
Abstract
The cadherin switch has profound consequences on cancer invasion and metastasis. The endothelial-specific vascular endothelial cadherin (VE-cadherin) has been demonstrated in diverse cancer types including breast cancer and is supposed to modulate tumor progression and metastasis, but underlying mechanisms need to be better understood. First, we evaluated VE-cadherin expression by tissue microarray in 392 cases of breast cancer tumors and found a diverse expression and distribution of VE-cadherin. Experimental expression of fluorescence-tagged VE-cadherin (VE-EGFP) in undifferentiated, fibroblastoid and E-cadherin-negative MDA-231 (MDA-VE-EGFP) as well as in differentiated E-cadherin-positive MCF-7 human breast cancer cell lines (MCF-VE-EGFP), respectively, displayed differentiation-dependent functional differences. VE-EGFP expression reversed the fibroblastoid MDA-231 cells to an epithelial-like phenotype accompanied by increased β-catenin expression, actin and vimentin remodeling, increased cell spreading and barrier function and a reduced migration ability due to formation of VE-cadherin-mediated cell junctions. The effects were largely absent in both MDA-VE-EGFP and in control MCF-EGFP cell lines. However, MCF-7 cells displayed a VE-cadherin-independent planar cell polarity and directed cell migration that both developed in MDA-231 only after VE-EGFP expression. Furthermore, VE-cadherin expression had no effect on tumor cell proliferation in monocultures while co-culturing with endothelial cells enhanced tumor cell proliferation due to integration of the tumor cells into monolayer where they form VE-cadherin-mediated cell contacts with the endothelium. We propose an interactive VE-cadherin-based crosstalk that might activate proliferation-promoting signals. Together, our study shows a VE-cadherin-mediated cell dynamics and an endothelial-dependent proliferation in a differentiation-dependent manner.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metastasis is responsible for the majority of the cancer deaths in humans and can be categorized into a series of steps. During the initial stage of hematogenous metastasis, cells leave the primary tumor and enter the bloodstream, a process that occurs mostly via capillaries and post-capillary venules (Wyckoff et al. 2000). Cells that survive in the circulatory system can settle in different organs and form new tumors (metastatic tumors). However, the underlying mechanisms through which tumor cells are able to escape from the tumor nodule and pass the vascular endothelium in either direction are not fully understood. A critical protein in this process is epithelial cadherin (E-cadherin) whose loss of expression or function usually contributes to metastasis (Herzig et al. 2007). Down-regulation of E-cadherin has been shown to promote the disruption of epithelial cell–cell contacts and release of invasive tumor cells from the primary tumor (Hazan et al. 2004).
Cadherins belong to a large family of cell adhesion molecules that mediate cell–cell adhesion and contribute to control cell signaling, proliferation, migration, and polarity (Parri and Chiarugi 2010). Specifically, cadherins are trans-membrane cell adhesion receptors that mediate calcium-dependent cell–cell adhesion in a tissue-specific manner (Gumbiner 2005). The amino-terminal, extracellular domain of classical cadherins forms adhesion dimers/multimers between adjacent cells (Giepmans and van Ijzendoorn 2009) while the short carboxyl-terminal domain interacts indirectly with both the actin cytoskeleton, forming adherens junctions and intermediate filaments at desmosomes, respectively (Cavallaro and Christofori 2004). These interactions between cadherins and the cytoskeleton is important for maintaining tissue integrity in cells organized in sheet-forming layers such as epithelium.
Tumor development is frequently associated with a cadherin switch defined by losing E-cadherin and gaining other cadherin expression including N-cadherin (Rezaei et al. 2012), type II cadherins such as osteoblast cadherin (cadherin-11), or vascular endothelial cadherin (VE-cadherin) (Breier et al. 2014; van Roy 2014). There is evidence that the cadherin switch is accompanied by functional changes in cell adhesion, signal transduction, and subsequent tumor malignancy (Bill and Christofori 2015; Christofori 2003). Under physiological conditions, VE-cadherin is specifically expressed in endothelial cells (Lampugnani et al. 1992), while an aberrant expression of VE-cadherin has been documented in sarcoma or highly aggressive melanoma cells. Aberrant VE-cadherin expression was shown to increase tumor progression, metastasis, and seemed to be responsible for vasculogenic mimicry in melanoma (Hendrix et al. 2001; van der Schaft et al. 2005). Furthermore, our recent investigation on the mouse mammary carcinoma cell line (Ep5ExTu) introduced VE-cadherin as another player regarding the cadherin switch in breast cancer including its expression in human mammary carcinomas (Breier et al. 2014; Labelle et al. 2008). Furthermore, functional analyses revealed that VE-cadherin expression in this mouse mammary carcinoma model could promote tumor cell proliferation and invasion by stimulating TGF-β signaling (Labelle et al. 2008). Recent reports demonstrate an interdependent dynamics between VE-cadherin and actin-driven junction-associated intermittent lamellipodia (JAIL) structures that control the VE-cadherin dynamics in cell culture (Abu Taha et al. 2014) and in angiogenesis (Cao et al.). These studies were able to demonstrate a direct relationship between the VE-cadherin concentration on the cell contacts and cell contact dynamics, a finding that could play an important role in the tumor progression of VE-cadherin-positive tumors. Thus, we investigate the mechanisms through which VE-cadherin modulates breast cancer tumor cell features.
We demonstrate significant correlations between VE-cadherin expression and tumor sizes (expressed as pT-stage) as well as lymph node stage and estrogen receptor expression. Mechanistic analyses of forced VE-cadherin expression in aggressive and poorly differentiated MDA-231 cells (Mbalaviele et al. 1996) revealed that VE-cadherin promotes multiple aspects of mesenchymal-to-epithelial transition (MET). In contrast, VE-cadherin expression in the weakly invasive and poorly metastatic, E-cadherin expressing breast cancer cell line MCF-7 (Hazan et al. 2000) had less functional impact on parameters that control the invasive potential of MCF-7 cells. These findings highlight a differentiated impact of VE-cadherin in breast cancer tumor progression.
Materials and methods
Tissue microarray on human breast cancer tissue and immunofluorescent staining of frozen tumor sections
The expression of VE-cadherin, estrogen and progesterone receptor, and HER-2/neu was analyzed in 392 formalin-fixed, paraffin-embedded specimens of invasive breast carcinoma using tissue microarrays as described elsewhere (Kallioniemi et al. 2001; Rezaei et al. 2012). Two cores of different locations represented each case. The clinicopathological features of breast cancer cases are summarized (Additional file 1, table 1a). Immune histochemical markers that were used for the studies are listed (Additional file 2). All staining results were scored semiquantitatively through the percentage estimation of tumor cells expressing the markers. The classification for each marker is listed (Additional file 1). A tumor was scored as VE-cadherin positive if 10% or more of the tumor cells were positive (cut off). According to the current guidelines for diagnosis and therapy of breast cancer, a tumor was scored as estrogen or progesterone receptor positive when at least 1% of the tumor cells were estrogen or progesterone receptors positive. The HER2 staining was classified according to the ASCO recommendations 2013 (Wolff et al. 2013). In cases with equivocal immunohistochemical results, fluorescence in situ hybridization was performed for the final HER-2 stage, according to the ASCO recommendations 2013 (Wolff et al. 2013).
For immune fluorescence staining 5 μm frozen sections were cut and air-dried. Sections were then fixed in 100% acetone for 10 min at − 20 °C and air-dried. Rehydrated sections were stained with VE-cadherin and E-cadherin antibodies and detected by donkey-anti-goat Alexa 568 and donkey-anti-mouse Alexa 488, respectively. Ethical approval for the study was granted from the research ethics committee of the Dresden University Hospital “Carl Gustav Carus” (No. 59032007).
Cell culture
The MCF-7 cell line was kindly provided by Prof. Martin Götte (Department of Gynecology and Obstetrics, Münster, Germany) and maintained in RPMI (Life technologies, CA) with 10% fetal calf serum (FCS). MDA-231 cells were obtained from ATCC and were cultured in Dulbecco’s modified Eagle's medium (Life technologies, CA) supplemented with 10% fetal calf serum (FCS). Human umbilical cord vein endothelial cells (HUVEC) were isolated as described elsewhere (Kronstein et al. 2012) according to the principles outlined in the Declaration of Helsinki; this was approved by the ethics boards of the WW-University of Muenster (2009-537-f-S).
Generation of VE-cadherin expressing cell lines
Viral particles were produced by transient co-transfection of 293T cells with the lenti-vector pFUGW encoding EGFP-tagged full-length VE-cadherin (VE-cad-EGFP), psPAX2, and pMD2G as described elsewhere (Abu Taha et al. 2014; Kronstein et al. 2012). MCF-7 and MDA-231 cells were transduced with the VE-cadherin encoding virus particles. Stable cells expressing VE-cadherin were selected by flow cytometry (CyFlow Space, Sysmex Partec, Münster, Germany) using VE-cadherin-EGFP as the label.
Cell proliferation assay
30 × 103 cells were plated in 96-well plates and labeled with bromodeoxyuridine (BrdU). After 24, 48 or 72 h, cell proliferation was quantified using a colorimetric immunoassay based on BrdU incorporation and according to manufacturer’s instructions (Roche Mannheim, Germany). For each independent experiment, six wells per condition were used and the optical density of each well was measured with the ELISA plate reader (Thermo Scientific™ Multiskan™) at 450 nm (reference wave length: 690 nm).
Label-free quantification of cell migration, morphology and growth using Zernike phase contrast and digital holographic microscopy
The effects of VE-cadherin expression on cell migration, motility, morphology, and growth were observed using in vitro wound assays (ibidi µDish Petri dishes with culture inserts and glass lids, ibidi GmbH, Martinsried, Germany). To that end, 3 × 104 of MCF-7 cells (MCF-EGFP and MCF-VE-EGFP) or MDA-231 cells (MDA-EGFP and MDA-VE-EGFP) were, respectively, seeded into the insert chambers and incubated for 24 h until reaching 60–80% confluence. After removing the inserts, the medium was carefully removed. The remaining cell layers were washed with medium to remove dead cells and were, afterwards, supplemented with 2 ml fresh medium.
Time-lapse observation of wound closure was performed label-free by either Zernike phase microscopy using a fully automated microscope (Observer Z1, Zeiss Göttingen), or by using a custom-built module for quantitative phase contrast imaging with digital holographic microscopy (DHM) (Kemper et al. 2006) which was attached to the same microscope (Odenthal-Schnittler et al. 2016).
Migration and motility were quantified by single cell tracking using custom-built C++-software, based on an algorithm as described elsewhere (Kemper et al. 2010). While DHM quantitative phase images were directly evaluated with the cell tracking software, the gray level images from Zernike phase microscopy time-lapse series were preprocessed by image inversion and corrected for background intensity using the software ImageJ (NIH, Bethesda, MD, USA). From the resulting single cell migration trajectories, the mean squared displacement (Sridharan et al. 2011) and the maximum migration distance were calculated. Moreover, using the freely available software Chemotaxis and Migration Tool V2.0 (http://www.ibidi.com), the directness and forward migration index were determined.
For quantification of cell morphology and growth quantitative DHM phase contrast, images were evaluated for the cell occupied area and average cell thickness through image segmentation-based analysis as described in Bettenworth et al. (2014) using the free software cell profiler (http://www.cellprofiler.org).
Adhesion and proliferation assays of tumor cells to vascular endothelium
To accomplish a separate cell count of tumor cells and endothelium cells, the breast cancer cells were labelled with 2.5 μg/mL DiI (Invitrogen) for 30 min according to the manufacturer’s instructions. Labeled cancer cells were plated on HUVEC cell monolayer and incubated for 1 h or 3 h at 37 °C. After a three times wash with phosphate buffered saline (PBS) samples were dissolved in 2% sodium dodecyl sulfate containing buffer. Fluorescence intensity was measured using Tecan GENios microplate reader (Tecan, Salzburg, Austria). Co-cultures and proliferation assay was performed by plating DiI-labelled cancer cells on HUVEC monolayers followed by incubation for 24, 48, 72 or 96 h. At appropriate time points, cells were trypsinized and the number of labeled cells was determined by flow cytometry using the CyFlow Cube 8 instrument (Partec, Münster, Germany).
RNA isolation and reverse transcription-PCR analysis
2 × 105 of MCF-7 or MDA-231 cells were seeded in 6-cm dishes 1 day before RNA isolation. Total RNA was isolated from cell lysates using a universal RNA Purification Kit, according to the manufacturer’s protocol (Roboklon, Berlin, Germany). Aliquots of 3 μg of total RNA were reverse transcribed using a Reverse Transcriptase Core Kit (Eurogentec, Seraing, Belgium). Real-time PCR reactions were performed using the Mastercycler ep Realplex (Eppendorf, Hamburg, Germany) and the amplifications were done using the SYBR Green PCR Kit (Qiagen, Valencia, CA, USA). All reactions were run in duplicates and Ct values were normalized against the GAPDH gene, using the delta–delta-Ct method.
Immune labeling and Western blot
Samples were fixed using 2% freshly prepared paraformaldehyde and processed for indirect immune fluorescence microscopy using goat anti-human VE-cadherin (Santa Cruz Biotechnology, Inc., Dallas, Texas USA), mouse anti-cadherin 11 (Invitrogen. Eugene, Oregon, USA), mouse anti-β-catenin (BD Biosciences, Bedford, MA, USA), mouse anti-E-cadherin (BD Biosciences, Bedford, MA, USA), and appropriate secondary antibodies such as donkey-anti-goat Alexa 568, donkey-anti-mouse Alexa 568, and donkey-anti-mouse Alexa 488 (Invitrogen. Eugene, Oregon, USA). After three times rinsing with PBS, cultures were mounted in Dako fluorescent mounting medium (Agilent Technologies, Dusseldorf, Germany) and images were acquired using a Zeiss LSM 780 confocal imaging system (Carl Zeiss, Jena, Germany) at the respective wavelengths.
Prior to Western blot analyses, total protein was determined in SDS-sample buffer using amido black staining protocol (Dieckmann-Schuppert and Schnittler 1997). Samples were separated on 10% SDS gel and transferred by wet blotting onto nitrocellulose membrane (Whatman, Dassel, Germany) before subsequently being incubated with the respective antibodies, washed three times, and incubated with IRDye Infrared Dyes (IRDYE 800CW donkey anti-goat, IRDYE 800CW donkey anti-mouse) antibodies. Bands were detected using a LI-COR scanner (LI-COR Biosciences, Lincoln, NE, USA). The average intensity of normalized values was plotted as a function of total the amount of protein.
Determination of barrier function by impedance spectroscopy and transwell filter assay
Trans-cellular electrical resistance (TER) in cell layers was determined at 37 °C using an impedance spectroscopy setup with TER analytical software (MOS-Technologies, Telgte, Germany) (Seebach et al. 2000). Impedance spectroscopy is a high sensitive biophysical method that allows continuous recording of the para-cellular barrier function in cell culture models. Because of its high sensitivity and accuracy, this method is capable of detecting even small changes in barrier function since it is independent of tracer size (Seebach et al. 2000; Wegener and Seebach 2014). In brief, cancer cells were cultured on measuring chambers (Kronstein et al. 2012) and TER was calculated from impedance spectroscopy measurements over the entire time period of 24 h.
Transwell filter permeability assay was performed at 37 °C by using fluorescein isothiocyanate (FITC)-labeled dextran (Sigma, Deisenhofen, Germany) as the tracer substance as described elsewhere (Wahl-Jensen et al. 2005). Briefly, MDA and MDA-VE-EGFP cells were grown on transwell filters with 3 µm pore size. After application of 0.5 µg/ml of FITC-dextran (average MW 4 kDa, neutral charge) to the upper compartment the fluorescence intensity was determined in the lower compartment by photometry after 30 and 60 min, respectively.
Statistics
For all cell culture experiments, statistical significance was determined using SigmaPlot and GraphPad Software (Prism v4.02). Statistical significance was evaluated by Student’s t test (Prism v4.02), with p value < 0.05 considered as statistically significant. The results of immunohistochemistry were analyzed by Chi-squared test by SPSS (SPSS, Munich, Germany). Data were considered significant by p < 0.05.
Results
Correlation between VE-cadherin expression and clinico-pathological features
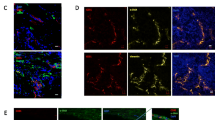
The expression of VE-cadherin in invasive human breast cancer tissue was evaluated and correlated with clincopathological features in special type (ST) (Ex: invasive lobular, invasive tubular, and invasive mucinous) and non-special type tumors (NST) (70% of all cases) by tissue microarray analyses. Since the appearance of VE-cadherin in the tumors was heterogeneous, we considered a tumor VE-cadherin positive if more than 10% of cells expressed VE-cadherin. More detailed analyses uncovered that 60% out of 392 investigated tumors were VE-cadherin positive. VE-cadherin localized in tumor cells at three subcellular sites; it was found in all cases in the cytoplasm, whereas the cell membrane localization occurred in 5% and the nuclear staining in 1% of the VE-cadherin-positive cells (Fig. 1a). These data suggest an increased turnover of VE-cadherin in the tumors as VE-cadherin in endothelium usually undergoes a cyclic turnover (Xiao et al. 2005). In addition, it appears that the proportion of the VE-cadherin-positive tumor cells in the tumors showed a tumor-size dependency. In particular, breast cancer tumors with a size < 2 cm (pT1) displayed 70% of VE-cadherin-positive cells while tumors with a size > 2 cm; (pT2-4) displayed 52% VE-cadherin-positive cells (p = 0.0003). This interesting result might relate to a VE-cadherin dependent control of cell proliferation and apoptosis in tumor cells, since VE-cadherin controls was shown to be involved in cell proliferation control in endothelium (Baumeister et al. 2005). Furthermore, it appears that the tumor differentiation impacts VE-cadherin expression as well. In particular, 82% of highly differentiated cases were VE-cadherin positive while 63% of the moderate differentiated tumors were VE-cadherin positive and only 53% of low differentiated tumors were found to express VE-cadherin (p = 0.0009). For further characterization of VE-cadherin expressing tumor cells, we looked for the relationship between VE-cadherin and estrogen receptor expression. The expression of estrogen receptor (ER) correlated with cytoplasmic VE-cadherin tumors in 65% of the cases while ER-negative tumors displayed 50% VE-cadherin positivity (p = 0.005) (Additional file 1, table 1a and table 1b). Since 392 cases were examined, we assume a moderate relationship between VE-cadherin and estrogen receptor expression. Since VE-cadherin is thought to modulate breast cancer cell invasiveness, we evaluated the invasive tumor front for VE-cadherin expression. Indeed, we found VE-cadherin-positive cells at the invasive tumor front and throughout the tumor (Fig. 1b). The epithelial mesenchymal transition (EMT) has been shown to be accompanied by a cadherin switch, which can include the loss of E-cadherin and the expression of other cadherins such as VE-cadherin in breast cancer cells. Thus, we performed a double immune fluorescent labeling of E-cadherin and VE-cadherin in 14 different breast cancer samples. We found VE-cadherin and E-cadherin co-expressed in the same tumors (Fig. 1c). Some parts of tumors were positive for both VE-cadherin and E-cadherin (Fig. 1c, left panel) while other parts were positive only for E-cadherin (Fig. 1c, middle panel) or VE-cadherin, respectively (Fig. 1c, right panel). Together the data give strong hints that VE-cadherin expression in breast cancer tumors depends on the tumor size and most importantly on the level of tumor differentiation.

VE-cadherin is expressed in human breast cancer tumors. a Representative immune histochemical labeling (IHC) of VE-cadherin in tumor tissues among 392 formalin-fixed, paraffin-embedded specimens of invasive breast carcinomas. Cytoplasmic VE-cadherin labeling was seen in all VE-cadherin expressing tumor cells. Some cases also displayed nuclear staining (1%) and membrane localization (5%). The boxed region in the merged figures is shown at higher magnification below. Bar 20 µm. b Two examples of invasion fronts of breast carcinomas (no special type) with weak-to-moderate VE-cadherin expression (left panel) and with strong VE-cadherin expression (right panel). Bar 100 µm. c Immune fluorescent staining of E-cadherin (green) and VE-cadherin (red) in human frozen section. Bar 30 µm. The boxed region in the merged figures is shown at higher magnification below Bar 10 μm
VE-cadherin-EGFP expression in MDA-231 and MCF cell lines modulates barrier function and morphology differentiation dependently
Based on the data obtained so far, we aim to investigate the mechanistic impact of VE-cadherin expression in two differently differentiated invasive human mamma carcinoma cell lines. In particular, we used the VE-cadherin-negative human breast carcinoma cell lines MCF-7 and MDA-231. While the E-cadherin-expressing MCF-7 cells display an epithelial-like morphology and are considered differentiated, the MDA-231 cell line is fibroblastoid-like, cadherin-11 positive, and poorly differentiated (Additional file 1, table 1c) (Nieman et al. 1999). For ectopic expression of VE-cadherin EGFP-tagged VE-cadherin was generated (VE-EGFP), which allows the identification of those cells easily. The construct was cloned into a lentivral vector (Abu Taha et al. 2014; Kronstein et al. 2012) and transduced in the respective cell lines. Due to the integrase-activity of the lentiviral vector, the transduced cells stably express the VE-EGFP that localized at the at cell junctions (MCF-VE-EGFP and MDA-VE-EGFP) (Fig. 2a), which demonstrates a functional VE-EGFP protein. VE-EGFP expressing MCF-VE-EGFP cells retained overall epithelial morphology (Fig. 2b), but a decrease in E-cadherin labeling at cell junctions was observed, which is in agreement with decreased total expression (Additional file 3a and 3b). In contrast, VE-EGFP expression in MDA-231 cells converted the fibroblastoid morphology into an epithelial-like phenotype (Fig. 2d) while decreasing cadherin-11 expression (Additional file 3c and 3d). Furthermore, recruitment of actin filaments to cell junctions in MDA-VE-EGFP cells facilitates cell junction stabilization in epithelium. Furthermore, VE-EGFP expression in MDA cells also reorganized vimentin intermediate filaments from a clumped, irregular patterning to a filamentous organized structure in the MDA-VE-EGFP expressing cells (Additional file 4a and 4b). The data are in agreement with the notion that VE-cadherin indirectly (via desmoplakin I and II) binds to vimentin (Valiron et al. 1996). The VE-cadherin-mediated change in morphological and cytoskeletal distribution is also reflected by changes in barrier function properties verified through the determination of transcellular electrical resistance (TER) which was determined from high sensitive and reliable impedance spectroscopy measurements. This technique allows the determination of barrier function by high accuracy and a time resolution of about 1 min (Seebach et al. 2000; Wegener and Seebach 2014). The TER increased during cell growth in MCF-VE-EGFP expressing and control cell lines (MCF-EGFP), but displayed no significant difference (Fig. 2c). In contrast, VE-EGFP expression in MDA-231 cells increased barrier function fourfold with a moderate TER-increase during cell growth (Fig. 2e). This result was totally confirmed using FITC-dextran as a tracer substance in a trans well filter (Fig. 2f), indicating that VE-cadherin expressed in undifferentiated MCF-7 tumor cells are able to form a competent barrier function. Together, the data demonstrate that VE-EGFP expression induces a barrier function accompanied by changes in actin and vimentin cytoskeletal organization and thus modulates the less differentiated tumor cells while the E-cadherin expressing and differentiated tumor cells displayed a moderate change only. The data suggest that the effect of VE-cadherin on the tumor cell response depends on the differentiation status of the tumor.

Ectopic expressed VE-cadherin in tumor cell lines forms cell junctions. a Immune localization of VE-cadherin in MCF-7 and MDA-231 cells expressing either green fluorescent protein (EGFP) (MCF-EGFP and MDA-EGFP) alone or VE-cadherin-EGFP (MCF-VE-EGFP and MDA-VE-EGFP). Human umbilical vein endothelial cells (HUVECs) were used as positive control for VE-cadherin expression. Nuclei are labeled by DAPI. Bar, 20 μm. b, d Phase contrast microscopy during cell growth of control cell lines (MCF-EGF and MDA-EGFP) and VE-cadherin expressing cell lines (MCF-VE-EGFP and MDA-EGFP). Bar, 50 μm. c, e Growth-dependent development of the trans cellular electrical resistance (TER) calculated from impedance spectroscopy measurements. f FITC-dextran permeability measured on confluent MCF-7 EGFP and MCF-VE EGFP cells cultured on a filter with a pore size of 3 μm (trans-well filter assay). FITC-dextran was applied to the upper compartment and the fluorescence intensity was measured in the lower compartment after 30 and 60 min, respectively. All data shown are based on three independent experiments. Mean values ± SDM is shown; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001
Impact of VE-cadherin expression on cell migration in MDA-231 and MCF-7 cells
Since cell migration is a key step in cancer invasion and metastasis, we investigated migration activity of the cell lines in a 2D migration assay. As expected from the morphological phenotype, the controls (MCF-EGFP) and MCF-VE-EGFP exhibited collective epithelial sheet migration (Fig. 3a). However, VE-cadherin expression in MCF-VE-EGFP decreased the directional migration of the cells (Fig. 3b, c), a phenomenon that might be related to competition between the E- and VE-cadherin. However, the overall cell velocity remained unchanged (Fig. 3a, d).

VE-cadherin expression in tumor cell lines modulates collective and directed sheet migration. Phase-contrast based time-lapse imaging of tumor cell migration of a MCF-7 cell lines (MCF-EGFP and MCF-VE-EGFP) and e MDA-231 cell lines (MDA-EGFP and MDA-VE-EGFP) in an in vitro scratch assay for 20 h. Quantification of the cell motility is indicated by b, f track plots, c, g forward migration index (x, y) directness and d, h cell velocity. p ≤ 0.05. Quantification was performed on 27 cells in each experiment. n = 3 independent experiments. Mean values ± SDM is shown; *p ≤ 0.05; ***p ≤ 0.001
In contrast, MDA-EGFP migration appears to be randomized with cells frequently migrating as individuals into the cell-free area, sometimes turning around and even spontaneously changing direction (Fig. 3e, f). The expression of VE-cadherin-EGFP led to the formation of cell contacts with the consequence of a directed sheet migration in the scratch assay (Fig. 3e–g). In addition, the overall cell velocity decreased by about 30% compared to the control cells (Fig. 3h). The observed change in the morphodynamic parameters corresponds to the phenomenon of VE-cadherin-mediated cell contact formation and can conclusively explain the altered migration properties of the MDA-cells. The effect on MCF-7 cells was restricted to the directness of cell migration while the migration velocity remained unchanged (Fig. 3d). To verify the effect of VE-cadherin to this end, we investigated the migration of MDA-VE-EGFP single cells and found an increase in single cell motility as well as (Additional file 5). The data indicate that the influence of VE-cadherin on cell migration depends on the differentiation status of the tumor cells.
In view of the changed directionality in VE-cadherin-expressing tumor cells, in particular the MDA-231 cells, we have considered the polar distribution of the rac-mediated protrusion formation. Using a scratch assay, the control cultures (MDA-EGFP) showed typical fibroblastoid structure with many randomized-appearing actin filament-positive protrusions that co-distributed with a Rac1 antibody (Additional file 6b). In contrast, MDA-VE-EGFP cells, however, changed the overall morphology through the formation of polarized Rac-1-positive actin-driven lamellipodia at the wound rim, confirming directed cell migration activity (Additional file 6a, 6b). The data indicate that VE-cadherin expression in breast cancer tumor cells has a significant impact on cell migration involving polarized Rac-1 activity.
Modulation of cell spreading and 3-D cell morphology by VE-cadherin expression
Next we investigated cell spreading and dynamic development of average cell thickness, which was shown to be changed during cell migration and during scratch healing by quantitative phase imaging using digital holographic microscopy (DHM) (Bettenworth et al. 2014). Quantitative phase imaging is a label-free method that can be used to dynamically measure cell thickness and cell surface area at high resolution. Compared to the controls (MCF-EGFP), MCF-VE-EGFP displayed a minor increase of cell thickness and induced a slightly more compact cell structure (Fig. 4a–c, Additional file 7a). In contrast, VE-cadherin expression in MDA-231 cells reduced the cell thickness by about 50% (Figs. 4d, f, 5e) and increased the surface area covered by single cells (see Additional file 7b). Data are also indicative for a VE-EGFP-mediated increase in cell spreading capabilities. Since cell spreading and motility depend on cell junction and cell substrate interactions, we quantitatively investigated vinculin expression, a protein component of focal contacts and cell junctions (Sumida et al. 2011). Increased vinculin expression was indeed found in MCF-VE-EGFP and MDA-VE-EGFP when compared to control cells (Fig. 4h, i). Furthermore, immune fluorescent staining localized vinculin in MCF-VE-EGFP cells in focal adhesion, and in MDA-VE-EGFP cells in focal adhesion and cell junctions in irregular pattern (Fig. 4g). In summary, the data indicate that VE-cadherin expression in MDA-231 modulates not only cell adhesion, but also cell substrate and cell spreading capabilities, parameters that may impact the adhesive properties of the tumor cells.

Tumor cell migration and 3-D cell morphology of tumor cells tested in a scratch assay. a, d Tumor cells as indicated were cultured until confluence. After setting a scratch, time-lapse recordings acquired by digital holographic microscopy (DHM) was performed. Bar, 30 µm. b, e Time course of wound closure over a period of 25 h. c, f The average cell layer thickness was calculated during wound closure over 25 h. g Immune fluorescence localization of vinculin in control and VE-cadherin expressing cell lines as indicated. The boxed region in the merged figures is shown at higher magnification to the right. Nuclei are staining with DAPI (blue). Bar, 20 μm. h, i Western blot-based quantitative analyses of vinculin related to tubulin as internal control. Mean values ± SDM is shown; *p ≤ 0.05

VE-cadherin modulates expression of β-catenin, cyclin-D and C-myc. a Immune fluorescence labeling of β-catenin in the MCF-7 and MDA-231 cells expressing either EGFP or VE-cadherin-EGFP as indicated. Nuclear DNA staining by DAPI (blue). Bar, 20 μm. b The diagrams demonstrate the relative β-catenin protein amount based by Western blotting and mRNA levels determined by rtPCR as indicated. d, e The diagrams show c-myc and cyclin D1 mRNA expression levels, analyzed by Western blotting. Data shown are based on three independent experiments. f Cell migration of tumor cells as indicated in the absence and presence of 10 µM Y27632. Analyses of migration was based on phase-contrast time-lapse recordings by manual tracking of the nuclei using Fiji “Manual Tracking” plugin and the “Chemotaxis and Migration” Tool. A mean of 50 cells of each group was tracked over a time period of 12 h. Mean values ± SDM is shown; **p ≤ 0.01; ***p ≤ 0.001
VE-cadherin expression in tumor cell modulates cell junction formation but does not impact cell proliferation
It has been shown that VE-cadherin expression in endothelial cells significantly decreases β-catenin translocation to the nucleus, thereby reducing its transcriptional activities in endothelium (Giampietro et al. 2012; Taddei et al. 2008). On the other hand, the expression of VE-cadherin in VE-cadherin-free cells leads to a strong β-catenin expression, as shown, for example, in CHO cells (Breviario et al. 1995; Kronstein et al. 2012). Same phenomenon could be observed in MDA-VE-EGFP cells that exhibited and dramatic increase in expression of β-catenin while mRNA level remained constant (Fig. 5a–c). The MDA-VE-EGFP but not the MDA-EGFP cells formed VE-cadherin-mediated cell junctions with recruitment of β-catenin (Fig. 5a). In MCF-7 cell lines, β-catenin was already highly expressed and did not increase upon expression of VE-cadherin (Fig. 5a–c). Furthermore, MCF-7 cells did not significantly modulate the overall organization of cell junctions, a phenomenon that most likely relates to the endogenous E-cadherin expression that recruited β-catenin to the junctions already. However, β-catenin is an essential player in Wnt signaling that is involved in control of proliferation. Surprisingly, VE-cadherin expression in MCF-7 and nor MDA-231 cells did not influence cell proliferation (see Additional file 8a and 8b) irrespective that the mRNA of the cell cycle regulators cyclin-D1 and c-myc (Swarbrick et al. 2005), both are involved in several proliferation controlling pathways including Wnt/β-catenin signaling (Ozaki et al. 2005), were significantly down-regulated in the MDA-VE-EGFP cells (Fig. 5d, e). However, previous work demonstrated that down-regulation of cyclin D1 and c-myc increased migration capacity in breast cancer and has been linked to unfavorable prognostic features (Alfano et al. 2010; Lehn et al. 2010; Liu et al. 2012; Tobin et al. 2011). Other work demonstrated that cyclin acts in fibroblasts via inhibition of the Rho-activated kinase II (ROCK II) (Li et al. 2006). Thus, we treated the cells with 10 µM ROCK inhibitor Y27632 and found a tendency of increased migration velocity in all cases, but the data remained insignificant (Fig. 5f). Thus, the expression of cyclin D1 and c-myc somehow seems to facilitate cell dynamics in VE-cadherin-expressing tumor cells but the effect most likely requires additional mechanisms.
VE-cadherin expressing breast cancer cell lines integrate into endothelial monolayers and form adherens junction-like cell contacts with the endothelium, which in turn facilitate tumor cell proliferation
In endothelium VE-cadherin mediates homotypic adherens junction type intercellular junctions. Thus, we aimed to investigate whether VE-cadherin expressing tumor cells might be able to directly interact with cultured vascular endothelial cells. Indeed, VE-cadherin expression in both MCF-7 and MDA-231 cells significantly increased the adhesion of cancer cells to HUVEC monolayer compared to EGFP expressing cells (Fig. 6a). Furthermore, co-culturing MCF-VE-EGFP and MDA-VE-EGFP cells with HUVEC increased cancer cell proliferation (Fig. 5b, c, e). This effect was independent of soluble mediators secreted by HUVEC, as HUVEC-conditioned medium had no effect on tumor cell proliferation (Fig. 5d, f). Importantly we documented that MCF-VE-EGFP and MDA-VE-EGFP become incorporated into the endothelial cell monolayers, forming VE-cadherin-based tumor–endothelial cell junctions (Fig. 6a). These junctions seem to be competent as a-E-catenin displayed a colocalization with VE-cadherin (Fig. 6b, c). The data document a direct junction-based interaction between cancer cells and HUVEC, a fact that might be responsible for tumor cell proliferation due to VE-cadherin promoting signaling. This idea is further supported by appearance of VE-cadherin/α-E-catenin containing vesicle-like structures in the tumor cells that might be due to endocytosis (Fig. 6b, c). We further evaluated the impact of VE-cadherin in a human breast cancer cell line, SUM149PT, that natively expresses VE-cadherin. Silencing of VE-cadherin in this cell line by short hairpin RNA targeting human VE-cadherin mRNA reduced the adhesion of cells to HUVEC cultures by approximately 15% (data not shown). Together, the results further support the significant impact of VE-cadherin on tumor cell adhesion to endothelial cells, which seems to provide the conditions of tumor proliferation via VE-cadherin.

Cell junction formation of VE-cadherin-expressing tumor cells with HUVEC monolayer facilitate tumor cell proliferation. a To assess breast cancer adhesion capacity to HUVEC monolayer, control cell line (MCF-EGFP and MDA-EGFP) and VE-cadherin expressing cell lines (MCF-VE-EGFP and MDA-VE-EGFP) were stained with DiI and added to HUVEC monolayers. Microplate reader measured the intensity of the fluorescent signal in the cell lysates. b, c VE-cadherin-EGFP expressing tumor cells (green) were added to HUVEC monolayer for around 20 h and subsequently labeled for VE-cadherin (red) and α-E-catenin (magenta) as indicated. Both VE-cadherin-EGFP expressing cell lines integrate into endothelial monolayer and form competent cell junctions as visible by co-distributed labels. c, e Quantitative analysis of proliferation of MCF-7 cell lines (MCF-EGFP and MCF-VE-EGFP) and MDA-231 cell lines (MDA-EGFP and MDA-VE-EGFP) that were co-cultured with endothelium (integrated) or d, f treated with conditioned medium, as indicated. *VE-cadherin-EGFP expressing tumor cells, Bar 10 µm, Mean values ± SDM is shown; **p ≤ 0.01; ***p ≤ 0.001
Discussion
Changes in cell adhesion and the cadherin switch play a critical role in cancer progression and metastasis (Hazan et al. 2004). In particular, loss of E-cadherin expression in cancer cells and the acquisition of N-cadherin are believed to be one of the major events involved in tumor progression and metastasis (Ziober et al. 2006). In this study, we focused on the functional impact of VE-cadherin, a novel player in the cadherin switch, in human breast carcinoma. Based on the patient data, we detected VE-cadherin expression in 60% of breast cancer specimens. Thus, the analysis of a large number of human tumors (392) allowed a more precise determination of VE-cadherin expression in breast cancer as compared to our previous study, where we observed 27 VE-cadherin-positive tumors out of 37 (73%) (Labelle et al. 2008). However, cytoplasmic VE-cadherin occurred at much higher frequency compared to membranous labeling (5%), a result indicative for a higher turn-over due to internalization, and/or proteolysis/degradation of the protein, resulting in a loss of adhesion function. A comparable phenomenon has been reported for E-cadherin during tumor progression (David and Rajasekaran 2012), demonstrating a release of soluble E-cadherin ectodomain which has been correlated with enhanced cancer cell invasion (David and Rajasekaran 2012). It is tempting to speculate that the VE-cadherin protein on breast cancer cells is similarly cleaved to release an extracellular domain, a process that was described during apoptosis (Herren et al. 1998). Interestingly, glycosylated VE-cadherin has been indicated as a serum biomarker for metastatic breast cancer, in particular in estrogen receptor-positive breast cancers with vascular invasion (Fry et al. 2013, 2016). These observations are consistent with the hypothesis that a soluble VE-cadherin ectodomain is released from both breast cancer and endothelial cells in tumor vessels. However, further studies are required to address this issue.
During tumor development, a few tumor types such as melanomas and certain sarcomas have been reported to aberrantly express VE-cadherin, which may be responsible for tubular structure formation and vasculogenic mimicry (Hendrix et al. 2001; van der Schaft et al. 2005). In a mouse breast cancer model, we have previously shown that VE-cadherin is induced in breast cancer cells during EMT early in tumor progression (Labelle et al. 2008). However, here we demonstrate that a number of breast cancer tumor cells expressed both E-cadherin and VE-cadherin. Since E-cadherin was described to modulate tumor progression (Bill and Christofori 2015; Christofori 2003), we aimed to investigate if VE-cadherin expression differently changes the cell behavior in an E-cadherin- and differentiation-dependent manner. Indeed we found significant differences in the impact of VE-cadherin ectopic expressed in MDA-231 and MCF-cells. This includes cell migration, cell spreading, barrier function, and tumor cell/endothelial cell interactions; all processes critical to metastasis. Since endothelial cells are of mesenchymal origin, expression of VE-cadherin in epithelium might reflect de- or re-differentiation. Indeed, exogenous expression of VE-cadherin in human MDA-231 cells which were derived from aggressive advanced tumors, initiated a shift from a fibroblastoid to a more epithelial-like phenotype, a process that resemble mesenchymal-to-epithelial transition (MET), a process critical in metastasis (Christofori 2003). The morphological and functional changes induced by VE-cadherin in MDA-231 cells are due to the formation of functionally competent cell junctions as indicated by the upregulation of barrier function properties, coordinated sheet migration and integration of tumor cells into the HUVEC monolayer.
The most impressive changes induced by VE-cadherin expression were found in MDA-231 that displayed an increased ß-catenin expression that recruited to VE-cadherin, which together indicates a competent cell junction complex. This complex is linked to the actin filament and intermediate filaments system that in turn stabilizes cell junctions and contribute to cell shape change (Nelson and Chen 2003). Additionally, the VE-cadherin induced reorganization of the intermediate filament protein vimentin into a filamentous structure is a further indicator for a mesenchymal differentiation process and probably also stabilizes entire cells and cell junctions (Schnittler et al. 1998; Valiron et al. 1996). The lower effect of VE-cadherin expression that we observed in the well-differentiated MCF-7 tumor cell line could be associated with the co-expression of VE- and E-cadherin, irrespective of slightly diminished expression levels of E-cadherin in MCF-VE-EGFP. Our previous work has shown a down-regulation of E-cadherin expression and induction of VE-cadherin expression during TGF-ß1-mediated EMT (Labelle et al. 2008). However, it remains questionable if E-cadherin and VE-cadherin compete for expression in human breast cancer tumors because our analyses of various human breast cancer cell lines revealed frequent co-expression of both cadherins (own unpublished observations of GB), and we observed only a moderate down regulation of E-cadherin after VE-cadherin expression in MCF-7 cells.
Changes in intercellular adhesive properties of tumor cells and their interactions with the ECM contribute to their spread, survival, and subsequent growth at metastatic sites (Bendas and Borsig 2012; Givant-Horwitz et al. 2004). VE-cadherin is a major component of endothelial adherens junctions (Giannotta et al. 2013) and its dynamics plays a key role in the maintenance of vascular integrity (Abu Taha et al. 2014; Giannotta et al. 2013; Breslin et al. 2016; Seebach et al. 2015). In endothelial cells, VE-cadherin regulates cytoskeletal tension, cell spreading, and focal adhesions by stimulating RhoA (Nelson et al. 2004). Similarly, in both MCF-7 and MDA-231 cells, forced VE-cadherin expression results in functional intercellular cell adhesion. However, this effect is more prominent in MDA-231 cells lacking E-cadherin expression when compared to MCF-7 cells. VE-cadherin expression in MDA-231 cells not only resulted in tight, epithelial-like cell junctions, but also influenced its interaction with the ECM. In endothelial cells, vinculin, a molecule found at cell junctions and cell substrate adhesion sites is suggested to associate with VE-cadherin and focal contacts to control force-dependent remodeling (Huveneers et al. 2012; Goldmann 2016).
Since vinculin was upregulated in MDA-231 cells upon VE-cadherin expression it seems reasonable to assume that vinculin contributes to dynamics of both intercellular adhesion and cell–matrix adhesions in cancer cells, which in turn might modulate cell migration abilities. Indeed, we demonstrated that the expression of VE-cadherin in MDA-231 cells changes the migratory behavior of cells from individual non-directional migration to collective unidirectional migration, yet we did not observe an increase in directional migration for single cells. Thus, we conclude that the intercellular adhesion sites provide key structures in controlling cell migration directionality during sheet migration in tumor cells.
Another important function of adherens junctions is the control of cell proliferation by contact inhibition (Morris and Huang 2016). Collective cell migration of cranial neural crest (CNC) is mediated via Wnt/PCP signaling, the activation of small Rho GTPase RhoA, and the inhibition of cell protrusions on the contact side (Becker et al. 2013). Stabilization of ß-catenin at the cell junctions is critical in the canonical Wnt signaling pathway (Van Mater et al. 2003) and causes two effects. Firstly, the total amount of β-catenin expression is up-regulated and secondly the Wnt/β-catenin signaling is decreased. Importantly, through the expression of VE-cadherin in MDA-231 cells, recruitment and stabilization of ß-catenin to the junction and was shown. As a result of Wnt/beta-catenin signaling down regulation, levels of c-myc and cyclin D1, known regulators of cell proliferation and cell cycle progression, were significantly decreased. However, VE-cadherin expression had no effect on breast cancer cell proliferation. Aside from their role in regulating cell cycles, cyclin D1 and c-myc have been shown to regulate cell motility and invasiveness in cancer cells. For instance, the repression of c-myc or cyclin D1 in human breast cancer cells induces an increase in cell motility that is linked to unfavorable prognostic features (40–43). In contrast, the expression of VE-cadherin allows the tumor cells to integrate into endothelial monolayer by forming competent junctions. This is verified by junction recruitment of both β-catenin and α-E-catenin that in turn can connect to the actin cytoskeleton. Intriguingly, such a connection significant increased the tumor cell proliferation, a phenomenon that most likely involves VE-cadherin controlled cell signaling. However, further studies are essentially required to better understand this phenomenon.
In conclusion, a key finding of this study is that the impact of VE-cadherin on cancer cell behavior depends on the respective stage of cell differentiation. The impact of VE-cadherin was higher in the undifferentiated, aggressive and E-cadherin-negative MDA-231 cell line, compared to the differentiated, poorly metastatic MCF-7 cell line which has maintained expression of E-cadherin. Thus, it appears that the E-cadherin status may influence the impact of VE-cadherin on tumor cell behavior. The MET-like morphological and functional changes that were induced by VE-cadherin in the human breast cancer cells were mostly related to the formation of cell junctions paired with the Wnt signaling pathway that, together, regulate cell motility. Importantly, VE-cadherin promoted also collective migration of E-cadherin-negative MDA-231 breast cancer cells. Only VE-cadherin expressing cell integrated into endothelial monolayer and developed functional competent cell junctions that furthermore control tumor cell proliferation. These changes may represent a critical mode through which VE-cadherin could promote cancer cell invasion and eventually metastasis. Taken together, this study provides novel evidence for an important role of VE-cadherin in human breast cancer progression.
References
Abu Taha A, Taha M, Seebach J, Schnittler HJ (2014) ARP2/3-mediated junction-associated lamellipodia control VE-cadherin-based cell junction dynamics and maintain monolayer integrity. Mol Biol Cell 25(2):245–256. https://doi.org/10.1091/mbc.E13-07-0404
Alfano D, Votta G, Schulze A, Downward J, Caputi M, Stoppelli MP, Iaccarino I (2010) Modulation of cellular migration and survival by c-Myc through the downregulation of urokinase (uPA) and uPA receptor. Mol Cell Biol 30(7):1838–1851. https://doi.org/10.1128/MCB.01442-09
Baumeister U, Funke R, Ebnet K, Vorschmitt H, Koch S, Vestweber D (2005) Association of Csk to VE-cadherin and inhibition of cell proliferation. EMBO J 24(9):1686–1695
Becker SF, Mayor R, Kashef J (2013) Cadherin-11 mediates contact inhibition of locomotion during Xenopus neural crest cell migration. PloS One 8(12):e85717. https://doi.org/10.1371/journal.pone.0085717
Bendas G, Borsig L (2012) Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int J Cell Biol 2012:676731. https://doi.org/10.1155/2012/676731
Bettenworth D, Lenz P, Krausewitz P, Bruckner M, Ketelhut S, Domagk D, Kemper B (2014) Quantitative stain-free and continuous multimodal monitoring of wound healing in vitro with digital holographic microscopy. PLoS One 9(9):e107317. https://doi.org/10.1371/journal.pone.0107317
Bill R, Christofori G (2015) The relevance of EMT in breast cancer metastasis: correlation or causality? FEBS Lett 589(14):1577–1587. https://doi.org/10.1016/j.febslet.2015.05.002
Breier G, Grosser M, Rezaei M (2014) Endothelial cadherins in cancer. Cell Tissue Res 355(3):523–527. https://doi.org/10.1007/s00441-014-1851-7
Breslin JW, Daines DA, Doggett TM, Kurtz KH, Souza-Smith FM, Zhang XE, Wu MH, Yuan SY (2016) Rnd3 as a novel target to ameliorate microvascular leakage. J Am Heart Assoc. https://doi.org/10.1161/JAHA.116.003336 (ARTN e003336)
Breviario F, Caveda L, Corada M, Martin-Padura I, Navarro P, Golay J, Introna M, Gulino D, Lampugnani MG, Dejana E (1995) Functional properties of human vascular endothelial cadherin (7B4/cadherin-5), an endothelium-specific cadherin. Arterioscler Thromb Vasc Biol 15(8):1229–1239
Cao J, Ehling M, März S, Seebach J, Tarbashevich K, Sixta T, Pitulescu ME, Werner A, Flach B, Montanez E, Raz E, Adams RH, Schnittler H (2017) Polarized actin and VE-cadherin dynamics regulate junctional remodelling and cell migration during sprouting angiogenesis. Nat Commun (final revision)
Cavallaro U, Christofori G (2004) Multitasking in tumor progression: signaling functions of cell adhesion molecules. Ann N Y Acad Sci 1014:58–66
Christofori G (2003) Changing neighbours, changing behaviour: cell adhesion molecule-mediated signalling during tumour progression. EMBO J 22(10):2318–2323. https://doi.org/10.1093/emboj/cdg228
David JM, Rajasekaran AK (2012) Dishonorable discharge: the oncogenic roles of cleaved E-cadherin fragments. Cancer Res 72(12):2917–2923. https://doi.org/10.1158/0008-5472.can-11-3498
Dieckmann-Schuppert A, Schnittler H-J (1997) A simple assay for quantification of protein in tissue sections, cell cultures, and cell homogenates, and of protein immobilized on solid surfaces. Cell Tissue Res 288(1):119–126. https://doi.org/10.1007/s004410050799
Fry SA, Sinclair J, Timms JF, Leathem AJ, Dwek MV (2013) A targeted glycoproteomic approach identifies cadherin-5 as a novel biomarker of metastatic breast cancer. Cancer Lett 328(2):335–344. https://doi.org/10.1016/j.canlet.2012.10.011
Fry SA, Robertson CE, Swann R, Dwek MV (2016) Cadherin-5: a biomarker for metastatic breast cancer with optimum efficacy in oestrogen receptor-positive breast cancers with vascular invasion. Br J Cancer 114(9):1019–1026. https://doi.org/10.1038/bjc.2016.66
Giampietro C, Taddei A, Corada M, Sarra-Ferraris GM, Alcalay M, Cavallaro U, Orsenigo F, Lampugnani MG, Dejana E (2012) Overlapping and divergent signaling pathways of N-cadherin and VE-cadherin in endothelial cells. Blood 119(9):2159–2170. https://doi.org/10.1182/blood-2011-09-381012
Giannotta M, Trani M, Dejana E (2013) VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell 26(5):441–454. https://doi.org/10.1016/j.devcel.2013.08.020
Giepmans BN, van Ijzendoorn SC (2009) Epithelial cell-cell junctions and plasma membrane domains. Biochimica et biophysica acta 1788(4):820–831. https://doi.org/10.1016/j.bbamem.2008.07.015
Givant-Horwitz V, Davidson B, Reich R (2004) Laminin-induced signaling in tumor cells: the role of the M(r) 67,000 laminin receptor. Cancer Res 64(10):3572–3579. https://doi.org/10.1158/0008-5472.CAN-03-3424
Goldmann WH (2016) Role of vinculin in cellular mechanotransduction. Cell Biol Int 40(3):241–256. https://doi.org/10.1002/cbin.10563
Gumbiner BM (2005) Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol 6(8):622–634
Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA (2000) Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol 148(4):779–790
Hazan RB, Qiao R, Keren R, Badano I, Suyama K (2004) Cadherin switch in tumor progression. Ann N Y Acad Sci 1014:155–163
Hendrix MJ, Seftor EA, Meltzer PS, Gardner LM, Hess AR, Kirschmann DA, Schatteman GC, Seftor RE (2001) Expression and functional significance of VE-cadherin in aggressive human melanoma cells: role in vasculogenic mimicry. Proc Natl Acad Sci USA 98(14):8018–8023
Herren B, Levkau B, Raines EW, Ross R (1998) Cleavage of beta-catenin and plakoglobin and shedding of VE-cadherin during endothelial apoptosis: evidence for a role for caspases and metalloproteinases. Mol Biol Cell 9(6):1589–1601
Herzig M, Savarese F, Novatchkova M, Semb H, Christofori G (2007) Tumor progression induced by the loss of E-cadherin independent of beta-catenin/Tcf-mediated Wnt signaling. Oncogene 26(16):2290–2298. https://doi.org/10.1038/sj.onc.1210029
Huveneers S, Oldenburg J, Spanjaard E, van der Krogt G, Grigoriev I, Akhmanova A, Rehmann H, de Rooij J (2012) Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J Cell Biol 196(5):641–652. https://doi.org/10.1083/jcb.201108120
Kallioniemi OP, Wagner U, Kononen J, Sauter G (2001) Tissue microarray technology for high-throughput molecular profiling of cancer. Hum Mol Genet 10(7):657–662
Kemper B, Carl D, Höink A, von Bally G, Bredebusch I, Schnekenburger J (2006) Modular digital holographic microscopy system for marker free quantitative phase contrast imaging of living cells. Proc SPIE 6191:61910T
Kemper B, Bauwens A, Vollmer A, Ketelhut S, Langehanenberg P, Muthing J, Karch H, von Bally G (2010) Label-free quantitative cell division monitoring of endothelial cells by digital holographic microscopy. J Biomed Opt 15(3):036009. https://doi.org/10.1117/1.3431712
Kronstein R, Seebach J, Grossklaus S, Minten C, Engelhardt B, Drab M, Liebner S, Arsenijevic Y, Taha AA, Afanasieva T, Schnittler HJ (2012) Caveolin-1 opens endothelial cell junctions by targeting catenins. Cardiovasc Res 93(1):130–140. https://doi.org/10.1093/cvr/cvr256
Labelle M, Schnittler HJ, Aust DE, Friedrich K, Baretton G, Vestweber D, Breier G (2008) Vascular endothelial cadherin promotes breast cancer progression via transforming growth factor beta signaling. Cancer Res 68(5):1388–1397
Lampugnani MG, Resnati M, Raiteri M, Pigott R, Pisacane A, Houen G, Ruco LP, Dejana E (1992) A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J Cell Biol 118(6):1511–1522
Lehn S, Tobin NP, Berglund P, Nilsson K, Sims AH, Jirstrom K, Harkonen P, Lamb R, Landberg G (2010) Down-regulation of the oncogene cyclin D1 increases migratory capacity in breast cancer and is linked to unfavorable prognostic features. Am J Pathol 177(6):2886–2897. https://doi.org/10.2353/ajpath.2010.100303
Li Z, Wang C, Jiao X, Lu Y, Fu M, Quong AA, Dye C, Yang J, Dai M, Ju X, Zhang X, Li A, Burbelo P, Stanley ER, Pestell RG (2006) Cyclin D1 regulates cellular migration through the inhibition of thrombospondin 1 and ROCK signaling. Mol Cell Biol 26(11):4240–4256. https://doi.org/10.1128/MCB.02124-05
Liu H, Radisky DC, Yang D, Xu R, Radisky ES, Bissell MJ, Bishop JM (2012) MYC suppresses cancer metastasis by direct transcriptional silencing of alphav and beta3 integrin subunits. Nat Cell Biol 14(6):567–574. https://doi.org/10.1038/ncb2491
Mbalaviele G, Dunstan CR, Sasaki A, Williams PJ, Mundy GR, Yoneda T (1996) E-cadherin expression in human breast cancer cells suppresses the development of osteolytic bone metastases in an experimental metastasis model. Cancer Res 56(17):4063–4070
Morris SL, Huang S (2016) Crosstalk of the Wnt/beta-catenin pathway with other pathways in cancer cells. Genes Dis 3(1):41–47. https://doi.org/10.1016/j.gendis.2015.12.003
Nelson CM, Chen CS (2003) VE-cadherin simultaneously stimulates and inhibits cell proliferation by altering cytoskeletal structure and tension. J Cell Sci 116(Pt 17):3571–3581. https://doi.org/10.1242/jcs.00680
Nelson CM, Pirone DM, Tan JL, Chen CS (2004) Vascular endothelial-cadherin regulates cytoskeletal tension, cell spreading, and focal adhesions by stimulating RhoA. Mol Biol Cell 15(6):2943–2953
Nieman MT, Prudoff RS, Johnson KR, Wheelock MJ (1999) N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J Cell Biol 147(3):631–644
Odenthal-Schnittler M, Schnittler HJ, Kemper B (2016) Online quantitative phase imaging of vascular endothelial cells under fluid shear stress utilizing digital holographic microscopy. Proc SPIE 9718:97180U
Ozaki S, Ikeda S, Ishizaki Y, Kurihara T, Tokumoto N, Iseki M, Arihiro K, Kataoka T, Okajima M, Asahara T (2005) Alterations and correlations of the components in the Wnt signaling pathway and its target genes in breast cancer. Oncol Rep 14(6):1437–1443
Parri M, Chiarugi P (2010) Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal CCS 8:23. https://doi.org/10.1186/1478-811X-8-23
Rezaei M, Friedrich K, Wielockx B, Kuzmanov A, Kettelhake A, Labelle M, Schnittler H, Baretton G, Breier G (2012) Interplay between neural-cadherin and vascular endothelial-cadherin in breast cancer progression. Breast Cancer Res BCR 14(6):R154. https://doi.org/10.1186/bcr3367
Schnittler HJ, Schmandra T, Drenckhahn D (1998) Correlation of endothelial vimentin content with hemodynamic parameters. Histochem Cell Biol 110(2):161–167
Seebach J, Dieterich P, Luo F, Schillers H, Vestweber D, Oberleithner H, Galla HJ, Schnittler HJ (2000) Endothelial barrier function under laminar fluid shear stress. Lab Invest 80(12):1819–1831
Seebach J, Abu Taha A, Lenk J, Lindemann N, Jiang XY, Brinkmann K, Bogdan S, Schnittler HJ (2015) The CellBorderTracker, a novel tool to quantitatively analyze spatiotemporal endothelial junction dynamics at the subcellular level. Histochem Cell Biol 144(6):517–532. https://doi.org/10.1007/s00418-015-1357-8
Sridharan S, Mir M, Popescu G (2011) Simultaneous optical measurements of cell motility and growth. Biomed Opt Express 2(10):2815–2820. https://doi.org/10.1364/BOE.2.002815
Sumida GM, Tomita TM, Shih W, Yamada S (2011) Myosin II activity dependent and independent vinculin recruitment to the sites of E-cadherin-mediated cell-cell adhesion. BMC Cell Biol 12:48. https://doi.org/10.1186/1471-2121-12-48
Swarbrick A, Akerfeldt MC, Lee CS, Sergio CM, Caldon CE, Hunter LJ, Sutherland RL, Musgrove EA (2005) Regulation of cyclin expression and cell cycle progression in breast epithelial cells by the helix-loop-helix protein Id1. Oncogene 24(3):381–389. https://doi.org/10.1038/sj.onc.1208188
Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E (2008) Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol 10(8):923–934. https://doi.org/10.1038/ncb1752
Tobin NP, Sims AH, Lundgren KL, Lehn S, Landberg G (2011) Cyclin D1, Id1 and EMT in breast cancer. BMC Cancer 11:417. https://doi.org/10.1186/1471-2407-11-417
Valiron O, Chevrier V, Usson Y, Breviario F, Job D, Dejana E (1996) Desmoplakin expression and organization at human umbilical vein endothelial cell-to-cell junctions. J Cell Sci 109(Pt 8):2141–2149
van Roy F (2014) Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nat Rev Cancer 14(2):121–134. https://doi.org/10.1038/nrc3647
Van Mater D, Kolligs FT, Dlugosz AA, Fearon ER (2003) Transient activation of beta-catenin signaling in cutaneous keratinocytes is sufficient to trigger the active growth phase of the hair cycle in mice. Genes Dev 17(10):1219–1224. https://doi.org/10.1101/gad.1076103
van der Schaft DW, Hillen F, Pauwels P, Kirschmann DA, Castermans K, Egbrink MG, Tran MG, Sciot R, Hauben E, Hogendoorn PC, Delattre O, Maxwell PH, Hendrix MJ, Griffioen AW (2005) Tumor cell plasticity in Ewing sarcoma, an alternative circulatory system stimulated by hypoxia. Cancer Res 65(24):11520–11528. https://doi.org/10.1158/0008-5472.CAN-05-2468
Wahl-Jensen VM, Afanasieva TA, Seebach J, Stroher U, Feldmann H, Schnittler HJ (2005) Effects of Ebola virus glycoproteins on endothelial cell activation and barrier function. J Virol 79(16):10442–10450
Wegener J, Seebach J (2014) Experimental tools to monitor the dynamics of endothelial barrier function: a survey of in vitro approaches. Cell Tissue Res 355(3):485–514. https://doi.org/10.1007/s00441-014-1810-3
Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, Allred DC, Bartlett JM, Bilous M, Fitzgibbons P, Hanna W, Jenkins RB, Mangu PB, Paik S, Perez EA, Press MF, Spears PA, Vance GH, Viale G, Hayes DF (2013) Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol Off J Am Soc Clin Oncol 31(31):3997–4013. https://doi.org/10.1200/JCO.2013.50.9984
Wyckoff JB, Jones JG, Condeelis JS, Segall JE (2000) A critical step in metastasis: in vivo analysis of intravasation at the primary tumor. Cancer Res 60(9):2504–2511
Xiao K, Garner J, Buckley KM, Vincent PA, Chiasson CM, Dejana E, Faundez V, Kowalczyk AP (2005) p120-Catenin regulates Clathrin-dependent endocytosis of VE-cadherin. Mol Biol Cell 16(11):5141–5151
Ziober AF, Falls EM, Ziober BL (2006) The extracellular matrix in oral squamous cell carcinoma: friend or foe? Head Neck 28(8):740–749. https://doi.org/10.1002/hed.20382
Acknowledgements
The authors are grateful for highly sufficient assistance by Annelie Ahle, Vesna Bojovic and Christine Schimp. We thank Martin Muermann for MS editing and to the “Biobank” of University Cancer Center Dresden (UCC) for supporting the preparation of tissue microarray. We also thank Anke Klawitter and Thomas Brock for sharing unpublished results. This work was supported by grants from the German Research Council to H.S. (DFG INST 2105/24-1 and SCHN 430/6-2). The Excellence Cluster Cells In Motion (CIM) of the WWU-Münster, flexible fund to H.S (FF-2014-15).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None of the authors have financial or other types of competing interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rezaei, M., Cao, J., Friedrich, K. et al. The expression of VE-cadherin in breast cancer cells modulates cell dynamics as a function of tumor differentiation and promotes tumor–endothelial cell interactions. Histochem Cell Biol 149, 15–30 (2018). https://doi.org/10.1007/s00418-017-1619-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-017-1619-8