
Abstract
Purpose
To give an overview over cell therapeutic approaches to immunosuppression in clinical kidney transplantation. A focus is on myeloid suppressor cell therapy by mitomycin C-induced cells (MICs).
Methods
Literature review with an emphasis on already existing therapies.
Results
Several cell therapeutic approaches to immunosuppression and donor-specific unresponsiveness are now being tested in early phase I and phase II trials in clinical kidney transplantation. Cell products such as regulatory T cells or regulatory macrophages, or other myeloid suppressor cell therapies, may either consist of donor-specific, third-party, or autologous cell preparations. Major problems are the identification of the suppressive cell populations and their expansion to have sufficient amount of cells to achieve donor unresponsiveness (e.g., with regulatory T cells). We show a simple and safe way to establish donor unresponsiveness in living-donor kidney transplantation by MIC therapy. A phase I clinical trial is now under way to test the safety and efficacy of this cell therapeutic approach.
Conclusions
Cell therapeutic approaches to immunosuppression after kidney transplantation may revolutionize clinical transplantation in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Kidney transplantation represents the optimal treatment for patients with chronic kidney disease stage 4 or 5. Compared to dialysis, kidney transplant recipients have a survival advantage and improvement in quality of life [1]. According to a recent analysis of the United Network of Organ Sharing (UNOS) database, during a 25-year period, a total of 1,372,969 life years were saved by kidney transplantation compared to patients who were waitlisted but received no allograft [2]. This number resulted in 4.4 life years saved per patient during the observation period. Analysis of more recent procedures or of younger patients may bring even more impressive figures. Long-term survival of kidney graft recipients, however, is limited by two major problems: (1) death with a functioning graft due to the side effects of immunosuppressive therapy and (2) graft loss often due to chronic antibody-mediated rejection. Therefore, there is still an urgent need for more powerful immunosuppressive tools that at the same time have fewer side effects than currently applied quadruple immunosuppressive therapies. Ideally, one should achieve selective (donor) unresponsiveness of the graft recipient against the donor without the use of broad immunosuppression and full responsiveness of the patient’s immune system against bacteria, viruses, and other pathogens—i.e., tolerance.
Transplantation tolerance
In the absence of powerful immunosuppression, organ transplantation primes the activation of the innate and adaptive immune system leading to an immune response specifically directed against the graft alloantigens, resulting in graft injury and destruction. Previous exposure to alloantigens by pregnancy, blood transfusion, or prior transplantation may even lead to accelerated immune reactions via cellular and antibody-mediated mechanisms. Since the pioneering work from Medawar and colleagues in 1953, it is well known that allogenic cells may not only stimulate but also inhibit immune responses [3]. After injection of allogeneic bone marrow cells into newborn mice, the recipients became tolerant to subsequently transplanted skin of the same donor. Since then, our understanding of the mechanisms of the regulation of the immune system is steadily improving. Today, tolerance is believed to be the result of the net balance of regulatory and effector mechanisms in the patient’s immune system. Many years after Medawar’s first efforts to induce donor-specific unresponsiveness, it has been shown that multiple blood transfusions (containing allogeneic cells) from third-party donors were able to inhibit the rejection process of subsequent kidney transplants [4]. At that time, still before the introduction of powerful immunosuppression, e.g., by cyclosporine, 1-year graft survival in non-transfused patients was 23 % while it was strikingly 87 % (P < 0.001) in patients who had received more than 10 transfusions. This “tolerogenic” effect of multiple transfusions may be attributable to a higher content of polymorphonuclear leukocytes in these early preparations since the graft-protective effect was lost later when packed red cells, i.e., leucocyte-depleted preparations were administered. In recent years, transfusions of packed red cells were rather linked to recipient sensitization and premature graft loss than to a graft-protective effect. In a recent Swiss study, Marti and colleagues could demonstrate that donor antigen-specific transfusions may reduce rejection episodes even when applied together with powerful immunosuppression [5]. Instead of packed red cells, they used either whole blood or mononuclear blood cells from buffy coats. A total of 61 potential recipients of living-donor kidney allografts received two donor-specific transfusions at a 2-month interval. After one or two donor-specific transfusions, a total of six patients developed a positive complement-dependent cytotoxicity (CDC) T cell crossmatch and therefore were not transplanted. Patients with donor-specific transfusions showed better graft and fewer rejection episodes compared to matched control patients in Western Europe and to living-donor kidney transplant recipients in other Swiss transplant centers or Western Europe.
The recipient’s immune system and regulatory T cells
The ability of the immune system to discriminate between self and non-self is the basis of allorecognition. Allografts induce alloimmune responses because of the recognition of non-self-antigens from the foreign tissue by recipient T cells. Once activated and expanded, T cells exhibit effector functions that result in destruction of graft tissue. Animals experimentally deprived of T cells do not reject allografts. However, T cells are not only powerful effector cells but also main regulators of autoimmune responses. Autoreactive T cells with high affinity for self-antigens are deleted in the thymus, whereas self-reactive T cells that escape thymus censorship have the potential to induce autoimmunity if they are not controlled by peripheral regulatory mechanisms. It is known that regulatory T cells (Tregs) play an important role in maintaining self-tolerance and are essential for the restoration of the immune homeostasis after antigenic stimulation. Two major Treg cell types can be distinguished: naturally occurring Tregs (nTreg), which develop in the thymus and express a broad repertoire of α/β T cell receptors (TCRs) with specificities for both self and non-self-antigens, and induced Tregs (iTregs), which arise in the periphery by conversion of conventional cluster of differentiation (CD) 4+-T cells after immunological challenge [6]. A unique cell marker that differentiates nTregs from iTregs has not yet been found. Functionally active Tregs are characterized by a constitutive, strong expression of the interleukin (IL)-2 receptor alpha chain (CD25) and a low or negative expression of the IL-7 receptor alpha chain (CD127) [7]. Furthermore, these cells express the transcription factor forkhead box (Fox) P3, which is essential for their function. The mechanisms of immunoregulation by Tregs can be divided into those that target T cells (e.g., cytolysis, suppressive cytokines, IL-2 consumption) and those that target antigen-presenting cells (APCs) (e.g., reduced costimulation or antigen presentation) [8]. Human Tregs have been shown to express several other markers such as cytotoxic T lymphocyte antigen 4 (CTLA4) or human leucocyte antigen-DR (HLA-DR). The expression of CTLA4 by Tregs can inhibit APC activity, thereby preventing the development of effector T cells [9]. HLA-DR+-Tregs potentially affect the suppressive activity of the total Treg pool [10]. The disappearance of this Treg subset represents a strong indicator for acute rejection processes [11]. On the other hand, also antigen-presenting cells (APCs) have the capability to induce, maintain, or increase the number of Tregs that in turn cause the generation of new tolerogenic APCs [12]. Upon encounter to Tregs, all major subpopulations of APCs, i.e., dendritic cells (DCs), B cells, and monocytes/macrophages, respond by downregulation of their antigen-presenting function, upregulation of immunosuppressive molecules, and secretion of immunosuppressive cytokines [9]. Dendritic cells and macrophages are capable of both stimulating and suppressing T cell-mediated responses according to their state of activation [13, 14]. Immature DCs and macrophages present self and harmless antigens under non-inflammatory conditions. Antigen presentation without costimulation inactivates effector T cells. In this way, antigen presentation by non-activated myeloid APCs contributes to the maintenance of steady-state self-tolerance [15, 16]. Furthermore, distinct populations of Tregs and tolerogenic APCs act synergistically to maintain an immunological balance [17].
Cell therapeutic approaches to transplantation tolerance
Cell therapeutic approaches to immunosuppression and donor-specific unresponsiveness are a double-edged sword. The same cell type may either inhibit or trigger an immune response depending on a whole series of circumstances such as age, number of administered cells, concomitant immunosuppressive therapy, and state of the patient’s immune system. The only cell therapeutic approaches for tolerance induction that are effective in clinical transplantation combine kidney with bone marrow transplantation after myeloablative or non-myeloablative conditioning regimens [17–19]. Such approaches are logistically challenging, bear many potential side effects, and are extremely costly, and therefore limited to only few selected patients. Alternative cell therapeutic strategies to minimize immunosuppression are mainly composed of immunoregulatory cell populations. These preparations can further be divided into groups of cell populations that are donor antigen-specific and those that are donor-unrelated, derived from third-party donors or autologous cell preparations. Though most cell therapies demonstrated at least some immunosuppressive properties in in vitro andpreclinical studies, convincing proof for their clinical efficacy in kidney transplant recipients is still pending. Asdiscussed below, the transfer of cell-based products into clinical trials needs to obey strict governmentalregulations. Even after proof of effectiveness in human kidney transplantation, the introduction of the majority of cell-based immunoregulatory approaches into clinical routine would not be easy due to logistical hurdles, especially tedious processes of separation and long-lasting cultivation of the particular therapeutic cell populations under conditions of good manufacturing practice (GMP) [15, 20, 21]. Another significant dilemma is the need for additional immunosuppressive therapies. For example, immunosuppression by calcineurin inhibitors may either have direct toxic effects on the administered regulatory cell populations or may interfere with the formation of a tolerogenic state of the recipient’s immune system [22]. This is of particular interest, since most cell-based immunoregulatory therapies are aimed at a minimization rather than complete avoidance of immunosuppressive medication. A further issue consists in the half-life of the administered cell populations. Most cells, especially of allogeneic origin, have a rather short life span after administration and may not be detectable in the recipient’s circulation within only a few hours to days. It is possible, however, that in some cases, this short time is sufficient to induce a tolerogenic state in the recipient and that the immunosuppressive effect is not limited to the life span of the therapeutic cell population itself.
Combined kidney and bone marrow transplantation
Successful clinical tolerance has been achieved by combining kidney and bone marrow transplantation. During this procedure, a mixed chimerism is established in the kidney recipient at least for a certain time period, suppressing the recipient’s effector T cell responses while at the same time, regulatory T cell responses are established. The Stanford group recently published their extended experience in the treatment of 38 HLA-matched and mismatched patients by combined living-donor kidney and CD34+-enriched hematopoietic stem cell transplantation [23]. Conditioning of these patients consisted of post-transplant total lymphoid irradiation (10 doses of 80 or 120 cGy) and anti-thymocyte globulin (1.5 mg/kg b.w. at 5 daily doses). Nineteen of 22 HLA-matched patients had persistent chimerism for at least 6 months with 16 of the 19 patients successfully weaned from immunosuppressive medication. The Boston group published their extended experience in 10 patients with combined kidney and bone marrow transplantation after non-myeloablative conditioning in July 2014 [19]. All patients developed a transient chimerism with seven patients being off immunosuppressive therapy for 4 years or more. Conditioning was modified during the study and consisted of cyclophosphamide (60 mg/kg intravenously on days −5 and −4); a humanized anti-CD2 monoclonal antibody (0.1 mg/kg on day −2 and 0.6 mg/kg on days −1, 0, and +1); cyclosporine A (5 mg/kg intravenously on day −1); and thymic irradiation (700 cGy on day −1). More recently, rituximab (375 mg2 on days −7, −2, +5, and +12) and prednisone (2 mg/kg from day 0 and tapered to post-transplant day +20) were added to prevent development of donor-specific antibodies. Cyclosporine A was replaced by tacrolimus that was tapered until month 8. On the day of kidney transplant, unprocessed donor bone marrow (2–3 × 108 cells/kg) was intravenously infused in patients. At latest follow-up, four patients were off immunosuppressive therapy while two patients had lost their graft. One patient (patient 3) suffered from early acute antibody-mediated rejection and one patient (patient 10) had pyelonephritis followed by severe T cell-mediated rejection BANFF IIB and had retransplantation after 3.3 years. At Northwestern University, 31 patients were enrolled and 25 were transplanted in a phase IIb tolerance trial in HLA-mismatched living-donor kidney transplantation [24]. Conditioning consisted of fludarabine (30 mg/kg on days −4, −3, and −2); cyclophosphamide (50 mg/kg on days −3 and +3); and 200 cGy total body irradiation (200 cGy on day −1). Tacrolimus and mycophenolate mofetil at a regular dose were given from day −3 and continued after transplantation. Immunosuppression was tapered from months 6 and discontinued at months 12 after transplantation if persistent whole blood and T cell macrochimerism was persistent. Seventeen patients with a follow-up of more than 18 months segregated into patients with either persistent chimerism (12 of 17), transient chimerism (4 of 17), and patients who never developed a chimerism (1 of 17). Twelve of 17 patients were successfully weaned from immunosuppressive medication. Unfortunately, in above-mentioned studies, little information is given on the side effects of the invasive therapies.
Induction therapy with mesenchymal stem cells
Current immunosuppressive treatment usually consists of a tripe drug regimen together with potent antibody induction therapy. New induction agents with enhanced efficacy and reduced side effects are desirable. Tan and colleagues recently published results of a prospective randomized study that investigated the use of autologous mesenchymal stem cells (MSCs) as an induction agent for living-donor kidney transplantation [25]. MSCs represent a non-hematopoietic cell population that inhibits T cell proliferation and monocyte differentiation and regulates natural killer cell and B cell functions. Therefore, MSCs are thought to exhibit immunosuppressive properties [26]. During the study, 159 end-stage renal disease patients transplanted with a related living donor were divided into three treatment arms: group A, MSC treatment (1–2 × 106 cells/kg body weight, on days 0 and 14) plus standard dose cyclosporine; group B, MSC treatment plus reduced dose cyclosporine (80 %); and group C, basiliximab (a monoclonal anti-CD25 antibody) instead of MSC induction therapy. There was no significant difference in graft or patient survival and acute rejection after 12 months. Interestingly, four rejections in group C were classified as steroid-resistant while no such rejection was observed in groups A and B. Kidney function was better in MSC-treated patients at year 1 and the hazard ratio for opportunistic infections in MSC—as compared to basiliximab-treated patients—was 0.42 (95 % confidence interval (CI) 0.2–0.85; P = 0.02) while no significant side effects of MSC therapy were noticeable. As a note of caution, overall rejection in recipients of a living-related kidney transplant during the first year after transplantation was with 26 % rather high and there are still concerns with respect to a possible malignant transformation of MSCs.
Regulatory T cells (Tregs)
Regulatory T cells are essential for achieving transplantation tolerance. In animal models, the transfer of Treg cells has demonstrated to be a promising strategy for controlling acute and chronic allograft rejection [21]. But these preclinical animal studies have shown that a drastic change of conventional T (Tconv) cells to Treg balance is needed to control transplant rejection using Treg cell therapy [20]. To alter this equilibrium in favor to Tregs, reduction of Tconv cells or expansion of Tregs is possible strategies... is a possible strategy, or the ..., or the combination of both. To receive the requested billions of Treg cells, it is necessary to expand them in culture by stimulating freshly isolated Tregs through the CD3 and CD28 molecules [27, 28]. The dose of Tregs that will give the optimal immunosuppression in solid organ transplantation is not known, but based on Treg therapy studies in type 1 diabetes and hematopoietic stem cell transplantation, a dose of 1 × 106 to 20 × 106/kg of bodyweight seems to be necessary [29]. Some authors recommend a simultaneous deletion of host donor-reactive T cells with thymoglobulin induction to improve the Treg to Tconv-cell balance, and finally, to reduce the number of required Tregs [20, 30]. A further strategy to optimize Treg therapy is to use donor alloantigen-reactive Tregs. These Tregs are 10–100 times more effective at suppressing Tconv-cell proliferation to alloantigens than polyclonal Tregs [31]. Alternative to isolating preexisting Tregs for ex vivo expansion, Tconv cells can be converted to Tregs during ex vivo expansion with the addition of transforming growth factor (TGF)-beta together with rapamycin or all-trans retinoic acid [20]. But further experiments focusing on the stability and commitment of ex vivo induced Tregs are needed before they can be considered as therapeutic Tregs in humans. In particular, since it is known that the total Treg pool consists of many phenotypic and functional different subpopulations which were classified based on expression of immunologic markers, sites of differentiation, mechanisms of function, and lineage plasticity [32].
To date, a few clinical studies have infused Treg cells as a GvHD prophylaxis in patients receiving hematopoietic stem cell transplantation and as a therapy in patients with GvHD [33–35]. No safety concerns were reported and a lower incidence of GvHD was observed in comparison to the results of historical controls [33]. Data for Treg cell therapy in solid organ transplantation are not published so far. But a phase I/II international multicenter clinical trial of Treg therapy for renal transplantation patients is currently in progress (The ONE Study) [36]. Another clinical study (ThRIL) will be evaluating the safety and efficacy of Treg therapy in liver transplantation [37].
Myeloid regulatory cell products
Myeloid-derived suppressor cells (MDSCs) occur in tumor patients where they compromise the patient’s immune response. Whereas these cells may be deleterious in cancer patients, they might be useful for controlling allograft rejection in organ transplantation. MDSCs accumulate due to tumor factors which activate immature myeloid cells and concomitantly block their maturation to dendritic (DCs) or other cells [38]. Several groups attempt to reproduce this process in vitro to get a suppressive cell population for the treatment of allograft rejection and autoimmune diseases [39, 40]. Myeloid suppressor cells have been produced by the treatment with IL-10 (DC10 cells), rapamycin (Rapa-DCs), culture with GM-CSF (Tol-DCs), or culture with dexamethasone and vitamin D. Myeloid suppressor cells may be of donor, recipient (autologous), or third-party origin and exert their effects by various mechanisms. While myeloid suppressor cells were tested for the induction of tolerance in rodent heart and kidney transplantation models, their role in the induction of tolerance in clinical transplantation is so far unclear.
Regulatory macrophages
An approach to immunosuppression after kidney transplantation by macrophages that have a regulatory phenotype is proposed by the Regensburg group. CD14+ monocytes were exposed for 7 days to M-CSF, human serum, and IFN-gamma [41]. These regulatory macrophages (Mregs) have a distinct morphology and do not stimulate allogenic T cell proliferation in vitro. A first use in patients was performed during an individual treatment attempt in two living-donor kidney transplant recipients. Both patients were successfully transplanted while still on tacrolimus monotherapy [42]. One year post-transplant, both patients showed a gene expression pattern in their peripheral blood that is consistent with the gene signature obtained in the IOT-RISET trial. The proof of the safety and efficacy of Mreg therapy, however, is pending and will be tested within the One Study [36].
Mitomycin-induced donor blood cells (MICs) for induction of donor-specific tolerance in organ transplantation
Dendritic cells (DCs) are the most potent professional, antigen-presenting cells (APCs). They have been recognized as effective initiators of specific immune responses against foreign antigens but play also a key role in maintaining central and peripheral tolerance towards self-antigens [13, 43]. Immunoregulatory APCs may be of great value for particular clinical applications such as the treatment of rejection episodes in organ transplantation or deleterious immune reactions in autoimmune diseases. The antibiotic and chemotherapeutic agent mitomycin C (MMC) was shown to cause non-immunogenic apoptotic cell death in tumor cells and was therefore identified as a candidate substance to abrogate the immunogenicity of allogeneic DCs [44]. If MMC could render allogenic DCs tolerogenic, they might be an elegant tool to control transplant rejection. To test this hypothesis, mature DCs were generated from rat bone marrow in vitro and treated with MMC at the end of culture. MMC-treated DCs (MMC-DCs) lost their stimulatory capacity. Proliferation of allogeneic T lymphocytes was not induced by MMC-DCs in vitro. Once suppressed, T cells could not be restimulated with untreated, freshly generated mature DCs of the same donor but with mature DCs from a third-party donor, pointing to an active donor-specific immunosuppressive mechanism [45, 46]. In a next step, MMC-DCs were examined in a heterotopic heart transplantation model. One week before surgery, recipient rats received either 1 × 106 mitomycin-treated DCs or naïve mature DCs generated from the same allogeneic donor strain. While the heart was rejected within approximately 1 week by untreated recipients, pretreatment with naïve mature DCs caused accelerated rejection. In contrast, recipients receiving a single injection of MMC-DCs 7 days prior to transplantation showed heart allograft survival that was significantly prolonged up to 1 month. The suppressive effect induced by the treatment with MMC-DCs was donor-specific since cardiac allografts of a third-party donor were rejected. The conversion of rat DCs into tolerogenic cells after MMC treatment was attributable to the downregulation of stimulatory cell surface receptor molecules such as CD80, CD86, as well as the intercellular adhesion molecule (ICAM)-1 [45, 46], a phenomenon that was not reproducible in MMC-treated mature DCs of human origin. Comprehensive gene expression analysis of in vitro generated human DCs revealed that MMC treatment modulated the expression levels of various apoptotic and immunoregulatory genes [47].
Though DCs were a promising tool for immunological interventions, there are still concerns for their clinical application. Standardized in vitro generation of DCs under GMP conditions in accordance with rigorous governmental regulations is labor-intensive, costly, and not easy to achieve. Most importantly, manipulated DCs may, under certain circumstances, regain their stimulatory capacity with negative consequences when transferred to patients. Peripheral mononuclear blood cells (PBMCs) were identified as a suitable alternative. They can easily be harvested from peripheral blood in sufficient number and quality. In an allogeneic rat heart transplantation model, a single preoperative injection of donor-derived MMC-treated PBMCs (mitomycin-induced donor blood cells, MICs) significantly prolonged allograft survival without additional immunosuppression. Allograft survival correlated with the number of administered MICs. When the maximum dose of 1 × 108 MICs was given, 50 % of animals had long-term acceptance of the allograft with more than 70 days survival. In contrast, administration of untreated PBMCs or heart transplantation from a third-party donor led to accelerated rejection (Fig. 1). Prophylactic immunomodulation with MICs solely targets the unwanted immune responses against the allograft, a central prerequisite for their application in clinical transplantation [48]. In composite tissue rat hind limb allotransplantation, MICs significantly prolonged allograft survival when given at the day of surgery [49]. Pretreatment of recipients with donor-derived MMC-treated splenocytes strongly attenuated acute rejection in a heart transplantation model in mice [50]. MICs were also tested in a large animal kidney transplantation model in the pig. Even the low dose of 1 × 108 MICs significantly prolonged mean allograft survival when compared to untreated kidney recipients [48].

Induction of donor-specific tolerance by prophylactic administration of donor-derived mitomycin C-induced peripheral mononuclear blood cells (MICs) in an allogeneic heart transplantation in the rat. a A rat heart (strain Dark Agouti, DA) heterotopically transplanted into the abdomen of an allogeneic recipient (Piebald Virol Glaxo, PVG) will be rejected within 8 to 10 days. b Transfusion of the recipient with peripheral blood cells of the donor shortly incubated with the chemotherapeutic agent mitomycin C (MICs) 1 week prior to transplantation (day −7) leads to significant prolongation of allograft survival and even induces tolerance of the transplant in 50 % of the recipient animals. c The induced tolerance is donor-specific, since only those allografts are protected from rejection, which are derived from the same rat strain as the original blood donor strain (PVG). Transplants from a further, third-party donor strain (Brown Norway, BN) are rejected in a normal fashion as in untreated controls shown in panel a
Additional studies revealed that monocytes were the relevant cell population responsible for the suppressive effects. When the monocytes were depleted from the PBMCs, the tolerogenic properties were lost [48]. When MMC-treated monocytes were cultured in the presence of certain cytokines, the resulting myeloid cells exhibited an immature phenotype resembling that of myeloid-derived suppressor cells (MDSCs). Treatment of monocytes with MMC blocks their differentiation towards stimulatory DCs and at the same time, confers them immunosuppressive competence, resulting in a highly effective regulatory myeloid cell type. Following MIC administration, increased numbers of CD4+CD25+FoxP3+ regulatory T cells (Tregs) were found in the peripheral blood and spleen of treated animals. In addition, increased Foxp3+ cell infiltration was seen in the rat heart allograft, suggesting a central role of Tregs in the induction of tolerance. Via adoptive transfer of peripheral blood cells from tolerant MIC-treated rats (graft acceptance for at least 70 days) into naïve syngeneic animals, it was possible to confer the cellular tolerogenic state to the naïve recipients. Approximately 50 % of the recipients developed operational tolerance (more than 70 days without rejection episode) towards heart allografts from the same original donor. Depletion of CD4+CD25+Tregs before transfer abrogated the protective effect [48].
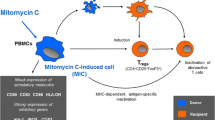
These observations led to the hypothesis of the mode of action of MIC-induced donor-specific tolerance (Fig. 2). As a consequence of MMC treatment, monocytes within the PBMCs are converted into tolerogenic cells, so-called MMC-induced cells (MICs). MICs are characterized by reduced expression of stimulatory molecules but increased expression of various immunosuppressive genes (Fig. 2). Following injection, MICs accumulate in secondary lymphatic organs. MICs inactivate antigen-specific T cells and induce inhibitory CD4+CD25+FoxP3+ Tregs. As MMC causes apoptosis in the target cells, uptake of these apoptotic MIC cells by APCs blocks their maturation and confers them an immunosuppressive phenotype with the potential to control immune responses and support Treg formation (Fig. 2).

Mode of action of mitomycin C-treated peripheral blood cells (MICs) for the induction of donor-specific tolerance in allogeneic organ transplantation. Short incubation of peripheral mononuclear blood cells (PBMCs) with mitomycin C (MMC) induces the generation of tolerogenic myeloid cells (MICs). These cells are characterized by low expression of immunostimulatory surface molecules, such as CD80, CD83, CD86, and HLA-DR, as well as the upregulation of immunosuppressive genes, such as arginase-1 (arg-1), inducible nitric oxide synthase (iNOS), interleukin (IL)-10, transforming growth factor (TGF)-β, cyclooxygenase (COX)-2, and the transcription factor C/EBPβ. MICs directly inactivate alloreactive T lymphocytes and induce the development of CD4+CD25+FoxP3+ regulatory T cells (Tregs) capable of suppressing harmful immune responses. In addition, MMC induces apoptosis in its target cells. MMC-treated apoptotic donor cells are taken up by recipient antigen-presenting cells (e.g., immature dendritic cells) preventing their maturation towards immunostimulatory cells. In turn, these immature myeloid cells exhibit an immunosuppressive phenotype inhibiting immune activation and promoting Treg formation
Based on the promising results of the preclinical investigations, MICs were applied for the first time in humans as an individual unique emergency treatment to a young patient with relapse of acute lymphoblastic leukemia (ALL) with recurrent therapy-resistant rejection of haploidentical stem cell transplants. At the time when first signs of rejection (of the third transplant) appeared, the patient received, at a weekly interval, two transfusions of MICs from the father (109 and 2 × 109), prepared from CD3/CD19-depleted donor blood cells. No acute complications attributable to MIC treatment were observed. Subsequently, continuously decreasing percentages of autologous NK , T, and B cells were noted, resulting in a stable, complete hematopoietic donor chimerism for more than 1 year. The patient received no further immunosuppressive treatment but was supported with additional donor stem cell boosts, one mesenchymal stem cell transfusion, and one administration of CMV-specific donor T cells [48]. Even though this clinical case does not allow us to draw any conclusion about the effectiveness of tolerogenic MIC therapy, it clearly demonstrates that MICs can be easily generated and safely translated into the clinic for human treatment.
The TOL-1 phase I study
To test the MIC therapy for the induction of donor-specific tolerance, a single-center phase I clinical study in living-donor kidney transplantation will start in summer 2015. A brief description of the study design is given in Fig. 3. In addition to MIC therapy, all patients will receive standard immunosuppressive therapy.

Protocol for the TOL-1 Study on MIC therapy. Seven days before transplantation, PBMCs are retrieved from the kidney donor. After incubation of PBMCs with Mitomycin C for 30 min, cells are washed. 1.5 × 108 MICs per kilogram bodyweight are infused to the recipient. Seven days later, recipients receive a kidney allograft from the same donor
Advanced therapy medicinal products and European Community regulations
In the last years, immunotherapies, particularly cellular therapies, increasingly gained interest for the treatment of cancer patients and autoimmune diseases and for purposes of regenerative medicine. However, in order to make these new innovative cellular therapies available for patient applications in form of authorized therapies on the European or national markets or for use in clinical trials or compassionate treatment programs, a strict regulatory framework established in the European Community (EC) and in its member states has to be fulfilled. For the entire EC, the European Medicines Agency (EMA) with its advisory Committee for Advanced Therapies (CAT), represents the competent authority. In Germany, the Paul-Ehrlich-Institut (PEI) is the national competent authority (NCA) for biological medicinal products whereas the regional authorities (Regierungspräsidium, RP) are responsible for the supervision of the entire manufacturing process in order to ensure quality, efficacy, and safety of medicinal products.
The development of an investigational medicinal product (IMP) for innovative cellular therapies according to the EC and national regulations is time-consuming and cost-intensive. First of all, intensive preclinical studies have to be performed. When proof of concept has been shown, the investigational cellular approach can be translated from bench to bedside at a good manufacturing practice (GMP) unit in compliance with the legal requirements. This means that only GMP grade substances, reagents, and materials are allowed to be used. The manufacturing process, all devices, the GMP clean room facility, and the personnel have to be established, trained, and validated. External service providers and laboratories have to be audited and particular agreements reached and contracts concluded. Most importantly, various applications regarding the use of the advanced therapy medicinal products (ATMP) preparation have to be filed at the appropriate governmental and institutional authorities to eventually obtain (1) the approval to perform a clinical phase I study (at the PEI), (2) the manufacturing license (at the RP), and (3) an ethical vote (from the ethics committee at the study site). Therefore, the preparation of a detailed study protocol, an investigator’s brochure (IB), an investigational medicinal product dossier (IMPD), extensive validation documents, and numerous standard operating procedures (SOPs) is required.
When preparing the documents for an investigational medicinal product (IMP), one has to determine whether the cellular approach is considered as an advanced therapy medicinal product (ATMP). The classification of a medicinal product is of major importance because it determines the procedure of authorization, i.e., for ATMPs, a centralized marketing authorization enabling a free movement of ATMPs within the EC. Based on the novelty and complexity of ATMPs, special harmonized regulations have been established in the EC [Regulation (EC) no. 1394/2007] and introduced into the national law of the German Medicinal Products Act (Arzneimittelgesetz, AMG). For developers of ATMPs within Germany, the Innovation Office at the PEI offers regulatory and scientific advice for the classification and development of ATMPs and provides contact to the EMA, to the coordinating centers for clinical trials (Koordinierungszentren für Klinische Studien, KKS), to the institute for quality and efficiency in health care (Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen, IQWiG), and to the Federal Joint Committee (Gemeinsamer Bundesausschuss, GBA).
Furthermore, not only regulatory requirements have to be fulfilled for the availability of medicinal products for patients but also on the long run, financial and economic support by the health insurance funds (Gesetzliche Krankenversicherung, GKV) have to be provided. The GBA, as the highest decision-making body in Germany of physicians, dentists, hospitals, and health insurance funds (GKV), issues directives specifying which services in medical care are reimbursed by the health insurance funds. Decision of reimbursement might, among others, also be based on scientific reports of the independent study organization IQWiG, which evaluates benefits and harms of medical interventions like the administration of ATMPs.
More and more innovative cellular therapies are evolving and within this context, the regulations and directives in the EC augment. Hence, it is a long and tedious way from the original idea of a cell-based therapeutic through its proof of concept and translation into GMP conform manufacturing up to the final ATMP approval which might take one decade or longer. This constitutes an enormous challenge in a global world which is currently changing almost at an annual basis. Therefore, it is extremely difficult for both academy and industry to make a sound prognosis of future development of products.
Conclusion
Several cell therapeutic approaches for the immunosuppression after solid organ transplantation are now tested in early phase clinical trials. The transfer of cell therapeutic approaches from bench to bedside, however, is challenging. Cell therapeutic approaches must show their safety and efficacy and the application of these therapies must be feasible in clinical routine. An elegant approach to immunosuppression by MIC cells had recently been introduced and is now tested in a clinical phase I study.
References
Wolfe RA, Ashby VB, Milford EL, Ojo AO, Ettenger RE, Agodoa LY, Held PJ, Port FK (1999) Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med 341(23):1725–1730
Rana A, Gruessner A, Agopian VG, Khalpey Z, Riaz IB, Kaplan B, Halazun KJ, Busuttil RW, Gruessner RW (2015) Survival benefit of solid-organ transplant in the United States. JAMA Surg 150(3):252–259
Billingham RE, Brent L, Medawar PB (1953) Actively acquired tolerance of foreign cells. Nature 172(4379):603–606
Opelz G, Graver B, Terasaki PI (1981) Induction of high kidney graft survival rate by multiple transfusion. Lancet 1(8232):1223–1225
Marti HP, Henschkowski J, Laux G, Vogt B, Seiler C, Opelz G, Frey FJ (2006) Effect of donor-specific transfusions on the outcome of renal allografts in the cyclosporine era. Transpl Int 19(1):19–26
Schmetterer KG, Neunkirchner A, Pickl WF (2012) Naturally occurring regulatory T cells: markers, mechanisms, and manipulation. FASEB J 26(6):2253–2276
Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, Gottlieb PA, Kapranov P, Gingeras TR, de St F, Groth B et al (2006) CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med 203(7):1701–1711
Shevach EM (2009) Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 30(5):636–645
Mahnke K, Bedke T, Enk AH (2007) Regulatory conversation between antigen presenting cells and regulatory T cells enhance immune suppression. Cell Immunol 250(1–2):1–13
Schaier M, Seissler N, Schmitt E, Meuer S, Hug F, Zeier M, Steinborn A (2012) DR(high+)CD45RA(−)-Tregs potentially affect the suppressive activity of the total Treg pool in renal transplant patients. PLoS One 7(3), e34208
Schaier M, Seissler N, Becker LE, Schaefer SM, Schmitt E, Meuer S, Hug F, Sommerer C, Waldherr R, Zeier M et al (2013) The extent of HLA-DR expression on HLA-DR(+) Tregs allows the identification of patients with clinically relevant borderline rejection. Transpl Int 26(3):290–299
Hsu SM, Mathew R, Taylor AW, Stein-Streilein J (2014) Ex-vivo tolerogenic F4/80(+) antigen-presenting cells (APC) induce efferent CD8(+) regulatory T cell-dependent suppression of experimental autoimmune uveitis. Clin Exp Immunol 176(1):37–48
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392(6673):245–252
Broichhausen C, Riquelme P, Geissler EK, Hutchinson JA (2012) Regulatory macrophages as therapeutic targets and therapeutic agents in solid organ transplantation. Curr Opin Organ Transplant 17(4):332–342
Hutchinson JA, Geissler EK (2015) Now or never? The case for cell-based immunosuppression in kidney transplantation. Kidney Int
Steinman RM (2012) Decisions about dendritic cells: past, present, and future. Annu Rev Immunol 30:1–22
Andre S, Tough DF, Lacroix-Desmazes S, Kaveri SV, Bayry J (2009) Surveillance of antigen-presenting cells by CD4+ CD25+ regulatory T cells in autoimmunity: immunopathogenesis and therapeutic implications. Am J Pathol 174(5):1575–1587
Kawai T, Cosimi AB, Spitzer TR, Tolkoff-Rubin N, Suthanthiran M, Saidman SL, Shaffer J, Preffer FI, Ding R, Sharma V et al (2008) HLA-mismatched renal transplantation without maintenance immunosuppression. N Engl J Med 358(4):353–361
Kawai T, Sachs DH, Sprangers B, Spitzer TR, Saidman SL, Zorn E, Tolkoff-Rubin N, Preffer F, Crisalli K, Gao B et al (2014) Long-term results in recipients of combined HLA-mismatched kidney and bone marrow transplantation without maintenance immunosuppression. Am J Transplant 14(7):1599–1611
Tang Q, Bluestone JA (2013) Regulatory T-cell therapy in transplantation: moving to the clinic. Cold Spring Harb Perspect Med 3(11)
Wood KJ, Bushell A, Hester J (2012) Regulatory immune cells in transplantation. Nat Rev Immunol 12(6):417–430
Gallon L, Traitanon O, Yu Y, Shi B, Leventhal JR, Miller J, Mas V, L X, Mathew JM (2015) Differential effects of calcineurin and mammalian target of rapamycin inhibitors on alloreactive Th1, Th17, and Regulatory T Cells. Transplantation
Scandling JD, Busque S, Shizuru JA, Lowsky R, Hoppe R, Dejbakhsh-Jones S, Jensen K, Shori A, Strober JA, Lavori P et al (2015) Chimerism, graft survival, and withdrawal of immunosuppressive drugs in HLA matched and mismatched patients after living donor kidney and hematopoietic cell transplantation. Am J Transplant 15(3):695–704
Leventhal JR, Elliott MJ, Yolcu ES, Bozulic LD, Tollerud DJ, Mathew JM, Konieczna I, Ison MG, Galvin J, Mehta J et al (2015) Immune reconstitution/immunocompetence in recipients of kidney plus hematopoietic stem/facilitating cell transplants. Transplantation 99(2):288–298
Tan J, Wu W, Xu X, Liao L, Zheng F, Messinger S, Sun X, Chen J, Yang S, Cai J et al (2012) Induction therapy with autologous mesenchymal stem cells in living-related kidney transplants: a randomized controlled trial. JAMA 307(11):1169–1177
Nauta AJ, Fibbe WE (2007) Immunomodulatory properties of mesenchymal stromal cells. Blood 110(10):3499–3506
Schliesser U, Streitz M, Sawitzki B (2012) Tregs: application for solid-organ transplantation. Curr Opin Organ Transplant 17(1):34–41
Juvet SC, Whatcott AG, Bushell AR, Wood KJ (2014) Harnessing regulatory T cells for clinical use in transplantation: the end of the beginning. Am J Transplant 14(4):750–763
van der Net JB, Bushell A, Wood KJ, Harden PN (2015) Regulatory T cells: first steps of clinical application in solid organ transplantation. Transpl Int
Putnam AL, Brusko TM, Lee MR, Liu W, Szot GL, Ghosh T, Atkinson MA, Bluestone JA (2009) Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes 58(3):652–662
Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G (2011) Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Transl Med 3(83):83ra42
Edozie FC, Nova-Lamperti EA, Povoleri GA, Scotta C, John S, Lombardi G, Afzali B (2014) Regulatory T-cell therapy in the induction of transplant tolerance: the issue of subpopulations. Transplantation 98(4):370–379
Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, Defor T, Levine BL, June CH, Rubinstein P et al (2011) Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood 117(3):1061–1070
Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, Del Papa B, Zei T, Ostini RI, Cecchini D et al (2011) Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 117(14):3921–3928
Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, Marek N, Mysliwska J, Hellmann A (2009) First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol 133(1):22–26
Geissler EK (2012) The ONE Study compares cell therapy products in organ transplantation: introduction to a review series on suppressive monocyte-derived cells. Transplant Res 1(1):11
Safinia N, Vaikunthanathan T, Fraser H, Scotta C, Lechler R, Lombardi G (2014) A GMP Treg expansion protocol restores Treg suppressor function in end-stage liver disease; implications for adoptive transfer therapy. Gut 63:A92–A93
Ferrer IR, Hester J, Bushell A, Wood KJ (2014) Induction of transplantation tolerance through regulatory cells: from mice to men. Immunol Rev 258(1):102–116
Wu T, Zhao Y, Zhao Y (2014) The roles of myeloid-derived suppressor cells in transplantation. Expert Rev Clin Immunol 10(10):1385–1394
Rosborough BR, Raich-Regue D, Turnquist HR, Thomson AW (2014) Regulatory myeloid cells in transplantation. Transplantation 97(4):367–379
Hutchinson JA, Riquelme P, Geissler EK, Fandrich F (2011) Human regulatory macrophages. Methods Mol Biol 677:181–192
Hutchinson JA, Riquelme P, Sawitzki B, Tomiuk S, Miqueu P, Zuhayra M, Oberg HH, Pascher A, Lutzen U, Janssen U et al (2011) Cutting edge: immunological consequences and trafficking of human regulatory macrophages administered to renal transplant recipients. J Immunol 187(5):2072–2078
Steinman RM, Banchereau J (2007) Taking dendritic cells into medicine. Nature 449(7161):419–426
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N et al (2007) Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 13(1):54–61
Jiga LP, Bauer TM, Chuang JJ, Opelz G, Terness P (2004) Generation of tolerogenic dendritic cells by treatment with mitomycin C: inhibition of allogeneic T-cell response is mediated by downregulation of ICAM-1, CD80, and CD86. Transplantation 77(11):1761–1764
Jiga LP, Ehser S, Kleist C, Opelz G, Terness P (2007) Inhibition of heart allograft rejection with mitomycin C-treated donor dendritic cells. Transplantation 83(3):347–350
Terness P, Oelert T, Ehser S, Chuang JJ, Lahdou I, Kleist C, Velten F, Hammerling GJ, Arnold B, Opelz G (2008) Mitomycin C-treated dendritic cells inactivate autoreactive T cells: toward the development of a tolerogenic vaccine in autoimmune diseases. Proc Natl Acad Sci U S A 105(47):18442–18447
Kleist C, Sandra-Petrescu F, Jiga L, Dittmar L, Mohr E, Greil J, Mier W, Becker LE, Lang P, Opelz G et al (2015) Generation of suppressive blood cells for control of allograft rejection. Clin Sci (Lond) 128(9):593–607
Radu CA, Kiefer J, Horn D, Kleist C, Dittmar L, Sandra F, Rebel M, Ryssel H, Koellensperger E, Gebhard MM et al (2012) Mitomycin-C-treated peripheral blood mononuclear cells (PBMCs) prolong allograft survival in composite tissue allotransplantation. J Surg Res 176(2):e95–e101
Liu L, Wang F, Zheng Y, Yuan X, Wang D, Zeng W, He X, Wang C, Deng S (2014) Pretreatment of transfused donor splenocytes and allografts with mitomycin C attenuates acute rejection in heart transplantation in mice. Transplant Proc 46(4):1169–1174
Funding
The TOL-1 study is funded by a grant from the German government (EXIST-Forschungstransfer: TolerogenixX, 03EFB BW56).
Conflicts of Interest
None.
Ethical approval
All procedures will perform in the planned TOL-1 study involving human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent will obtain from all individual participants included in the study.
Author information
Authors and Affiliations
Corresponding authors
Additional information
All authors drafted and critically revised the manuscript.
Rights and permissions
About this article

Cite this article
Morath, C., Schmitt, A., Zeier, M. et al. Cell therapy for immunosuppression after kidney transplantation. Langenbecks Arch Surg 400, 541–550 (2015). https://doi.org/10.1007/s00423-015-1313-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00423-015-1313-z