responsible for the protective effects of this compound in stroke (see additional discussion in “Neuronal VRAC and its potential impact on volume control, excitability, and excitotoxic cell death”). Altogether, animal data support the major pathological significance for VRAC in stroke and suggest that it represents a promising therapeutic target in this disorder [104, 129]. This notion led to designation of VRAC as the “frenemy” of the brain.
Another example of human disease that is strongly associated with astrocytic swelling is hyponatremia—the most prevalent fluid-electrolyte disorder encountered in clinical practice. It is defined as a drop in blood serum Na+ levels from 145 to 150 mM to lower than 135 mM, with matching changes in systemic osmolarity. Hyponatremia is frequent in hospitalized patients and elderly individuals in long-term care and usually develops due to improper retention of water [3, 67, 204]. Although all tissues are affected by the osmotic change, the brain represents the main target organ. In its most severe forms, acute hyponatremia is life-threatening because it causes dramatic swelling of the brain tissue. Because the brain is confined by the rigid skull, tissue expansion compresses blood vessels to cause deficits in cerebral circulation and eventually leads to herniation of the brainstem. Milder reductions in blood Na+ levels produce a variety of neurological symptoms—such as nausea, confusion, headaches, and hallucinations—and elevate the risk of death in comorbid states [67, 162, 204].
The neurological manifestations of hyponatremia may be related to the swelling-activated release of glutamate, aspartate, and other neuroactive substances via VRAC. In model microdialysis experiments performed in the rat cortex, local perfusion of hypoosmotic medium triggered elevation in extracellular levels of several VRAC-permeable amino acids, much like what is seen in cultured cells [75, 93] (see Fig. 2e). Strangely enough, hyponatremia causes rather selective swelling of astrocytes and their processes surrounding blood vessels [123, 167, 208]. In vivo observations suggest that cortical pyramidal neurons do not express aquaporins and due to low water permeability of their membranes do not extensively swell under hypoosmotic conditions, and the same is true for neuronal processes and both synaptic and dendritic structures [12, 192]. We found that swelling can modify neurotransmitter signaling by acting on astrocytes via at least two mechanisms [93]. Normally, astrocytes maintain low extracellular glutamate levels due to uptake via the Na+-dependent glutamate transporters. In pathology, opening of VRAC shunts neurotransmitters to the extracellular space, causing neuronal excitation [75]. This “noxious” or “toxic” excitatory amino acid release can be partially buffered via re-uptake by the Na+-dependent glutamate transporters, but this compensatory mechanism is not sufficient to protect against pathological degrees of cell swelling [187]. Additionally, astrocytic swelling inhibits glutamine synthetase, the enzyme which converts the excitatory glutamate to inert glutamine, which is returned to neurons to complete the glutamate-glutamine cycle [93].
Yet another syndrome that disproportionally affects the CNS and is strongly associated with astrocytic swelling is hyperammonemia—systemic increases in ammonia levels above the threshold of ~100 μM, which lead to hyperammonemic encephalopathy (see for example [143] and reviews [23, 33, 144]). Most frequently, hyponatremia develops due to chronic liver disease or acute failure of the liver function. However, other conditions—such as inherited deficiencies of the urea cycle, certain bacterial infections of the gut, and Reye’s syndrome—may also lead to the same type of neuropathology [33, 59]. High blood ammonia levels approaching 1 mM and brain ammonia levels within the range of 1–5 mM trigger severe brain edema that is frequently lethal due to severe ischemia and herniation of the brain [23, 33, 144]. These complications are the result of increasing intracranial pressure due to net accumulation of organic osmolytes and osmotically obligated water. The main mechanism of osmolyte accumulation is fixation of ammonia and production of glutamine by the astrocytic ATP-dependent glutamine synthetase [33, 144]. The osmotic burden due to the enhanced activity of glutamine synthetase is further accentuated by accumulation of brain alanine via the work of the alternative detoxification mechanism incorporating glutamate dehydrogenase and alanine aminotransferase [41, 201]. Actions of astrocytic glutamine are not restricted to its direct osmotic effects, since this amino acid is also thought to induce cellular pathology indirectly, via oxidative and nitrosative stress and impaired mitochondrial function (the “Trojan horse” hypothesis [8]).
In acute hyperammonemia, cell swelling is nearly exclusively restricted to astrocytes and is likely largely mediated by the activity of glutamine synthetase in these cells [102, 143, 144]. Cell swelling and metabolic changes in astrocytes dramatically dysregulate metabolism of numerous neurotransmitters and lead to a plethora of neurological deficits. Among the relevant changes, elevated extracellular glutamate (which was directly observed in microdialysis experiments) and ensuing activation of NMDA receptors (confirmed via protective actions of selective NMDA antagonists) play a major role in hyperexcitation, seizures, tissue damage, and mortality [26, 59, 83]. It is tempting to link the ammonia-induced glutamate release and excitotoxicity to swelling-activated VRAC (similar to that discussed above for hyponatremia). However, this is rather an overly simplistic and speculative view. The strong experimental support for contribution of VRAC is currently lacking, and other mechanisms for neurotransmitter release have also been proposed (see for example [69] and review [137]).
Unlike the acute syndrome, chronic hyperammonemias are less dangerous but much more common in patients with chronic liver disease. In these, and related animal models, astrocytic swelling is not obvious (“low-grade” cellular edema), but rather astrocytes show progressive morphological changes characterized as Alzheimer type II astrocytic degeneration, with nuclear enlargement, prominent nucleoli, and distinct margination of chromatin [58, 59, 76]. Some of the associated changes in brain physiology and neurotransmitter homeostasis and signaling may be related to moderate astrocytic swelling (and, by extension, to VRAC activity). However, despite the fact that such connection has been proposed and continues to be discussed [76], it has not been thoroughly evaluated. Overall, it is clear that robust changes in brain physiology that occur during chronic hyperammonemic encephalopathy cannot be reduced to the effects of cellular swelling. The alternative concepts and views have been recently reviewed in [58].
As already mentioned in the “Introduction: general properties and physiological significance of VRAC,” brain cells, including astrocytes, express the ubiquitous isoform of Na+,K+,2Cl− cotransporter—NKCC1, which is typically activated by cellular shrinkage and mediates a regulatory accumulation of inorganic ions and RVI (reviewed in [176]). High expression of astrocytic NKCC1 allows astroglial cells to keep their cytosolic Cl− levels high, above the electrochemical equilibrium for this anion [176]. In primary rat astrocytes and rat glioblastoma C6 cells, NKCC1 is paradoxically activated by cell swelling [130, 131] and can be also potently stimulated by high extracellular [K+], both of which can be expected during extensive neuronal activity, epileptic episodes, or ischemia [122]. The resulting net uptake of Na+/K+/Cl− may induce physiological cell swelling or sustain pathological cell volume increase (see [129] for detailed discussion). The diuretic compound bumetanide, which selectively blocks NKCCs, or genetic deletion of NKCC1 prevents high [KCl]out-induced cell swelling and the associated release of excitatory amino acids, which is presumed to occur via VRAC and contribute to tissue damage [177, 195, 196]. In a rat model of focal ischemia, bumetanide reduces stroke infarction volumes by >50 %, while the NKCC-null mice show 30–45 % lessening of their infarction volumes and up to 50 % reduction in brain edema [36, 212]. Neuroprotective properties of the NKCC blocker bumetanide are not restricted to stroke models but have also been reported in acute liver failure [100] and traumatic brain injury [99]. The caveat of these in vivo studies is associated with expression of the NKCC1 in numerous cell types, including endothelial cells. Therefore, the suggested contributions of NKCC1 to the development of brain edema and neurodegeneration may not be limited to astrocytes [32, 145, 146].
Besides the rare situations of acute osmotic stress and several abovementioned brain pathologies, little is known about what functional impact astrocytic VRAC has in the healthy brain. One clear exception is related to the role of astrocytes in the hypothalamic neuroendocrine structures. The supraoptic and the paraventricular nuclei of the hypothalamus contain magnocellular neurons, whose main function is regulation of the whole-body water-electrolyte homeostasis [29, 90]. These cells accomplish this task via regulated secretion of the antidiuretic hormones vasopressin and oxytocin, both of which reduce water secretion (or rather increase water reabsorption) in the kidney. Magnocellular neurons sense changes in systemic osmolarity via (i) input from [Na+]-sensitive and osmosensitive neurons in circumventricular structures (the subfornical organ and the organum vasculosum lamina terminalis), (ii) engaging their own intrinsic osmosensing mechanisms, but also (iii) receiving signals from the surrounding specialized astrocytes which contain extremely high levels of taurine in the cytosol. These astrocytes sense small decreases in systemic osmolarity and, once swollen, release taurine via VRAC or a VRAC-like channel [31, 44]. In turn, taurine suppresses activity of magnocellular neurons by acting as the endogenous ligand for the inhibitory glycine receptors and reduces the release of vasopressin and oxytocin [91]. Downstream of this area, in the neurohypophysis, another population of specific astrocytes (pituicytes) acts presynaptically, also via the release of taurine, and inhibits secretion of vasopressin and oxytocin at this level. Interestingly, in addition to their systemic functions, vasopressin and oxytocin regulate taurine secretion from pituicytes, creating a loop of paracrine intercellular communication [170].
In a much similar fashion, astrocytes in other brain areas may also release taurine, glutamate, and other neuroactive substances, which contribute to astrocyte-neuron communication. Although attractive, this idea has not been thoroughly tested. We know that in the CNS, oscillatory activities in neuronal networks cause fluctuations in the volume of extracellular and intracellular space (see for example [11, 89] and review in [197]). Such fluctuations are thought to involve swelling of astrocytes due to the net accumulation of K+, glutamate, and accompanying ions. However, it is not clear if the degree of physiological swelling (estimated at ~5 % of the “normal” cell volume) is sufficient to stimulate VRAC. To date, this conceptual problem was addressed only in vitro by studies which discovered the phenomenon of VRAC modulation by G-protein-coupled receptors (GPCRs) in non-swollen cells. The impetus for these studies was created by an early report in hepatoma cells, which proposed that release of ATP and stimulation of purinergic P2Y receptors serve as an obligatory autocrine signal linking cell swelling to VRAC opening [207]. Although the obligatory nature of ATP signaling had not been confirmed for brain cells, or any other cell types, we and others found that activation of P2Y receptors in astrocyte cultures by ATP and other purinergic agonists produces limited, Ca2+-dependent stimulation of VRAC [5, 42, 132, 135, 199]. The ATP-induced increases in VRAC activity and osmolyte release were smaller than those induced by cellular swelling and involved distinct signaling mechanisms. ATP signaling cascades integrate P2Y1 and P2Y2/P2Y4 receptors, release of Ca2+ from the intracellular stores, activation of classical PKCα and βI, and additionally Ca2+/calmodulin-dependent protein kinase II [135, 175]. According to our current working model, purinergic signaling activates or modulates a small fraction of VRAC channels that are already open or “primed” for opening (present in the membrane?) via phosphorylation of VRAC itself or its auxiliary protein. Although ATP effects are measurable in non-swollen cells, the main, larger pool of VRAC molecules remains inaccessible for purinergic stimulation. A concurrent hypotonic cell swelling taps into this volume-sensitive pool and acts synergistically with the purinergic stimulus, presumably by making the VRAC protein accessible to phosphorylation or delivering phosphorylated proteins to the plasmalemma [135].
Following the discovery of the purinergic modulation of VRAC activity in non-swollen astrocytes, numerous studies described similar effects of the diverse GPCR receptors, linked to either Ca2+ or cAMP signaling, in the CNS cells and cells derived from many other tissues. There are too many to list, but a good summary of the relevant publications can be found in at least three reviews on this topic [64–66]. In the context of astrocyte physiology, the main point here is that VRAC opening may occur even in the absence of astrocytic swelling and lead to the release of glutamate and other gliotransmitter molecules. This idea dovetails with numerous reports that astrocytes signal to neurons by liberating glutamate, d-serine, and/or ATP. Thus far, the main mechanism that was ascribed to this process is the Ca2+-dependent release from the vesicular pool (reviewed in [15, 80, 152]). It has been debated that alternative mechanisms of gliotransmission, including those mediated by VRAC, are also possible [61, 72]. Unfortunately, due to the unknown molecular nature of VRAC and the general lack of selective pharmacological and molecular tools, testing VRAC contribution to gliotransmission in complex systems and in vivo has been nearly impossible. Due to the recent discoveries, we are about to see a dramatic change in this situation (see “A whole new game: the discovery of LRRC8 proteins and its impact on CNS research” for further discussion).
Neuronal VRAC and its potential impact on volume control, excitability, and excitotoxic cell death
In neurons or cells of neuronal origin, swelling-activated VRAC or VRAC-like currents were reported in rat sympathetic neurons [112]; mouse cortical, hypothalamic, and cerebellar granular neurons [95, 155, 183]; neuroblastoma cell lines [20]; and hippocampal CA1 pyramidal neurons recorded in situ [215]. In addition to electrophysiology studies, several groups explored the swelling-activated release of taurine and other organic osmolytes in relationship to the RVD process [20, 138, 188, 189]. Compared to astrocytes and non-CNS cells, there are fewer studies in primary neuronal cells because they are more difficult to isolate, cultivate, and patch. Nevertheless, an analysis of existing literature reveals several unique features of VRAC and volume regulation in neuronal cells. One peculiarity of neuronal VRAC is its insensitivity to the potent VRAC inhibitor tamoxifen [95, 96, 112, 215]. Whether the differences in pharmacologic profile point to the existence of a distinct swelling-activated Cl− anion channel in neuronal cells is presently unknown.
Another striking aspect of neuronal cell volume regulation is that pyramidal neurons in the cortex and hippocampus—whether acutely isolated or studied in situ and in vivo—are highly resilient to osmotic swelling due to very low water permeability of their membranes [4, 12, 13]. Recent in vivo work demonstrated that neuronal processes and dendritic and synaptic structures also do not show appreciable swelling when exposed to osmotic gradients as large as 100 mOsm [192]. The in vivo and in situ studies were made possible with the introduction of transgenic animals expressing green fluorescent protein (GFP) or its variants under the cell-type-specific promoters. Using two-photon confocal microscopy through a small cranial window, it is now possible to monitor changes in cellular volume and morphology at the single-cell level in anesthetized animals. However, the abovementioned resistance to osmotic challenge is not universal for all types of neurons. Osmotic swelling and effective RVD have been reported in cerebellar granular cells [189], sympathetic neurons [112], hippocampal neurons [214], and neuroblastoma cells [10, 20, 116]. Osmosensitive hypothalamic neurons readily swell but regulate their volume at extremely slow rates [183, 214]. Therefore, depending on their type, neuronal cells may exert dramatic differences in osmotic behavior and VRAC activation, particularly in vivo, where changes in the extracellular and intracellular osmolarity tend to develop much slower than in model in vitro experiments employing hypoosmotic media.
The functional significance of VRAC in neurons is usually considered in terms of cell volume regulation and development and resolution of pathological cell swelling. Being the excitable cells, neurons extensively utilize the transmembrane ionic currents as a tool of the trade. High-frequency stimulation typically leads to net accumulation of Na+ and Cl− and swelling of somata and axonal processes and dendritic spines (see for example [14, 63, 203]). Douglas Fields proposed that in the axonal regions, the activity-dependent swelling may be functionally important for communication between axons and surrounding glial cells and involve release of neuroactive molecules, presumably via VRAC [62]. Under physiological and supraphysiological conditions, changes in neuronal volume are quite small which make it difficult to differentiate them from changes in the surrounding glial cells. Nevertheless, some inferences can be made based off the existing literature. For example, Haj-Yasein et al. [71] analyzed the high-frequency-stimulation-induced shrinkage of the extracellular space (which is proportional to cell swelling) in the hippocampus of animals carrying deletion of astrocytic water channel aquaporin-4. In the hippocampal areas enriched with astrocytic processes, high-frequency stimulation triggered extracellular space shrinkage that was highly influenced by deletion on aquaporin-4, meaning it was related to astrocytes. On the contrary, in the areas devoid of astrocytic processes, changes in the extracellular space (cell swelling) were more profound and completely independent of aquaporin-4, pointing to their neuronal origin [71].
In vitro and in situ, neuronal swelling is largely driven by Na+ accumulation via the glutamatergic AMPA and NMDA receptor channels [96, 203]. VRAC is thought to serve as a critical partner in this process because it provides a route for coupled Cl− influx. In cortical neurons, structurally diverse VRAC blockers strongly attenuate the NMDA-induced increase in volume of somata and profound swelling (“bidding”) of dendritic spines [96]. However, after NMDA is removed, surviving neurons tend to recover their volume and reverse dendritic bidding in a process that is also sensitive to VRAC inhibitors [96]. This reveals the Janus face of VRAC in neuronal cell volume regulation: depending on the state of membrane Na+ permeability and directionality of cation fluxes, VRAC either promotes pathological cell swelling or assists in cell volume recovery (RVD). How harmful are neuronal swelling and VRAC activity? Apparently very much so, because in the neurons exposed to excitotoxic levels of NMDA, VRAC blockers reduce neuronal death by 80–95 % [96]. These data fit well with older reports which found that when neurons die upon exposure to glutamate, NMDA, or kainate, the first (acute) phase of cell death was prevented by reducing extracellular concentration of Cl− [172]. The acute glutamate-induced cell death is followed by a delayed phase of neuronal demise, which involves apoptosis or apoptosis-like processes [38, 117]. These later stages of neurodegeneration require intracellular [Ca2+] increases but are largely insensitive to extracellular Na+ and Cl− levels. Nevertheless, they also may rely on VRAC, because in many cell types, fluxes of inorganic and organic anions via VRAC, which accompany K+ efflux via K+ channels, are thought to mediate obligatory apoptotic volume decrease or AVD [25, 87, 149]. To what extent VRAC and AVD contribute to delayed neuronal cell death remains to be established.
Other processes of similar nature, which may be highly relevant to VRAC activity, are spreading depolarization and spreading depression. Spreading depolarization and spreading depression are waves of robust depolarization of neurons and glial cells which occur focally and then spread through the brain tissue at a rate of 2–6 mm/min [49]. These take place frequently in humans and in animal models during migraines and after traumatic brain injuries, hemorrhagic events, and strokes, and exert a powerful negative influence on neurological outcomes [49, 50, 121, 210]. Early model studies in brain slices linked spreading depression to waves of glutamate release to the extracellular space, which likely accelerate and propagate massive depolarization. Glutamate release during spreading depression was attributed to the activation of VRAC, based on its inhibition by NPPB and insensitivity to extracellular Ca2+; the latter would be necessary for release from the neuronal vesicular pool [19]. Due to the lack of specific pharmacological inhibitors and molecular biology tools, involvement and significance of VRAC in spreading depression have not been thoroughly tested, but this is something that awaits additional studies. Besides the already-mentioned glutamate release, VRAC can serve as a route for water flux in aquaporin-deficient neurons and neuronal processes because this channel is thought to conduct water [140].
Finally, I would like to briefly touch on a possibility of interplay of VRAC and KCC in the regulation of neuronal excitability and neuronal volume under physiological and pathological conditions. As already mentioned in the “Introduction: general properties and physiological significance of VRAC,” the majority of mature neurons in the adult brain have very low intracellular Cl− (<10 mM), which is maintained at this level due to the constant work of the KCC2 [126]. The KCC2 differs from all other KCC isoforms because it continues to work in non-swollen cells, while other KCCs are activated by cell swelling [101]. Low intracellular Cl− allows neurons to regulate their excitability via actions of the inhibitory neurotransmitters, such as GABA or glycine. Upon stimulation of GABA and glycine receptor channels, inward-directed Cl− fluxes hyperpolarize mature neurons and reduce their excitability [126]. Since, as I already mentioned, high-frequency neuronal stimulation leads to neuronal swelling, it is easy to imagine that opening of VRAC in a swelling-dependent (or swelling-independent) manner can cause inhibition of neuronal activity, acting as the GABA-insensitive equivalent of GABA receptors. This is something which has never been experimentally tested but remains a possibility. The second point here is that KCCs and VRAC may cooperate in neuronal volume regulation. Due to low intracellular Cl− and the inward-directed electrochemical gradient, VRAC function in RVD would be dependent on alternative anions and overall reduced. The swelling-activated KCC activity is also impacted by low intracellular Cl− levels and likely insufficient for effective and timely cell volume regulation. Therefore, cooperative actions of KCC and VRAC may be needed to support volume homeostasis. It is pure speculation, but this may explain why mutations in swelling-activated KCCs, such as KCC3, result in severe neurological phenotypes, possibly via dysregulation of cell volume control (see discussion in [101]).
VRAC in cell proliferation, migration, phagocytosis, and release of neuroactive substances in microglial cells
Microglial cells originate from monocyte precursors, which populate the brain at very early stages of development, and then become “isolated” from the rest of the body by emergence of the blood-brain barrier [139]. Therefore, in terms of their functions and properties, these cells are closer to monocytes/macrophages than to other glial cells or neurons. The ion channel expression profile of microglia is tailored to sustain their functions: migration, proliferation, phagocytosis, and production of reactive oxygen species by the membrane NADPH oxidase. There have been very few electrophysiology studies focusing on swelling-activated Cl− channels in microglia, and the literature is somewhat controversial. Two key publications came from the laboratory of Lyanne Schlichter [53, 186]. In their experiments, which were carried out in primary rat microglia, induction of hypoosmotic swelling led to activation of outwardly rectifying VRAC-like Cl− currents. However, these currents differed from the “classical” VRAC profile in at least two respects: low single-channel conductance and insensitivity to high concentrations of extracellular ATP [53]. Our group recorded swelling-activated Cl− currents in the same cell type and, to the contrary, found that the biophysical profile, average current densities, and high sensitivity to DCPIB were all consistent with expression of the canonical VRAC [74] (see representative traces in Fig. 2c). Additionally, Eder et al. [54] discovered stretch-activated Cl− currents in the murine microglial cells. Although moderate outward rectification of these currents and their sensitivity to the broad spectrum Cl− channel blockers SITS, flufenamic acid, and NPPB resembled VRAC properties, VRAC is known to be insensitive to membrane stretch. Clearly, more work is needed to address the molecular nature of Cl− channel(s) in microglia, including studies utilizing molecular biology techniques.
Although the molecular nature of swelling- and stretch-activated Cl− channels in microglia is unknown, there is little doubt about their functional significance. Broad-spectrum Cl− channel blockers completely suppressed proliferation of microglial cells [186], reversibly inhibited formation of ramified processes [54] and lamellipodia [217], prevented phagocytosis of bacteria and microspheres [53, 68], and limited cytokine-stimulated cell migration [165]. Furthermore, Cl− channel inhibitors blocked cell volume regulation in hypoosmotically swollen microglia and caused cell swelling when applied under isoosmotic conditions [53]. The latter finding is consistent with relatively high resting Cl− permeability in unstimulated microglia which is conducive for the net KCl uptake [53]. Importantly, the functional significance of microglial Cl− channels was tested in situ, in a slice model of microglial response to brain injury [84]. In the latter study, extension of microglial processes towards the site of injury was completely blocked by the extracellular media with low Cl−, and by the broad-spectrum VRAC inhibitors DIDS, NPPB, and tamoxifen. The take-home message from this brief literature analysis is that Cl− channels are critical for microglial proliferation and the processes that require polarized activation of ion channels, such as migration, cell shape changes, and phagocytosis. Although the experimental evidence for connection between these cellular functions and VRAC is not conclusive, such an idea is consistent with findings in other cell types [87, 110].
A topic which requires separate attention in the context of the present discussion is the potential contribution of microglia to pathological release of excitatory amino acids. In our past work, we measured microglial release of glutamate, traced with its non-metabolized analog d-[3H]aspartate, in cells which were exposed to either hypoosmotic media or zymosan. In microglia (and macrophages), zymosan activates Toll-like receptors and triggers production of reactive oxygen species. Both cell swelling and zymosan-stimulated release of glutamate and their actions were additive [74]. In all cases, we found that release of the radiotracer was completely suppressed with DCPIB, strongly suggesting that VRAC is involved. These data are consistent with two electrophysiology reports in primary and immortalized microglia, which estimated the relative permeability for glutamate and aspartate to be 0.12–0.18 and 0.22, respectively, as normalized to Cl− permeability [53, 185]. In another more recent study, we measured d-[3H]aspartate release in microglial cells during ATP-stimulated migration and found that chemotaxis also causes the DCPIB-sensitive efflux of excitatory amino acids [48]. Taken together, these data suggest that activation and migration of microglial cells are associated with release of potentially toxic glutamate and aspartate. In pathologies such as stroke and traumatic brain injury, microglial cells migrate towards the site of injury or inflammation in large numbers to assist in phagocytosis of dying cells and tissue remodeling. VRAC or VRAC-like channels are active participants and the driving force in many of these processes. Yet, due to the release of glutamate, microglial VRAC may also contribute to the excitotoxic death of neuronal cells, feeding into the frenemy theme of this review.
VRAC pharmacology in the context of probing its biological functions
One of the biggest challenges of studying VRAC physiology and functions was and is the lack of selective pharmacological agents, which block or modulate its activity. This is certainly a problem for several if not the majority of Cl− channels as a class [141, 205]. Many of the inhibitors mentioned in this review such as NPPB, DIDS, SITS, and flufenamic acid inhibit numerous Cl− channels, and their potency in inhibiting VRAC may vary widely among cell types (for details and discussion, see [7, 142, 148]). Over the years, a number of studies utilized tamoxifen as “more selective” and one of the most potent inhibitors. This compound was originally discovered as the blocker of estrogen receptors and is extensively used as a treatment for estrogen-receptor-sensitive breast cancer. In the concentration range of 1–10 μM, tamoxifen inhibits VRAC by >80–90 % in many, but not all, cell types [7]. However, in the similar concentration range, tamoxifen also blocks several types of cationic, Na+ and K+ channels [9, 73, 82] and activates the BK type of K+ channels [45, 46]. To make matters worse, tamoxifen possesses antioxidant properties [40] and potently inhibits neuronal (nNOS) and inducible (iNOS) isoforms of nitric oxide synthase (NOS) [151]. The latter two actions of this agent created a particular problem for interpreting the neuroprotective effect of tamoxifen in stroke, a disease that includes oxidative stress and excessive NO production as important components of its pathology [47, 120].
Needless to say, leading laboratories in the field were constantly in search for more selective anion channel blockers. One of the agents that partially discriminate between VRAC and other Cl− channels is phloretin, which was originally identified as an inhibitor of the Na+-dependent glucose transporters [179]. At a concentration of 100 μM, phloretin blocks VRAC currents by 80 % or more, but either does not affect or very weakly suppresses several other types of Cl− channels [57]. Another highly touted and widely utilized compound is DCPIB. It was found to strongly discriminate VRAC from an extensive array of anion and cation channels in model and primary cell lines [43]. Consequently, this inhibitor became the staple for probing VRAC functions in cells, more complex experimental systems, and in vivo. In the CNS, this agent was found to block VRAC in astrocytes [2, 118], neurons and neuroblastoma cells [35, 78], and microglial cells [74, 185]. Furthermore, DCPIB potently protected the brain against ischemic damage in a rodent model of stroke [216]. The latter finding is considered the strongest evidence to date for pathological significance of VRAC in the brain. However, as it is the case for majority of pharmacological agents, over time, the selectivity of DCPIB has been questioned. Our group tested this agent for its ability to inhibit diverse glutamate permeability pathways and glutamate transporters. We found that in addition to VRAC, DCPIB also potently blocks glutamate release via connexin-43 hemichannels, and glutamate uptake by glial glutamate transporter GLT-1 [30]. Although not anticipated, these effects certainly follow the pattern of cross-inhibition of VRAC and astrocytic connexin hemichannels with numerous VRAC and connexin blockers, such as NPPB, tamoxifen, carbenoxolone, and others [21, 213]. Even more peculiar, DCPIB was found to enhance the activity of the two-pore domain K+ channels TREK-1 and TREK-2 in their native environment and after recombinant expression [128]. The effects of DCPIB on glutamate release via connexins and activation of K+ channels will have to be taken into consideration while interpreting the effects of DCPIB on cellular and tissue functions and stroke outcomes.
Limited selectivity of pharmacological tools has always been an issue in a cell-based search for VRAC functions. Still, this problem was not unsurmountable. Studies in cell models typically utilized several structurally unrelated inhibitors to increase confidence in conclusions and also included comparisons of biological phenomena with changes in transmembrane VRAC currents, which have distinct biophysical properties. In contrast, in vivo studies relied (and continue to rely) nearly exclusively on the use of pharmacological agents. Recent findings on the limited selectivity of DCPIB and an ever-expanding list of the off-target effects of other Cl− channel blockers led to stagnation of the research on VRAC physiology. The field was and still is waiting for more selective molecular biology tools.
A whole new game: the discovery of LRRC8 proteins and its impact on CNS research
The molecular nature of VRAC has remained an enigma for several decades. As already mentioned in the preceding text, this gap of knowledge created a significant barrier for progress in the field. Numerous laboratories attempted to identify VRAC, initially via heterologous expression in Xenopus oocytes or other expression systems, and later using RNA interference (RNAi)-driven downregulation of the candidate genes, which encoded known Cl− channels or proteins possessing Cl− channel homology. Several early molecular contenders and more recent VRAC candidates have all been discarded due to the “non-fitting” biophysical properties of the related channels, some limitations of employed experimental techniques, and/or because the newly identified proteins were found to encode VRAC “regulators” rather than VRAC itself. The various stages of the long and turbulent history of VRAC research have been summarized in several excellent reviews [142, 148, 157, 191, 193].
It is only now, retrospectively, that we understand why traditional methods were poorly suited for the identification of VRAC. Two recent independent studies used remarkably similar methodology to accomplish the long-awaited breakthrough. Research teams headed by Ardem Patapoutian in the USA and Tobias Stauber and Thomas Jentsch in Germany utilized small interfering RNA (siRNA) screens in HEK293 cells in conjunction with monitoring fluorescence of the iodide-sensitive mutant of the yellow fluorescent protein (YFP) to detect VRAC activity [164, 206]. In cells exposed to hypoosmotic medium, which additionally contained I−, opening of VRAC caused influx of I− into the cytosol and quenching of the YFP fluorescence. The latter change in the signal was registered using high-throughput microplate readers allowing for effective, genome-wide (or nearly genome-wide) screens for the siRNA species that knocked down endogenous VRAC in HEK293. Results of fluorescence experiments were further validated using an electrophysiology approach in HEK293 and several other cell lines. Both groups reached essentially the same conclusion that expression of the ubiquitous transmembrane protein leucine-rich repeat-containing 8A (LRRC8A) is indispensable for VRAC activity, while the other four members of the same protein family, LRRC8B–E, appeared to be expendable [164, 206]. However, Voss et al. [206] went one step further and, using the erase-and-replace approach, had proven that LRRC8A ought to heteromerize with at least one additional member of the LRRC8 family in order to create functional VRAC. Qiu et al. [164] renamed LRRC8A to SWELL1 to highlight its biological function. The brief summary of what is known about LRRC8A structure and multimeric composition of the LRRC8/VRAC channels is provided in Fig. 4.

Schematic representation of the predicted topology of LRRC8A and multimeric composition of the LRRC8 heteromeric complexes forming VRAC. a Transmembrane topology of the LRRC8A protein based on the model proposed by Abascal and Zardoya [1]. This diagram contains positions of known glycosylation sites (Y), four conserved cysteines (yellow circles), N-terminal leucine-rich repeat motifs (red cylinders), and putative phosphorylation sites for casein kinase II (CK), protein kinase C (PKC), and cAMP-/cGMP-dependent kinases (PKA/PKG) (color-coded multipoint stars, as indicated). The putative phosphorylation sites were determined using the LRRC8A sequence from NCBI Protein database (sequence NP_001120717.1) and ScanProsite phosphorylation probability software (Swiss Institute for Bioinformatics). b Multimeric composition of LRRC8 complexes based on pannexin homology and findings of Voss et al. [206]. Six subunits are proposed to form a channel with one central pore; LRRC8A (red) is an obligatory component of this complex and has to be heteromerized with at least one of the four other members of the same family (LRRC8B–E) to form a functional VRAC channel. The exact amount of each subunit in the heteromer is unknown
Identification of the LRRC8A and discovery of the complementary function for other LRRC8 proteins were very instructive for understanding why other research groups came up short in their search for VRAC identity. Overexpression of the LRRC8A alone reduced rather than increased endogenous VRAC currents, while overexpression of the LRRC8A in combination with its LRRC8 paralogs did not have any suppressive effects but neither did it increase the VRAC current densities [206]. This latter set of data and the presence of endogenous “background” VRAC currents in the majority of cells explain the failure of previous hetero-/overexpression studies. Additionally, the new data on the inability to upregulate VRAC currents by overexpression of the LRRC8 proteins point to the likelihood that VRAC activity requires additional auxiliary or regulatory proteins, which are yet to be discovered [191, 206]. An alternative approach to addressing the VRAC identity, which was heavily utilized in the past, involves rational genome screening for Cl− channel homologs and targeting such homologs using RNAi with a goal to downregulate endogenous VRAC. We now know that such an approach has been fruitless because the LRRC8 proteins do not have homology with any known Cl− channels, but are rather related to vertebrate pannexins and invertebrate innexins [1, 191].
As many others in the field, my research group was engaged in the quest (admittedly fruitless) for VRAC identity for a number of years. Therefore, with everything being in place, we were able to test if LRRC8A is important for VRAC function in brain cells within 2 weeks of the publication of study by Qiu et al. [164]. Because of the pathological significance of swelling-activated excitatory amino acid release, we started with testing if LRRC8A is essential for this process. We found that primary rat astrocytes express LRRC8A and that knockdown of astrocytic LRRC8A expression with several gene-specific siRNAs inhibits swelling-activated release of preloaded radiotracers d-[3H]aspartate and [14C]taurine by >70 % [92] (see representative data in Fig. 5). Findings with radiotracers were further validated for transport of the endogenous l-glutamate and taurine using HPLC [92]. In addition to LRRC8A, we quantified expression for the other four members of the LRRC8 family and found that in primary astrocytes, their expression levels (relative to LRRC8A) were 1 LRRC8A:~2 LRRC8B:~2 LRRC8C:~2 LRRC8D (and very low expression of LRRC8E) ([92], see Fig. 5a). The absence of the LRRC8E subunit may explain why, in astrocytes, VRAC currents show very modest time-dependent inactivation at highly negative potentials [2, 6] (see representative electrophysiology traces in Fig. 2a). Voss et al. [206] found in erase-and-replace experiments that coexpression of LRRC8A with LRRC8E generates VRAC currents with dramatically “accelerated” inactivation at positive potentials.

LRRC8 proteins are expressed in primary rat astrocytes and form swelling-activated glutamate release pathway. a Results of quantitative RT-PCR experiments quantifying mRNA expression levels for LRRC8A–E. Data are the mean values of LRRC8 expression levels in five astrocyte cultures, as normalized to the housekeeping genes GAPDH, RPL13a, and RPS20. b Swelling-activated glutamate release from primary rat astrocytes measured with the non-metabolizable glutamate analog d-[3H]aspartate. Astrocytes were transfected with either scrambled siRNA (siNC) or a mix of four siRNAs targeting the pore-forming LRRC8A (siLrrc8a_mix). Functional assays of glutamate release were performed 72 h after transfection. ***p < 0.001, swelling-activated release in control cells and cells treated with LRRC8A siRNA. #p < 0.05, release under basal conditions. Modified with permission from M.C. Hyzinski-Garcia et al. [92]
In addition to the conclusion that the LRRC8A-containing protein complexes are essential for swelling-activated (“pathological”) glutamate release in the astrocytes, our study provided potentially critical insights into the mechanisms of “physiological” release of small organic molecules in these cells. As already mentioned in the preceding text, astrocytes release numerous neuroactive substances (gliotransmitters) in response to stimulation with agonists activating GPCRs and stimulating increases in the intracellular Ca2+ levels, all occurring without changes in cellular volume. This latter mode of gliotransmitter release is generally believed to have a predominantly vesicular origin. Our prior work explored in great detail an alternative mechanism for the ATP-stimulated efflux of glutamate and taurine from non-swollen and swollen astrocytes [132, 133, 135, 175]. Although we found strong evidence for a contribution of VRAC, the already-mentioned problems with specificity of VRAC blockers placed these results in doubt. Knockdown of LRRC8A with siRNA completely suppressed the ATP-stimulated release of d-[3H]aspartate and [14C]taurine in non-swollen astrocytes [92]. These findings pave the way towards testing potential contributions of VRAC/LRRC8 channels to astrocyte-neuron communication in complex multicellular systems and in vivo.
Interestingly, the LRRC8A protein and other members of the same family were known long before the discovery of the LRRC8A-VRAC connection. The naturally occurring truncation mutation of the LRRC8A gene found in a human causes congenital agammaglobulinemia and the lack of peripheral B cells [184]. The patient was heterozygous, and both truncated and full-length proteins were detected in lymphocytes. In mice, introduction of the human mutated LRRC8A into hematopoietic cells caused defects of not only B but also T lymphocytes, thus partially recapitulating the human phenotype [184]. The truncated LRRC8A protein apparently fails to reach the plasma membrane, but it does not have dominant negative effects on VRAC currents in overexpression experiments [164, 206]. Therefore, the reasons for disease phenotype will have to be further explored. The recently published LRRC8A-deletion mouse (LRRC8A−/−) showed a much more severe phenotype, but paradoxically, the B lymphocytes were spared [109]. LRRC8A−/− animals have defects in thymus development and loss of T cell expansion and function. Furthermore, they show increased in utero mortality, are born well below the Mendelian ratio, and have a maximal life span of 16 weeks and strong histological and functional abnormalities in skeletal muscle tissue, skin and hair, kidney, and reproductive organs [109]. No gross abnormalities in the central nervous system were reported up until now. Heterozygous animals appeared to be normal [109]. The detailed analysis of thymocyte function revealed that in these cells, LRRC8A is associated with the lymphocyte-specific protein tyrosine kinase (LCK) and that LRRC8A deletion strongly decreases PI3 kinase and Akt signaling. This latter bit of information is very interesting because of an old report about defective RVD and strongly reduced VRAC currents in lck-deficient T cells [115].
After the discovery of LRRC8A, several other homologous proteins, LRRC8B–E, with very similar structure and topology were immediately identified [108]. Based on the preliminary expression work, all LRRC8 proteins, with the exception of LRRC8E, are enriched in the brain [108]. Although the individual functions of LRRC8 members remain poorly understood, LRRC8A and LRRC8D appear to be ubiquitous, while other members of the same family have more restrictive expression profile [1, 108]. LRRC8B was proposed to be related to peripheral stimulation of lymphocytes and monocytes due to its differential expression during this process [108]. This isoform is thought to also be enriched in the CNS, particularly in the forebrain [171]. Several other studies involving gene knockout implicated LRRC8C (FAD158) in adipocyte differentiation and development of obesity and insulin resistance [79, 202]. The just-released publication by Planells-Cases et al. [161] highlighted the intriguing role of LRRC8D in sensitivity and resistance of cancer cells to the chemotherapeutic agent cisplatin and related compounds. According to this latter study, although LRRC8D is dispensable for the formation of the “canonical” VRAC Cl− channel, it represents an essential part of the pore which is responsible for uptake of cisplatin. Downregulation of LRRC8D increased cisplatin resistance without dramatic impact on cell volume control and also modulated relative permeability to taurine [161]. This fascinating work echoes a number of older phenomenological observations indicating that downregulation of VRAC makes cancer cells less sensitive to cisplatin [113, 163]. Perhaps, LRRC8D-containing VRAC represents a distinct subset of LRRC8 heteromers. In addition to the few abovementioned functional studies targeting various LRRC8 members, large-scale expression studies linked several LRRC8s to prevalent CNS disorders. LRRC8A was found to be strongly reduced in mice lacking the serotonin transporter, which have a depression-related phenotype [94]. LRRC8B was found among the most downregulated genes in patients who died from hemorrhagic stroke [168]. For additional analysis of functional association of the LRRC8 genes and insights on the predicted LRRC8 structure, the reader may consult two recent reviews [1, 191]. Clearly, more work is expected to emerge in this area and future studies will help to clarify the role of the LRRC8 family proteins in brain physiology and pathological states.
Through the looking glass: future avenues for the LRRC8/VRAC research
Clearly, studies on the structure and functions of the LRRC8 proteins are in their early infancy. However, one can certainly appreciate the tremendous opportunities the discovery of the LRRC8-VRAC connection has brought to the field. More work is warranted to further validate the hypothesis that LRRC8A and its LRRC8B–E counterparts are the pore-forming subunits of VRAC, to decipher the 3D structure of the VRAC pore, and to uncover the exact stoichiometry of the LRRC8 subunits in heterohexameric VRAC complex(es). Using information on the LRRC8 proteins’ sequences, we can utilize site-directed mutagenesis for gaining knowledge on the mechanisms of cell-volume-dependent and cell-volume-independent VRAC gating. Numerous studies associated several types of protein kinases with activation or modulation of VRAC. By performing simple in silico analyses, similar to that shown in Fig. 4a, we can now identify putative sites for phosphorylation. These will help in designing experiments which will test whether LRRC8A is phosphorylated directly or if auxiliary proteins are involved. Arguably the easiest but most exciting and extensive work that lies in front of us is to determine the precise physiological roles of VRAC on the cellular, tissue, and whole-organism levels. The discovery of VRAC genes equipped us with all the might of the tools provided by molecular biology and molecular genetics. With use of the Crispr/Cas9 approach to gene editing, viral-driven manipulation of gene expression in vivo, and floxed LRRC8A animals for tissue-specific and conditional knockouts, we are likely to see dramatic improvements and revisions to our understanding of VRAC physiology within the next 2 to 3 years. This latter work is extremely important. Among other things, it will allow for discriminating physiological contributions of LRRC8/VRAC from other volume-sensitive Cl− channels in the CNS and elsewhere. For example, the bestrophin-1 Ca2+-activated Cl− channels are thought to contribute to tonic and agonist-stimulated release of glutamate, GABA, and other bioactive molecules from astrocytes [114, 147, 211], and in such a way possibly work in parallel with the LRRC8 channels. The same Best1 channels have also been found to be indispensable for cell volume regulation in retinal pigment epithelial cells [127]. Therefore, Best1 may play the role of a cell-type-specific functional homolog of VRAC. In astrocytes, cellular swelling and chemical ischemia activate not only classical VRAC but also additional, functionally distinct glutamate-permeable maxi anion channels, the molecular identity of which is yet to be found [119]. Some of these anion channels are considered in other contributions included in this special issue of the Pflügers Archiv. Finally, uncovering the mechanisms of VRAC activation will likely bring us closer to the last frontier in the field, to the Holy Grail of cell volume regulation—the molecular nature of cell volume sensor(s). It is a whole new game indeed.
Abbreviations
- CNS:
-
Central nervous system
- DCPIB:
-
4-[(2-Butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid
- GFP:
-
Green fluorescent protein
- GPCR:
-
G-protein-coupled receptor
- KCC:
-
K+,Cl− cotransporter
- LRRC8:
-
Leucine-rich repeat-containing 8
- NKCC:
-
Na+,K+,2Cl- cotransporter
- NOS:
-
Nitric oxide synthase
- NPPB:
-
5-Nitro-2-(3-phenylpropylamino)benzoic acid
- RVD:
-
Regulatory volume decrease
- RVI:
-
Regulatory volume increase
- VRAC:
-
Volume-regulated anion channel
- VSOAC:
-
Volume-sensitive organic osmolyte-anion channel
- VSOR:
-
Volume-sensitive outwardly rectifying Cl− channel
- YFP:
-
Yellow fluorescent protein
- NMDA:
-
N-methyl-D-aspartate
References
Abascal F, Zardoya R (2012) LRRC8 proteins share a common ancestor with pannexins, and may form hexameric channels involved in cell-cell communication. Bioessays 34(7):551–560
Abdullaev IF, Rudkouskaya A, Schools GP, Kimelberg HK, Mongin AA (2006) Pharmacological comparison of swelling-activated excitatory amino acid release and Cl- currents in rat cultured astrocytes. J Physiol 572(Pt. 3):677–689
Adrogue HJ, Madias NE (2000) Hyponatremia. N Engl J Med 342(21):1581–1589
Aitken PG, Borgdorff AJ, Juta AJ, Kiehart DP, Somjen GG, Wadman WJ (1998) Volume changes induced by osmotic stress in freshly isolated rat hippocampal neurons. Pflugers Arch 436(6):991–998
Akita T, Fedorovich SV, Okada Y (2011) Ca2+ nanodomain-mediated component of swelling-induced volume-sensitive outwardly rectifying anion current triggered by autocrine action of ATP in mouse astrocytes. Cell Physiol Biochem 28(6):1181–1190
Akita T, Okada Y (2011) Regulation of bradykinin-induced activation of volume-sensitive outwardly rectifying anion channels by Ca2+ nanodomains in mouse astrocytes. J Physiol 589(Pt 16):3909–3927
Akita T, Okada Y (2014) Characteristics and roles of the volume-sensitive outwardly rectifying (VSOR) anion channel in the central nervous system. Neuroscience 275:211–231
Albrecht J, Norenberg MD (2006) Glutamine: a Trojan horse in ammonia neurotoxicity. Hepatology 44(4):788–794
Allen MC, Newland C, Valverde MA, Hardy SP (1998) Inhibition of ligand-gated cation-selective channels by tamoxifen. Eur J Pharmacol 354(2-3):261–269
Altamirano J, Brodwick MS, Alvarez-Leefmans FJ (1998) Regulatory volume decrease and intracellular Ca2+ in murine neuroblastoma cells studied with fluorescent probes. J Gen Physiol 112(2):145–160
Amzica F, Neckelmann D (1999) Membrane capacitance of cortical neurons and glia during sleep oscillations and spike-wave seizures. J Neurophysiol 82(5):2731–2746
Andrew RD, Labron MW, Boehnke SE, Carnduff L, Kirov SA (2007) Physiological evidence that pyramidal neurons lack functional water channels. Cereb Cortex 17(4):787–802
Andrew RD, Lobinowich ME, Osehobo EP (1997) Evidence against volume regulation by cortical brain cells during acute osmotic stress. Exp Neurol 143(2):300–312
Andrew RD, MacVicar BA (1994) Imaging cell volume changes and neuronal excitation in the hippocampal slice. Neuroscience 62(2):371–383
Araque A, Parpura V, Sanzgiri RP, Haydon PG (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22(5):208–215
Arundine M, Tymianski M (2003) Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 34(4-5):325–337
Banderali U, Roy G (1992) Anion channels for amino-acids in Mdck cells. Am J Physiol 263(6):C1200–C1207
Barron KD, Dentinger MP, Kimelberg HK, Nelson LR, Bourke RS, Keegan S, Mankes R, Cragoe EJ Jr (1988) Ultrastructural features of a brain injury model in cat. I. Vascular and neuroglial changes and the prevention of astroglial swelling by a fluorenyl (aryloxy) alkanoic acid derivative (L-644,711). Acta Neuropathol (Berl) 75(3):295–307
Basarsky TA, Feighan D, MacVicar BA (1999) Glutamate release through volume-activated channels during spreading depression. J Neurosci 19(15):6439–6445
Basavappa S, Chartouni V, Kirk K, Prpic V, Ellory JC, Mangel AW (1995) Swelling-induced chloride currents in neuroblastoma cells are calcium dependent. J Neurosci 15(5 Pt 1):3662–3666
Benfenati V, Caprini M, Nicchia GP, Rossi A, Dovizio M, Cervetto C, Nobile M, Ferroni S (2009) Carbenoxolone inhibits volume-regulated anion conductance in cultured rat cortical astroglia. Channels (Austin) 3(5):323–336
Benveniste H, Drejer J, Schousboe A, Diemer NH (1984) Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem 43(5):1369–1374
Blei AT, Larsen FS (1999) Pathophysiology of cerebral edema in fulminant hepatic failure. J Hepatol 31(4):771–776
Blum AE, Walsh BC, Dubyak GR (2010) Extracellular osmolarity modulates G protein-coupled receptor dependent ATP release from 1321N1 astrocytoma cells. Am J Physiol Cell Physiol 298:C386–C396
Bortner CD, Cidlowski JA (1998) A necessary role for cell shrinkage in apoptosis. Biochem Pharmacol 56(12):1549–1559
Bosman DK, Deutz NE, Maas MA, van Eijk HM, Smit JJ, de Haan JG, Chamuleau RA (1992) Amino acid release from cerebral cortex in experimental acute liver failure, studied by in vivo cerebral cortex microdialysis. J Neurochem 59(2):591–599
Boulos AS, Deshaies EM, Dalfino JC, Feustel PJ, Popp AJ, Drazin D (2011) Tamoxifen as an effective neuroprotectant in an endovascular canine model of stroke. J Neurosurg 114(4):1117–1126
Bourque CW (2008) Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci 9(7):519–531
Bourque CW, Oliet SH (1997) Osmoreceptors in the central nervous system. Annu Rev Physiol 59:601–619
Bowens NH, Dohare P, Kuo YH, Mongin AA (2013) DCPIB, the proposed selective blocker of volume-regulated anion channels, inhibits several glutamate transport pathways in glial cells. Mol Pharmacol 83(1):22–32
Bres V, Hurbin A, Duvoid A, Orcel H, Moos FC, Rabie A, Hussy N (2000) Pharmacological characterization of volume-sensitive, taurine permeable anion channels in rat supraoptic glial cells. Br J Pharmacol 130(8):1976–1982
Brillault J, Lam TI, Rutkowsky JM, Foroutan S, O'Donnell ME (2008) Hypoxia effects on cell volume and ion uptake of cerebral microvascular endothelial cells. Am J Physiol Cell Physiol 294(1):C88–C96
Brusilow SW, Koehler RC, Traystman RJ, Cooper AJ (2010) Astrocyte glutamine synthetase: importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics 7(4):452–470
Cahalan MD, Lewis RS (1988) Role of potassium and chloride channels in volume regulation by T lymphocytes. Soc Gen Physiol Ser 43:281–301
Cheema TA, Pettigrew VA, Fisher SK (2007) Receptor regulation of the volume-sensitive efflux of taurine and iodide from human SH-SY5Y neuroblastoma cells: differential requirements for Ca2+ and protein kinase C. J Pharmacol Exp Ther 320(3):1068–1077
Chen H, Luo J, Kintner DB, Shull GE, Sun D (2005) Na(+)-dependent chloride transporter (NKCC1)-null mice exhibit less gray and white matter damage after focal cerebral ischemia. J Cereb Blood Flow Metab 25(1):54–66
Choi DW (1988) Glutamate neurotoxicity and diseases of the nervous system. Neuron 1(8):623–634
Choi DW (1992) Excitotoxic cell death. J Neurobiol 23(9):1261–1276
Crepel V, Panenka W, Kelly ME, MacVicar BA (1998) Mitogen-activated protein and tyrosine kinases in the activation of astrocyte volume-activated chloride current. J Neurosci 18(4):1196–1206
Custodio JB, Dinis TC, Almeida LM, Madeira VM (1994) Tamoxifen and hydroxytamoxifen as intramembraneous inhibitors of lipid peroxidation. Evidence for peroxyl radical scavenging activity. Biochem Pharmacol 47(11):1989–1998
Dadsetan S, Kukolj E, Bak LK, Sorensen M, Ott P, Vilstrup H, Schousboe A, Keiding S, Waagepetersen HS (2013) Brain alanine formation as an ammonia-scavenging pathway during hyperammonemia: effects of glutamine synthetase inhibition in rats and astrocyte-neuron co-cultures. J Cereb Blood Flow Metab 33(8):1235–1241
Darby M, Kuzmiski JB, Panenka W, Feighan D, MacVicar BA (2003) ATP released from astrocytes during swelling activates chloride channels. J Neurophysiol 89(4):1870–1877
Decher N, Lang HJ, Nilius B, Bruggemann A, Busch AE, Steinmeyer K (2001) DCPIB is a novel selective blocker of I(Cl, swell) and prevents swelling-induced shortening of guinea-pig atrial action potential duration. Br J Pharmacol 134(7):1467–1479
Deleuze C, Duvoid A, Hussy N (1998) Properties and glial origin of osmotic-dependent release of taurine from the rat supraoptic nucleus. J Physiol 507(Pt 2):463–471
Dick GM, Hunter AC, Sanders KM (2002) Ethylbromide tamoxifen, a membrane-impermeant antiestrogen, activates smooth muscle calcium-activated large-conductance potassium channels from the extracellular side. Mol Pharmacol 61(5):1105–1113
Dick GM, Rossow CF, Smirnov S, Horowitz B, Sanders KM (2001) Tamoxifen activates smooth muscle BK channels through the regulatory beta 1 subunit. J Biol Chem 276(37):34594–34599
Dirnagl U, Iadecola C, Moskowitz MA (1999) Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 22(9):391–397
Dohare P, Harrigan TJ, Abdullaev IF, Mongin AA (2011) Volume-regulated anion channels mediate glutamate release from migrating microglial cells. Program # PSM03-06 St. Louis, MO, American Society for Neurochemistry. Trans Am Soc Neurochem: CD-ROM
Dreier JP (2011) The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 17(4):439–447
Dreier JP, Woitzik J, Fabricius M, Bhatia R, Major S, Drenckhahn C, Lehmann TN, Sarrafzadeh A, Willumsen L, Hartings JA, Sakowitz OW, Seemann JH, Thieme A, Lauritzen M, Strong AJ (2006) Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain 129(Pt 12):3224–3237
Droogmans G, Maertens C, Prenen J, Nilius B (1999) Sulphonic acid derivatives as probes of pore properties of volume-regulated anion channels in endothelial cells. Br J Pharmacol 128(1):35–40
Droogmans G, Prenen J, Eggermont J, Voets T, Nilius B (1998) Voltage-dependent block of endothelial volume-regulated anion channels by calix[4]arenes. Am J Physiol 275(3 Pt 1):C646–C652
Ducharme G, Newell EW, Pinto C, Schlichter LC (2007) Small-conductance Cl(-) channels contribute to volume regulation and phagocytosis in microglia. Eur J Neurosci 26:2119–2130
Eder C, Klee R, Heinemann U (1998) Involvement of stretch-activated Cl- channels in ramification of murine microglia. J Neurosci 18(18):7127–7137
Ellis RJ (2001) Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci 26(10):597–604
Fabene PF, Weiczner R, Marzola P, Nicolato E, Calderan L, Andrioli A, Farkas E, Sule Z, Mihaly A, Sbarbati A (2006) Structural and functional MRI following 4-aminopyridine-induced seizures: a comparative imaging and anatomical study. Neurobiol Dis 21(1):80–89
Fan HT, Morishima S, Kida H, Okada Y (2001) Phloretin differentially inhibits volume-sensitive and cyclic AMP-activated, but not Ca-activated, Cl(-) channels. Br J Pharmacol 133(7):1096–1106
Felipo V (2013) Hepatic encephalopathy: effects of liver failure on brain function. Nat Rev Neurosci 14(12):851–858
Felipo V, Butterworth RF (2002) Neurobiology of ammonia. Prog Neurobiol 67(4):259–279
Feustel PJ, Jin Y, Kimelberg HK (2004) Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke 35(5):1164–1168
Fiacco TA, Agulhon C, McCarthy KD (2009) Sorting out astrocyte physiology from pharmacology. Annu Rev Pharmacol Toxicol 49:151–174
Fields RD (2011) Signaling by neuronal swelling. Sci Signal 4(155):tr1
Fields RD, Ni Y (2010) Nonsynaptic communication through ATP release from volume-activated anion channels in axons. Sci Signal 3(142):ra73
Fisher SK, Cheema TA, Foster DJ, Heacock AM (2008) Volume-dependent osmolyte efflux from neural tissues: regulation by G-protein-coupled receptors. J Neurochem 106(5):1998–2014
Fisher SK, Heacock AM, Keep RF, Foster DJ (2010) Receptor regulation of osmolyte homeostasis in neural cells. J Physiol 588(18):3355–3364
Franco R, Panayiotidis MI, de La Paz LD (2008) Autocrine signaling involved in cell volume regulation: the role of released transmitters and plasma membrane receptors. J Cell Physiol 216(1):14–28
Fraser CL, Arieff AI (1997) Epidemiology, pathophysiology, and management of hyponatremic encephalopathy. Am J Med 102(1):67–77
Furtner T, Zierler S, Kerschbaum HH (2007) Blockade of chloride channels suppresses engulfment of microspheres in the microglial cell line, BV-2. Brain Res 1184:1–9
Gorg B, Morwinsky A, Keitel V, Qvartskhava N, Schror K, Haussinger D (2010) Ammonia triggers exocytotic release of L-glutamate from cultured rat astrocytes. Glia 58(6):691–705
Grinstein S, Clarke CA, Dupre A, Rothstein A (1982) Volume-induced increase of anion permeability in human lymphocytes. J Gen Physiol 80(6):801–823
Haj-Yasein NN, Jensen V, Ostby I, Omholt SW, Voipio J, Kaila K, Ottersen OP, Hvalby O, Nagelhus EA (2012) Aquaporin-4 regulates extracellular space volume dynamics during high-frequency synaptic stimulation: a gene deletion study in mouse hippocampus. Glia 60(6):867–874
Hamilton NB, Attwell D (2010) Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci 11(4):227–238
Hardy SP, deFelipe C, Valverde MA (1998) Inhibition of voltage-gated cationic channels in rat embryonic hypothalamic neurones and C1300 neuroblastoma cells by triphenylethylene antioestrogens. FEBS Lett 434(3):236–240
Harrigan TJ, Abdullaev IF, Jourd’heuil D, Mongin AA (2008) Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: the role of NADPH oxidases. J Neurochem 106(6):2449–2462
Haskew-Layton RE, Rudkouskaya A, Jin Y, Feustel PJ, Kimelberg HK, Mongin AA (2008) Two distinct modes of hypoosmotic medium-induced release of excitatory amino acids and taurine in the rat brain in vivo. PLoS ONE 3(10):e3543
Haussinger D, Kircheis G, Fischer R, Schliess F, Vom DS (2000) Hepatic encephalopathy in chronic liver disease: a clinical manifestation of astrocyte swelling and low-grade cerebral edema? J Hepatol 32(6):1035–1038
Hawkins BT, Davis TP (2005) The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev 57(2):173–185
Hawkins EG, Dewey WL, Anitha M, Srinivasan S, Grider JR, Akbarali HI (2013) Electrophysiological characteristics of enteric neurons isolated from the immortomouse. Dig Dis Sci 58(6):1516–1527
Hayashi T, Nozaki Y, Nishizuka M, Ikawa M, Osada S, Imagawa M (2011) Factor for adipocyte differentiation 158 gene disruption prevents the body weight gain and insulin resistance induced by a high-fat diet. Biol Pharm Bull 34(8):1257–1263
Haydon PG, Carmignoto G (2006) Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev 86(3):1009–1031
Hazama A, Okada Y (1988) Ca2+ sensitivity of volume-regulatory K+ and Cl- channels in cultured human epithelial cells. J Physiol 402:687–702
He J, Kargacin ME, Kargacin GJ, Ward CA (2003) Tamoxifen inhibits Na+ and K+ currents in rat ventricular myocytes. Am J Physiol Heart Circ Physiol 285(2):H661–H668
Hermenegildo C, Marcaida G, Montoliu C, Grisolia S, Minana MD, Felipo V (1996) NMDA receptor antagonists prevent acute ammonia toxicity in mice. Neurochem Res 21(10):1237–1244
Hines DJ, Hines RM, Mulligan SJ, MacVicar BA (2009) Microglia processes block the spread of damage in the brain and require functional chloride channels. Glia 57(15):1610–1618
Hisadome K, Koyama T, Kimura C, Droogmans G, Ito Y, Oike M (2002) Volume-regulated anion channels serve as an auto/paracrine nucleotide release pathway in aortic endothelial cells. J Gen Physiol 119(6):511–520
Hoffmann EK (1978) Regulation of cell volume by selective changes in the leak permeabilities of Ehrlich ascites tumor cells. Alfred Benzon Symp XI:397–417
Hoffmann EK, Lambert IH, Pedersen SF (2009) Physiology of cell volume regulation in vertebrates. Physiol Rev 89(1):193–277
Hoffmann EK, Simonsen LO, Lambert IH (1984) Volume-induced increase of K+ and Cl- permeabilities in Ehrlich ascites tumor cells. Role of internal Ca2+. J Membr Biol 78(3):211–222
Holthoff K, Witte OW (1996) Intrinsic optical signals in rat neocortical slices measured with near-infrared dark-field microscopy reveal changes in extracellular space. J Neurosci 16(8):2740–2749
Hussy N, Deleuze C, Desarmenien MG, Moos FC (2000) Osmotic regulation of neuronal activity: a new role for taurine and glial cells in a hypothalamic neuroendocrine structure. Prog Neurobiol 62(2):113–134
Hussy N, Deleuze C, Pantaloni A, Desarmenien MG, Moos F (1997) Agonist action of taurine on glycine receptors in rat supraoptic magnocellular neurones: possible role in osmoregulation. J Physiol 502(Pt 3):609–621
Hyzinski-Garcia MC, Rudkouskaya A, Mongin AA (2014) LRRC8A protein is indispensable for swelling-activated and ATP-induced release of excitatory amino acids in rat astrocytes. J Physiol 592(22):4855–4862
Hyzinski-Garcia MC, Vincent MY, Haskew-Layton RE, Dohare P, Keller RW Jr, Mongin AA (2011) Hypoosmotic swelling modifies glutamate-glutamine cycle in the cerebral cortex and in astrocyte cultures. J Neurochem 118(1):140–152
Ichikawa M, Okamura-Oho Y, Shimokawa K, Kondo S, Nakamura S, Yokota H, Himeno R, Lesch KP, Hayashizaki Y (2008) Expression analysis for inverted effects of serotonin transporter inactivation. Biochem Biophys Res Commun 368(1):43–49
Inoue H, Mori S, Morishima S, Okada Y (2005) Volume-sensitive chloride channels in mouse cortical neurons: characterization and role in volume regulation. Eur J Neurosci 21(6):1648–1658
Inoue H, Okada Y (2007) Roles of volume-sensitive chloride channel in excitotoxic neuronal injury. J Neurosci 27(6):1445–1455
Jackson PS, Morrison R, Strange K (1994) The volume-sensitive organic osmolyte-anion channel VSOAC is regulated by nonhydrolytic ATP binding. Am J Physiol 267(5 Pt 1):C1203–C1209
Jackson PS, Strange K (1993) Volume-sensitive anion channels mediate swelling-activated inositol and taurine efflux. Am J Physiol 265(6 Pt 1):C1489–C1500
Jayakumar AR, Panickar KS, Curtis KM, Tong XY, Moriyama M, Norenberg MD (2011) Na-K-Cl cotransporter-1 in the mechanism of cell swelling in cultured astrocytes after fluid percussion injury. J Neurochem 117(3):437–448
Jayakumar AR, Valdes V, Norenberg MD (2011) The Na-K-Cl cotransporter in the brain edema of acute liver failure. J Hepatol 54(2):272–278
Kahle KT, Khanna AR, Alper SL, Adragna NC, Lauf PK, Sun D, Delpire E (2015) K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol Med 21(8):513–523
Kato M, Hughes RD, Keays RT, Williams R (1992) Electron microscopic study of brain capillaries in cerebral edema from fulminant hepatic failure. Hepatology 15(6):1060–1066
Kimelberg HK (1995) Current concepts of brain edema. Review of laboratory investigations. J Neurosurg 83(6):1051–1059
Kimelberg HK (2005) Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia 50(4):389–397
Kimelberg HK, Feustel PJ, Jin Y, Paquette J, Boulos A, Keller RW Jr, Tranmer BI (2000) Acute treatment with tamoxifen reduces ischemic damage following middle cerebral artery occlusion. Neuroreport 11(12):2675–2679
Kimelberg HK, Goderie SK, Higman S, Pang S, Waniewski RA (1990) Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J Neurosci 10(5):1583–1591
Kimelberg HK, Jin Y, Charniga C, Feustel PJ (2003) Neuroprotective activity of tamoxifen in permanent focal ischemia. J Neurosurg 99(1):138–142
Kubota K, Kim JY, Sawada A, Tokimasa S, Fujisaki H, Matsuda-Hashii Y, Ozono K, Hara J (2004) LRRC8 involved in B cell development belongs to a novel family of leucine-rich repeat proteins. FEBS Lett 564(1-2):147–152
Kumar L, Chou J, Yee CS, Borzutzky A, Vollmann EH, von Andrian UH, Park SY, Hollander G, Manis JP, Poliani PL, Geha RS (2014) Leucine-rich repeat containing 8A (LRRC8A) is essential for T lymphocyte development and function. J Exp Med 211(5):929–942
Lang F, Busch GL, Ritter M, Volkl H, Waldegger S, Gulbins E, Haussinger D (1998) Functional significance of cell volume regulatory mechanisms. Physiol Rev 78(1):247–306
Lauf PK, Bauer J, Adragna NC, Fujise H, Zade-Oppen AM, Ryu KH, Delpire E (1992) Erythrocyte K-Cl cotransport: properties and regulation. Am J Physiol 263(5 Pt 1):C917–C932
Leaney JL, Marsh SJ, Brown DA (1997) A swelling-activated chloride current in rat sympathetic neurones. J Physiol-London 501(3):555–564
Lee EL, Shimizu T, Ise T, Numata T, Kohno K, Okada Y (2007) Impaired activity of volume-sensitive Cl(-) channel is involved in cisplatin resistance of cancer cells. J Cell Physiol 211(2):513–521
Lee S, Yoon BE, Berglund K, Oh SJ, Park H, Shin HS, Augustine GJ, Lee CJ (2010) Channel-mediated tonic GABA release from glia. Science 330(6005):790–796
Lepple-Wienhues A, Szabo I, Laun T, Kaba NK, Gulbins E, Lang F (1998) The tyrosine kinase p56lck mediates activation of swelling-induced chloride channels in lymphocytes. J Cell Biol 141(1):281–286
Lippmann BJ, Yang R, Barnett DW, Misler S (1995) Pharmacology of volume regulation following hypotonicity-induced cell swelling in clonal N1E115 neuroblastoma cells. Brain Res 686(1):29–36
Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79(4):1431–1568
Liu HT, Akita T, Shimizu T, Sabirov RZ, Okada Y (2009) Bradykinin-induced astrocyte-neuron signalling: glutamate release is mediated by ROS-activated volume-sensitive outwardly rectifying anion channels. J Physiol 587(Pt 10):2197–2209
Liu HT, Tashmukhamedov BA, Inoue H, Okada Y, Sabirov RZ (2006) Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Glia 54(5):343–357
Lo EH, Dalkara T, Moskowitz MA (2003) Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 4(5):399–415
Luckl J, Dreier JP, Szabados T, Wiesenthal D, Bari F, Greenberg JH (2012) Peri-infarct flow transients predict outcome in rat focal brain ischemia. Neuroscience 226:197–207
MacVicar BA, Feighan D, Brown A, Ransom B (2002) Intrinsic optical signals in the rat optic nerve: role for K(+) uptake via NKCC1 and swelling of astrocytes. Glia 37(2):114–123
Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS (2000) Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 6(2):159–163
Manolopoulos VG, Voets T, Declercq PE, Droogmans G, Nilius B (1997) Swelling-activated efflux of taurine and other organic osmolytes in endothelial cells. Am J Physiol 273(1 Pt 1):C214–C222
Martin DL, Madelian V, Seligmann B, Shain W (1990) The role of osmotic pressure and membrane potential in K(+)-stimulated taurine release from cultured astrocytes and LRM55 cells. J Neurosci 10(2):571–577
Medina I, Friedel P, Rivera C, Kahle KT, Kourdougli N, Uvarov P, Pellegrino C (2014) Current view on the functional regulation of the neuronal K(+)-Cl(-) cotransporter KCC2. Front Cell Neurosci 8:27
Milenkovic A, Brandl C, Milenkovic VM, Jendryke T, Sirianant L, Wanitchakool P, Zimmermann S, Reiff CM, Horling F, Schrewe H, Schreiber R, Kunzelmann K, Wetzel CH, Weber BH (2015) Bestrophin 1 is indispensable for volume regulation in human retinal pigment epithelium cells. Proc Natl Acad Sci U S A 112(20):E2630–E2639
Minieri L, Pivonkova H, Caprini M, Harantova L, Anderova M, Ferroni S (2013) The inhibitor of volume-regulated anion channels DCPIB activates TREK potassium channels in cultured astrocytes. Br J Pharmacol 168(5):1240–1254
Mongin AA (2007) Disruption of ionic and cell volume homeostasis in cerebral ischemia: the perfect storm. Pathophysiology 14(3-4):183–193
Mongin AA, Aksentsev SL, Orlov SN, Kvacheva ZB, Mezen NI, Fedulov AS, Konev SV (1996) Swelling-induced activation of Na+,K+,2Cl− cotransport in C6 glioma cells: kinetic properties and intracellular signalling mechanisms. Biochim Biophys Acta 1285(2):229–236
Mongin AA, Aksentsev SL, Orlov SN, Slepko NG, Kozlova MV, Maximov GV, Konev SV (1994) Swelling-induced K+ influx in cultured primary astrocytes. Brain Res 655(1-2):110–114
Mongin AA, Kimelberg HK (2002) ATP potently modulates anion channel-mediated excitatory amino acid release from cultured astrocytes. Am J Physiol Cell Physiol 283(2):C569–C578
Mongin AA, Kimelberg HK (2003) Is autocrine ATP release required for activation of volume-sensitive chloride channels? J Neurophysiol 90(4):2791–2792
Mongin AA, Kimelberg HK (2005) Astrocytic swelling in neuropathology. In: Kettenmann H, Ransom BR (eds) Neuroglia, 2nd edn. Oxford University Press, Oxford/New York
Mongin AA, Kimelberg HK (2005) ATP regulates anion channel-mediated organic osmolyte release from cultured rat astrocytes via multiple Ca2+-sensitive mechanisms. Am J Physiol Cell Physiol 288(1):C204–C213
Mongin AA, Orlov SN (2001) Mechanisms of cell volume regulation and possible nature of the cell volume sensor. Pathophysiology 8(2):77–88
Montana V, Verkhratsky A, Parpura V (2014) Pathological role for exocytotic glutamate release from astrocytes in hepatic encephalopathy. Curr Neuropharmacol 12(4):324–333
Moran J, Morales-Mulia S, Hernandez-Cruz A, Pasantes-Morales H (1997) Regulatory volume decrease and associated osmolyte fluxes in cerebellar granule neurons are calcium independent. J Neurosci Res 47(2):144–154
Nakajima K, Kohsaka S (2001) Microglia: activation and their significance in the central nervous system. J Biochem (Tokyo) 130(2):169–175
Nilius B (2004) Is the volume-regulated anion channel VRAC a “water-permeable” channel? Neurochem Res 29(1):3–8
Nilius B, Droogmans G (2003) Amazing chloride channels: an overview. Acta Physiol Scand 177(2):119–147
Nilius B, Eggermont J, Voets T, Buyse G, Manolopoulos V, Droogmans G (1997) Properties of volume-regulated anion channels in mammalian cells. Prog Biophys Mol Biol 68(1):69–119
Norenberg MD (1977) A light and electron microscopic study of experimental portal-systemic (ammonia) encephalopathy. Progression and reversal of the disorder. Lab Invest 36(6):618–627
Norenberg MD, Rao KV, Jayakumar AR (2005) Mechanisms of ammonia-induced astrocyte swelling. Metab Brain Dis 20(4):303–318
O’Donnell ME (2014) Blood-brain barrier Na transporters in ischemic stroke. Adv Pharmacol 71:113–146
O’Donnell ME, Tran L, Lam TI, Liu XB, Anderson SE (2004) Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab 24(9):1046–1056
Oh SJ, Han KS, Park H, Woo DH, Kim HY, Traynelis SF, Lee CJ (2012) Protease activated receptor 1-induced glutamate release in cultured astrocytes is mediated by bestrophin-1 channel but not by vesicular exocytosis. Mol Brain 5:38
Okada Y (1997) Volume expansion-sensing outward-rectifier Cl- channel: fresh start to the molecular identity and volume sensor. Am J Physiol Cell Physiol 273(3 Pt 1):C755–C789
Okada Y, Maeno E, Shimizu T, Dezaki K, Wang J, Morishima S (2001) Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD). J Physiol 532(Pt 1):3–16
Olson JE, Li GZ (1997) Increased potassium, chloride, and taurine conductances in astrocytes during hypoosmotic swelling. Glia 20(3):254–261
Osuka K, Feustel PJ, Mongin AA, Tranmer BI, Kimelberg HK (2001) Tamoxifen inhibits nitrotyrosine formation after reversible middle cerebral artery occlusion in the rat. J Neurochem 76(6):1842–1850
Parpura V, Heneka MT, Montana V, Oliet SH, Schousboe A, Haydon PG, Stout RF Jr, Spray DC, Reichenbach A, Pannicke T, Pekny M, Pekna M, Zorec R, Verkhratsky A (2012) Glial cells in (patho)physiology. J Neurochem 121(1):4–27
Pasantes-Morales H, Moran J, Schousboe A (1990) Volume-sensitive release of taurine from cultured astrocytes: properties and mechanism. Glia 3(5):427–432
Pasantes-Morales H, Schousboe A (1989) Release of taurine from astrocytes during potassium-evoked swelling. Glia 2(1):45–50
Patel AJ, Lauritzen I, Lazdunski M, Honore E (1998) Disruption of mitochondrial respiration inhibits volume-regulated anion channels and provokes neuronal cell swelling. J Neurosci 18(9):3117–3123
Pedersen SF, Kapus A, Hoffmann EK (2011) Osmosensory mechanisms in cellular and systemic volume regulation. J Am Soc Nephrol 22(9):1587–1597
Pedersen SF, Klausen TK, Nilius B (2015) The identification of a volume-regulated anion channel: an amazing Odyssey. Acta Physiol (Oxf) 213(4):868–881
Phillis JW, O’Regan MH (1996) Mechanisms of glutamate and aspartate release in the ischemic rat cerebral cortex. Brain Res 730(1-2):150–164
Phillis JW, Song D, O’Regan MH (1997) Inhibition by anion channel blockers of ischemia-evoked release of excitotoxic and other amino acids from rat cerebral cortex. Brain Res 758(1-2):9–16
Phillis JW, Song D, O’Regan MH (1998) Tamoxifen, a chloride channel blocker, reduces glutamate and aspartate release from the ischemic cerebral cortex. Brain Res 780(2):352–355
Planells-Cases R, Lutter D, Guyader C et al. (2015) Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J. doi:10.15252/embj.201592409
Podesta MA, Faravelli I, Cucchiari D, Reggiani F, Oldani S, Fedeli C, Graziani G (2015) Neurological counterparts of hyponatremia: pathological mechanisms and clinical manifestations. Curr Neurol Neurosci Rep 15(4):536
Poulsen KA, Andersen EC, Hansen CF, Klausen TK, Hougaard C, Lambert IH, Hoffmann EK (2010) Deregulation of apoptotic volume decrease and ionic movements in multidrug-resistant tumor cells: role of chloride channels. Am J Physiol Cell Physiol 298(1):C14–C25
Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP, Patapoutian A (2014) SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157(2):447–458
Rappert A, Biber K, Nolte C, Lipp M, Schubel A, Lu B, Gerard NP, Gerard C, Boddeke HW, Kettenmann H (2002) Secondary lymphoid tissue chemokine (CCL21) activates CXCR3 to trigger a Cl- current and chemotaxis in murine microglia. J Immunol 168(7):3221–3226
Rasmusson RL, Davis DG, Lieberman M (1993) Amino acid loss during volume regulatory decrease in cultured chick heart cells. Am J Physiol 264(1 Pt 1):C136–C145
Risher WC, Andrew RD, Kirov SA (2009) Real-time passive volume responses of astrocytes to acute osmotic and ischemic stress in cortical slices and in vivo revealed by two-photon microscopy. Glia 57(2):207–221
Rosell A, Vilalta A, Garcia-Berrocoso T, Fernandez-Cadenas I, Domingues-Montanari S, Cuadrado E, Delgado P, Ribo M, Martinez-Saez E, Ortega-Aznar A, Montaner J (2011) Brain perihematoma genomic profile following spontaneous human intracerebral hemorrhage. PLoS ONE 6(2):e16750
Rossi DJ, Oshima T, Attwell D (2000) Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403(6767):316–321
Rosso L, Peteri-Brunback B, Poujeol P, Hussy N, Mienville JM (2004) Vasopressin-induced taurine efflux from rat pituicytes: a potential negative feedback for hormone secretion. J Physiol 554(Pt 3):731–742
Roth RB, Hevezi P, Lee J, Willhite D, Lechner SM, Foster AC, Zlotnik A (2006) Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics 7(2):67–80
Rothman SM (1985) The neurotoxicity of excitatory amino acids is produced by passive chloride influx. J Neurosci 5(6):1483–1489
Roy G, Malo C (1992) Activation of amino acid diffusion by a volume increase in cultured kidney (MDCK) cells. J Membr Biol 130(1):83–90
Roy G, Sauve R (1987) Effect of anisotonic media on volume, ion and amino-acid content and membrane potential of kidney cells (MDCK) in culture. J Membr Biol 100(1):83–96
Rudkouskaya A, Chernoguz A, Haskew-Layton RE, Mongin AA (2008) Two conventional protein kinase C isoforms, alpha and betaI, are involved in the ATP-induced activation of volume-regulated anion channel and glutamate release in cultured astrocytes. J Neurochem 105(6):2260–2270
Russell JM (2000) Sodium-potassium-chloride cotransport. Physiol Rev 80(1):211–276
Rutledge EM, Kimelberg HK (1996) Release of [3H]-D-aspartate from primary astrocyte cultures in response to raised external potassium. J Neurosci 16(24):7803–7811
Sabirov RZ, Kurbannazarova RS, Melanova NR, Okada Y (2013) Volume-sensitive anion channels mediate osmosensitive glutathione release from rat thymocytes. PLoS ONE 8(1):e55646
Sahagian BM (1965) Active glucose uptake by strips of guinea pig intestine; competitive inhibition by phlorhizin and phloretin. Can J Biochem 43(7):851–858
Sanchez-Olea R, Pena C, Moran J, Pasantes-Morales H (1993) Inhibition of volume regulation and efflux of osmoregulatory amino acids by blockers of Cl- transport in cultured astrocytes. Neurosci Lett 156(1-2):141–144
Sarkadi B, Mack E, Rothstein A (1984) Ionic events during the volume response of human peripheral blood lymphocytes to hypotonic media. I. Distinctions between volume-activated Cl- and K+ conductance pathways. J Gen Physiol 83(4):497–512
Sarkadi B, Mack E, Rothstein A (1984) Ionic events during the volume response of human peripheral blood lymphocytes to hypotonic media. II. Volume- and time-dependent activation and inactivation of ion transport pathways. J Gen Physiol 83(4):513–527
Sato K, Numata T, Saito T, Ueta Y, Okada Y (2011) V(2) receptor-mediated autocrine role of somatodendritic release of AVP in rat vasopressin neurons under hypo-osmotic conditions. Sci Signal 4(157):ra5
Sawada A, Takihara Y, Kim JY, Matsuda-Hashii Y, Tokimasa S, Fujisaki H, Kubota K, Endo H, Onodera T, Ohta H, Ozono K, Hara J (2003) A congenital mutation of the novel gene LRRC8 causes agammaglobulinemia in humans. J Clin Invest 112(11):1707–1713
Schlichter LC, Mertens T, Liu B (2011) Swelling activated Cl- channels in microglia: biophysics, pharmacology and role in glutamate release. Channels (Austin) 5(2):128–137
Schlichter LC, Sakellaropoulos G, Ballyk B, Pennefather PS, Phipps DJ (1996) Properties of K+ and Cl- channels and their involvement in proliferation of rat microglial cells. Glia 17(3):225–236
Schober AL, Mongin AA (2015) Intracellular levels of glutamate in swollen astrocytes are preserved via neurotransmitter reuptake and de novo synthesis: implications for hyponatremia. J Neurochem 135(1):176–185
Schousboe A, Moran J, Pasantes-Morales H (1990) Potassium-stimulated release of taurine from cultured cerebellar granule neurons is associated with cell swelling. J Neurosci Res 27(1):71–77
Schousboe A, Sanchez OR, Moran J, Pasantes-Morales H (1991) Hyposmolarity-induced taurine release in cerebellar granule cells is associated with diffusion and not with high-affinity transport. J Neurosci Res 30(4):661–665
Seki Y, Feustel PJ, Keller RW Jr, Tranmer BI, Kimelberg HK (1999) Inhibition of ischemia-induced glutamate release in rat striatum by dihydrokinate and an anion channel blocker. Stroke 30(2):433–440
Stauber T (2015) The volume-regulated anion channel is formed by LRRC8 heteromers—molecular identification and roles in membrane transport and physiology. Biol Chem 396(9-10):975–990
Steffensen AB, Sword J, Croom D, Kirov SA, MacAulay N (2015) Chloride cotransporters as a molecular mechanism underlying spreading depolarization-induced dendritic beading. J Neurosci 35(35):12172–12187
Strange K, Emma F, Jackson PS (1996) Cellular and molecular physiology of volume-sensitive anion channels. Am J Physiol 270(3 Pt 1):C711–C730
Stutzin A, Hoffmann EK (2006) Swelling-activated ion channels: functional regulation in cell-swelling, proliferation and apoptosis. Acta Physiol (Oxf) 187(1-2):27–42
Su G, Kintner DB, Flagella M, Shull GE, Sun D (2002) Astrocytes from Na(+)-K(+)-Cl(-) cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am J Physiol Cell Physiol 282(5):C1147–C1160
Su G, Kintner DB, Sun D (2002) Contribution of Na(+)-K(+)-Cl(-) cotransporter to high-[K(+)](o)-induced swelling and EAA release in astrocytes. Am J Physiol Cell Physiol 282(5):C1136–C1146
Sykova E (2004) Extrasynaptic volume transmission and diffusion parameters of the extracellular space. Neuroscience 129(4):861–876
Szatkowski M, Barbour B, Attwell D (1990) Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature 348(6300):443–446
Takano T, Kang J, Jaiswal JK, Simon SM, Lin JH, Yu Y, Li Y, Yang J, Dienel G, Zielke HR, Nedergaard M (2005) Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proc Natl Acad Sci U S A 102(45):16466–16471
Ternovsky VI, Okada Y, Sabirov RZ (2004) Sizing the pore of the volume-sensitive anion channel by differential polymer partitioning. FEBS Lett 576(3):433–436
Tofteng F, Hauerberg J, Hansen BA, Pedersen CB, Jorgensen L, Larsen FS (2006) Persistent arterial hyperammonemia increases the concentration of glutamine and alanine in the brain and correlates with intracranial pressure in patients with fulminant hepatic failure. J Cereb Blood Flow Metab 26(1):21–27
Tominaga K, Kondo C, Kagata T, Hishida T, Nishizuka M, Imagawa M (2004) The novel gene fad158, having a transmembrane domain and leucine-rich repeat, stimulates adipocyte differentiation. J Biol Chem 279(33):34840–34848
Uckermann O, Vargova L, Ulbricht E, Klaus C, Weick M, Rillich K, Wiedemann P, Reichenbach A, Sykova E, Bringmann A (2004) Glutamate-evoked alterations of glial and neuronal cell morphology in the guinea pig retina. J Neurosci 24(45):10149–10158
Upadhyay A, Jaber BL, Madias NE (2006) Incidence and prevalence of hyponatremia. Am J Med 119(7 Suppl 1):S30–S35
Verkman AS, Galietta LJ (2009) Chloride channels as drug targets. Nat Rev Drug Discov 8(2):153–171
Voss FK, Ullrich F, Munch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T, Jentsch TJ (2014) Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344(6184):634–638
Wang Y, Roman R, Lidofsky SD, Fitz JG (1996) Autocrine signaling through ATP release represents a novel mechanism for cell volume regulation. Proc Natl Acad Sci U S A 93(21):12020–12025
Wasterlain CG, Torack RM (1968) Cerebral edema in water intoxication. II. An ultrastructural study. Arch Neurol 19(1):79–87
Wehner F, Olsen H, Tinel H, Kinne-Saffran E, Kinne RKH (2003) Cell volume regulation: osmolytes, osmolyte transport, and signal transduction. Rev Physiol Biochem Pharmacol 148:1–80
Woitzik J, Hecht N, Pinczolits A, Sandow N, Major S, Winkler MK, Weber-Carstens S, Dohmen C, Graf R, Strong AJ, Dreier JP, Vajkoczy P (2013) Propagation of cortical spreading depolarization in the human cortex after malignant stroke. Neurology 80(12):1095–1102
Woo DH, Han KS, Shim JW, Yoon BE, Kim E, Bae JY, Oh SJ, Hwang EM, Marmorstein AD, Bae YC, Park JY, Lee CJ (2012) TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell 151(1):25–40
Yan Y, Dempsey RJ, Sun D (2001) Na+-K+-Cl- cotransporter in rat focal cerebral ischemia. J Cereb Blood Flow Metab 21(6):711–721
Ye ZC, Oberheim N, Kettenmann H, Ransom BR (2009) Pharmacological “cross-inhibition” of connexin hemichannels and swelling activated anion channels. Glia 57(3):258–269
Zhang Z, Bourque CW (2003) Osmometry in osmosensory neurons. Nat Neurosci 6(10):1021–1022
Zhang H, Cao HJ, Kimelberg HK, Zhou M (2011) Volume regulated anion channel currents of rat hippocampal neurons and their contribution to oxygen-and-glucose deprivation induced neuronal death. PLoS ONE 6(2):e16803
Zhang Y, Zhang H, Feustel PJ, Kimelberg HK (2008) DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Exp Neurol 210(2):514–520
Zierler S, Frei E, Grissmer S, Kerschbaum HH (2008) Chloride influx provokes lamellipodium formation in microglial cells. Cell Physiol Biochem 21(1-3):55–62
Acknowledgments
I am grateful to my colleagues, Dr. Richard W. Keller, Jr., and Alexandra L. Schober, for critical reading and helpful suggestions on the manuscript, as well as for help with figure preparation. The work in the author’s laboratory was supported by a grant from the National Institute for Neurological Disorders and Stroke (NS061953).
Author information
Authors and Affiliations
Corresponding author
Additional information
Frenemy (n.) is a portmanteau of “friend” and “enemy” that refers to one who pretends to be a friend but is actually an enemy. Frenemy may also denote the one who really is a friend but also a rival (Merriam-Webster Dictionary online accessed on 04/07/2015).
This article is published as a part of the special issue on “Molecular physiology of anion channels: dual function proteins and new structural motifs.”
Rights and permissions
About this article

Cite this article
Mongin, A.A. Volume-regulated anion channel—a frenemy within the brain. Pflugers Arch - Eur J Physiol 468, 421–441 (2016). https://doi.org/10.1007/s00424-015-1765-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-015-1765-6