
Abstract
Purpose
Microglia contribute to immune homeostasis of the retina, and thus act as a potential regulator determining successful repair or retinal stem cell transplantation. We investigated the interaction between human microglia and retinal progenitor cells in cell co-culture to further our exploration on developing a new therapeutic strategy for retinal degeneration.
Methods
Microglia and retinal progenitor cultures were developed using CD11b+ and CD133+, respectively, from adult donor retina. Microglia activation was developed using interferon-gamma and lipopolysaccharide. Retinal progenitor differentiation was analysed in co-culture with or without microglial activation. Retinal progenitor proliferation was analysed in presence of conditioned medium from activated microglia. Phenotype and function of adult human retinal cell cultures were examined using cell morphology, immunohistochemistry and real-time PCR.
Results
By morphology, neuron-like cells generated in co-culture expressed photoreceptor marker recoverin. Neurospheres derived from retinal progenitor cells showed reduced growth in the presence of conditioned medium from activated microglia. Delayed retinal progenitor cell migration and reduced cellular differentiation was observed in co-cultures with activated microglia. In independent experiments, activated microglia showed enhanced mRNA expression of CXCL10, IL-27, IL-6, and TNF-alpha compared to controls.
Conclusion
Adult human retina retains retinal progenitors or potential to reprogram cells to then proliferate and differentiate into neuron-like cells in vitro. Human microglia support retinal progenitor differentiation into neuron-like cells, but such capacity is altered following microglial activation. Modulating microglia activity is a potential approach to promote retinal repair and facilitate success of stem-cell transplantation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Retinal degeneration encompasses many conditions, resulting ultimately in photoreceptors or supporting cell (retinal pigment epithelium; RPE) death, and clinically these cells are not replaced readily. Retinal degenerations include monogenetic disorders such as retinitis pigmentosa, complex degenerative diseases such as age-related macular degeneration and metabolic and immune mediated disorders, contributing to retinal degeneration as seen in diabetic retinopathy or some forms of uveitis. In generating successful therapies to prevent outer retinal photoreceptor degeneration, gene therapy has been instrumental in improving outcomes for patients with monogenetic disorders [1–3], and RPE stem-cell transplants are currently ongoing in clinical trials [4, 5]. Various cell sources, including retinal progenitor, neural progenitor cells, mesenchymal stem cells, and retinal pigment epithelial cells, have been used in animal models [6]. More recently, utility of embryonic stem cells and induced pluripotent stem cells (iPS cells) have also moved into clinical trials [4, 5]. Nevertheless, stem/progenitor cell transplants into adult retina are arguably constrained by the limited capacity to integrate into the outer nuclear layer and differentiate into new photoreceptors, despite reports of successful integration of retinal progenitors into the degenerating mature retina [7]. One notion is that mature mammalian retina lacks the ability to accept and incorporate stem cells or promote photoreceptor differentiation [8]. During degeneration, both microglia and macrophages accumulate within the outer nuclear layer and photoreceptor outer segment debris [9, 10]. For example, following Mϋller stem-cell transplantation in a rat model, increased numbers of myeloid-derived cells were observed that might preclude successful integration [11].
Microglia, the resident macrophages in the brain and the retina, provide a primary immune defence mechanism contributing to neural homeostasis [12, 13]. Microglia are central to both homeostasis and progression of disease when activated [14, 15], and will contribute to retinal repair under certain conditions [16–18]. Our previous data demonstrated the presence of retinal progenitor cells in man [19], and that activated microglia inhibited the ability of retinal progenitor cells to generate neurospheres in vitro [20]. Despite such data, relatively little is known about the role of human microglia on retinal stem/progenitor cell behaviours in vitro and in vivo. This is partly due to the limited availability of appropriate technical approaches and the platforms to interrogate human retinal tissue interactions.
To elicit understanding of the role of human microglia, we wished to develop an in-vitro model using primary human cells to overcome the inherent limitations of animal models or established cell lines [21]. Firstly, we established a co-culture model where we successfully isolated adult human microglia and retinal progenitor cells, and secondly we explored the potential of these adult retinal progenitors to proliferate and to differentiate into photoreceptors; thereby generating an experimental human retinal tissue model to investigate the role of microglia.
Materials and methods
Adult microglia and retinal progenitor cell cultures
Post-mortem human eyes were obtained from Bristol Eye Bank (Bristol Eye Hospital), with ethical approval from Central and South Bristol Research and Ethics Committee (E5866) and research consent in compliance with the Helsinki Declaration. We obtained 87 paired eyes and 37 single eyes from 124 donors with an average age of 71 years (20 to 93 years; female 48, male 76). Retinae were processed to isolate microglia and retinal progenitor cells within 24 to 64 h, following enucleation as we have previously reported [22].
CD11b is a marker for retinal macrophage/microglia [23], and CD133 is a neural stem cell marker, which we have shown to be expressed on cells with retinal progenitor cell status [19]. Utilizing such phenotypic markers, CD11b-expressing cells (CD11b+cells) and CD133-expressing cells (CD133+ cells) were isolated from post-mortem retina using an established protocol [24]. Briefly, retina was dissociated using enzymes (trypsin and collagenase A) and mechanical trituration; non-specific binding sites on retinal cells were blocked using FcR block (Miltenyi Biotec); CD11b+ cells and CD133+ cells were isolated from the retinal cell suspensions respectively, using specific MACS MicroBeads according to manufacturer’s instructions (Miltenyi Biotec). Paired eyes gave rise to 2.6 × 105–7.2 × 105 CD11b+ cells, and 2.8 × 106–2.4 × 107 CD133+ cells.
CD11b+ cells were cultured to select microglia as previously described [24], seeded at 105 cells/ml in 6-well plates or T25 flasks in microglia medium (RPMI 1,640, GlutaMAX containing 5 % foetal calf serum, 1 % non-essential amino acid (NEAA )(Sigma), 1 % sodium pyruvate (Sigma–Aldrich), 0.45 % of Glucose (Sigma–Aldrich) and 10 ng/ml human macrophage colony-stimulating factor (M-CSF)(R & D system). Cell culture medium was changed every 3 days.
CD133+ cells were cultured to select retinal progenitor cells and seeded at 106 cells/ml in T25 or T75 flasks in retinal progenitor medium (DMEM/F12, GlutaMAX (Gibco) containing 1 % of N2 (PAA Laboratories), 2 % of B27 (Invitrogen), 5 ng/ml human FGF-2 and 10 ng/ml human EGF (Sigma–Aldrich). CD133+ cells were cultured without changing medium in the first 7 days to allow generation of neurospheres, followed by changing medium after centrifugation at 200 g for 5 min every 3 days.
Flow cytometric phenotype analysis of microglia and retinal progenitor cells
Archetypal ramified microglial morphology was observed in CD11b+ cell cultures. Confirmation via phenotype analysis of cells was provided via flow cytometry as previously reported [24]. Briefly, cell cultures were trypsinized using trypsin/EDTA (Sigma–Aldrich) and suspended in phosphate buffered saline (PBS) containing 2 % foetal calf serum (FCS) and 1 mM EDTA, pH 7.5. Cell suspension was blocked using Fc blocking reagent (Miltenyi Biotec) for 15 min at 4 °C and stained using APC-Cy7-labelled anti-human CD11b antibody, PE-Cy7- labelled anti-human CD45 antibody, and Alexa Fluor 647-labelled anti-human CD68 antibody or isotype controls (BD Biosciences). Compensation controls was achieved with BDTM CompBeads. Samples were acquired using BD LSR-II Flow Cytometry (BD Cytometry Systems) and data were analysed using Flowjo software (Tree Star). Isolated CD11b+ expressed Iba-1 by immunohistochemistry to further corroborate microbiological phenotype (Supplement Fig. 1).
Neurospheres were generated from CD133+ single cell cultures. Flow cytometric phenotype was established as previously described [24]. Briefly, neurospheres were washed by centrifugation at 200 g for 5 min to remove free cells and then dissociated using Neurosphere Dissociation Kits (Miltenyi Biotec) according to manufacturer’s instructions. Dissociated cells were prepared using Neurosphere Dissociation Kit (Miltenyi Biotec), and stained using PE-labelled anti-human CD133 antibody (Miltenyi Biotec), PE-Cy7-labelled anti-human CD45 antibody (BD Biosciences), and Alexa Flour 647-labelled anti-human CD68 antibody or isotype controls (BD Biosciences). The labelled dissociated neurosphere cells were acquired and analysed as described above.
Establishing co-cultures
To examine the role of cell–cell interaction between retinal progenitor cells (generating neurospheres) and microglia, we co-cultured retinal progenitor cells with activated microglia or non-activated microglia as comparison. Microglia were cultured for a variable period of 8–14 days in 6-well plates or T25 flasks, and further subcultured in 12-well plates, 1 ml/well. Prior to adding neurospheres, microglia medium was switched to co-culture medium (microglia medium without M-CSF). Neurospheres generated in CD133+ cell cultures for 10–14 days were washed by centrifugation at 200 g for 5 min to remove free cells and cell debris. Cell pellet was suspended in 0.5 ml of co-culture medium and loaded into the microglia cultures. The co-cultures were maintained for a further 14 to 21 days to determine retinal progenitor differentiation. In the co-culture, the majority of neurospheres closely aligned with microglia within 1 day. Co-culture medium was changed every 3 days.
To examine the role of soluble cytokines and chemokines secreted from activated microglia, we cultured retinal progenitor in presence of conditional medium from activated microglia compared to medium from non-activated microglia control. Retinal progenitor cells were grown in retinal progenitor medium for 10 days to generate neurospheres. Neurosphere cultures were loaded into the pre-separation filter (30 μm Miltenyi Biotech), and neurospheres retained within the reservoir of the filter were collected. The neurospheres were suspended in 1.6 ml of retinal progenitor medium, divided into two parts and transferred into two wells, 0.8 ml/well in 12-well plates. Conditioned medium from activated and non-activated microglia respectively was added at 0.2 ml/well into the two wells. Cell-culture medium was changed every 3 days by removing 0.5 ml of medium to avoid disturbing the neurospheres and adding 0.5 ml of new medium (0.1 ml of microglia conditioned medium and 0.4 ml of retinal progenitor medium). Neurospheres were cultured up to 14 days and observed and photographed at regular intervals. Neurosphere size was analysed using ImageJ software. Average neurosphere sizes were obtained in six randomized fields of neurosphere cultures in cultures with conditioned medium from activated microglia or non-activated microglia as comparison.
Microglia activation and functional analysis
Microglia were activated using interferon-gamma (IFN-γ) and lipopolysaccharide (LPS) as previously reported [20]. Microglia in culture were trypsinized and washed and divided into two equal parts and subcultured in 12-well plates. Microglia in one of the paired wells were treated using pre-determined concentrations of activation cytokines, 100U/ml of IFN-γ, and 5 ng/ml of LPS in microglia medium for 8 h, after which the medium was removed, cells washed, and replenished with co-culture medium. Microglia in the other paired wells were used as control, and treated using the same procedures without IFN-γ/LPS activation. After 48 h, conditioned medium was collected from the activated microglia and non-activated microglia control respectively, and used to supplement neurosphere culture.
Analysis of cell proliferation using bromodeoxyuridine (BrdU) incorporation
BrdU incorporation assay was employed for analysis of cell proliferation [25]. Neurospheres were generated from single cell CD133+ cells for 10 days. BrdU was added to the neurosphere cultures at 10 μM in retinal progenitor medium 24 h prior to analysis. Neurospheres were then dissociated using neurosphere dissociation kit (Miltenyi Biotec) and stained using APC BrdU Flow Kit (BD Bioscience) according to the manufacturer’s instructions. The stained cells were acquired using BD LSR-II Flow Cytometry (BD Cytometry Systems), and data was analysed using Flowjo software.
Immunofluorescent analysis of differentiated cells
Neuron-like cells were seen following co-culture of neurospheres with microglia. Cells were washed in phosphate buffered saline (PBS) and fixed using 2 % paraformaldehyde (PFA) at room temperature (RT) for 5 min, followed by permeabilization using 0.3 % Triton-100 at RT for 5 min. These cells were then exposed to 2 % bovine serum albumin (BSA) and 10 % normal rabbit serum in PBS block at RT for 30 min, and subsequently incubated with rabbit anti-human recoverin polyclonal antibody (Merck Millipore) at 1:500 and chicken anti-human tubulin-beta 3 IgY antibody (Merck Millipore) at 1:400 in PBS containing 0.2 % BSA and 0.03 % Triton-100 at 4 °C overnight. This was followed by incubation with rhodamine-labelled goat anti-rabbit antibody (Santa Cruz) at 1:200 and FITC- labelled donkey anti-chicken IgY antibody (Merck Millipore) at 1:300 at room temperature for 2 h. After each incubation step, cells were washed in PBS 3 times. Cells were mounted in Vector Shield mounting medium with DAPI (Vector Laboratories) and analysed using fluorescence microscopy (Leica, Germany).
Examination of mRNA expression using real-time PCR
The extent of mRNA expression by microglia, neurospheres or from the co-cultures was examined using TaqMan Gene Expression Assay (Applied Biosystems). Briefly, total RNA was isolated from the cells using Trizol Reagent according to the manufacture’s instructions (Life Technologies). To enhance RNA precipitation, 0.2 mg/ml of glycogen was added into isopropyl alcohol, and stored at −20 °C overnight. Total RNA was isolated from microglia cultures (152–296 ng RNA), neurosphere cultures (112–270 ng RNA) or co-cultures (282–662 ng RNA). RNA was treated with RQ1 RNase-free DNase (Promega). cDNA was generated using ImProm-IITM reverse transcription system (Promega). cDNA was amplified using TaqMan Universal PCR Master Mix (Applied Biosystems) and TaqMan Gene Expression Assays (Applied Biosystems) and StepOne™ Real-Time PCR System (Applied Biosystems). PCR primers information is detailed in Supplement Table 1. PCR reaction volume was 20 μl. Thermal cycling consisted of 50 °C for 2 min, 95 °C for 10 min and 60 cycles of 95 °C for 15 s and 60 °C for 1 min. In order to determine any difference in mRNA expression between the two groups (e.g., activated microglia vs non-activated microglia), the fold changes in expression of target genes relative to an internal control gene (house-keeping gene, GAPDH) were analysed using 2-ΔΔCt method [26]. ΔΔCt = (Ct,Target – Ct,GAPDH) activated microglia - (Ct,Target – Ct,GAPDH) microglia control.
Statistical analysis
Neurosphere size data and fold changes of gene expression between activated microglia group and non-activated microglia control are presented as mean + SD. Statistical comparisons were performed using the two-tailed Student’s t-test. A p-value less than 0.05 (p < 0.05) was considered significant.
Results
Development and validation of microglia and retinal progenitor cultures
CD11b MACS MicroBeads-selected retinal cells attached within hours following culture in microglia medium containing M-CSF. Attached cells displayed thin and rod-shaped cell morphology within a few days (Fig. 1a and b), and achieved the characteristic ramified morphology after 1 week (Fig. 1c and d). Cells used in co-culture or specific experiments were harvested after approximately 12 to 20 days in culture, when the majority of cells demonstrated ramified morphology. CD133 MACS MicroBeads-selected cells were cultured in suspension (Fig. 1e). Typical early neurosphere development occurred from day 7 of culture (Fig. 1f). Neurospheres then displayed increasing size up to 10–14 days in culture. Thereafter, neurospheres appeared congregated (Fig. 1e-h).

Isolation and culture of adult microglial and retinal progenitor cells. CD11b+ cells and CD133+ cells were isolated from a paired eyes of a donor (female, 81 years of age) using MACS CD11b MicroBeads and CD133 MiroBeads (Miltenyi Biotec), respectively. CD11b+ cells were cultured in microglia medium containing M-CSF (a–d). CD133+ cells were cultured in retinal progenitor medium containing N2, B27, EGF and FGF-2 (e–h). Photos were taken at day 1 (a, e), day 7 (b, f), day 14 (c, g) and day 21 (d, h). Scale bar = 50 μm
Microglia identity was confirmed by analysing CD45 and CD11b expression via flow cytometry [27]. Microglial isolation was confirmed: 93.3 % expressed CD45, 74.4 % expressed CD11b, and 24.4 % expressed CD68 (Fig. 2a). Dissociated neurosphere cells expressed CD133 (61 %), and did not express either CD45 or CD68 (Fig. 2b). Throughout, in order to enhance cultures and obtain sufficient cell numbers we adapted the culture technique by pooling retinal cells from two or three donors. This was on the observation that no cell loss or increased cell death was noted following pooling of donor retinal cells in this study and our previous studies.

Phenotype analysis of adult microglial and retinal progenitor cells using flow cytometry. Microglia cells developed using pooled CD11b+ cells from total four eyes of two donors (one female and one male; 65 and 64 years of age respectively) analysed using flow cytometry and displayed in top panel. Figures in each FACS plot show percentages of cells positive to specific cell membrane markers (CD11b and CD45) or intracellular marker (CD68). Neurosphere cultures developed using pooled CD133+ cells from total four eyes of two donors (both male, 74 and 63 years of age, respectively) analysed using flow cytometry and displayed in bottom panel. Blue curves represent results of using specific antibodies and red curves represent results of using isotype controls. Figures in each FACS plot show percentages of cells positive to specific cell membrane markers (CD133 and CD45) or intracellular marker (CD68)
Generation of morphologically neuron-like cells in co-culture of neurospheres with microglia
We examined retinal progenitor cell differentiation following co-culture with microglia in DMEM/F12 with GlutaMAX containing N2 and B27, or in co-culture medium containing the same components as the microglia medium minus M-CSF. Neurosphere-derived cells displayed vigorous cell differentiation in the co-culture medium in absence of exogenous specific neurotropic factors. Neurospheres were closely associated with microglia within hours following co-culture, and cells from neurospheres migrated along the top of microglia (Fig. 3a), and underwent differentiation into cells of neural cell morphology. Meanwhile, microglia underwent distinct cell morphological changes, which included loss of cell ramification and increased size of cell body (Fig. 3b–f). The majority of differentiated cells displayed bipolar or unipolar cell morphology (Fig. 3b–f). Differentiated cells formed cell-to-cell networks on top of underlying adherent microglia with large and flat cell body (Fig. 3b and f), and only rarely in absence of underlying microglia (Fig. 3e). Neuron-like cells expressed either recoverin and/or tubulin-beta 3 (Fig. 3g and h). In the dual expressing neuron-like cells, recoverin expression was confined to the cell body, while tubulin-beta 3 was along the axon to the synapse (Fig. 3g). Some differentiated cells demonstrated photoreceptor-like cell morphology, including cell body, segment region, and synapse. The cell body and segment region expressed recoverin, whereas only the synaptic region expressed tubulin-beta 3 (Fig. 3h).

Generation of neuron-like cells in co-culturing adult retinal progenitor cells (neurospheres) with microglia. Neurosphere and microglia cultures generated using pooled CD133+ cells and pooled CD11b+ cells, respectively, from total four eyes of two donors (both male and 69 years of age). Neurospheres were co-cultured with microglia for 14 days. Cell morphology was taken at day 1 (a) and day 14 b–f. Cells were stained for recoverin (red), tubulin-beta 3 (green) and DNA (blue) (g and h). Retinal progenitor cells attached on top of microglia at day 1 (a). Neuron-like cells are demonstrated by white arrows and microglial cells are noted by red arrow (c). A neuron-like cell is positive to recoverin (brown arrow), and a neuron-like cell is positive to tubulin-beta 3 (red arrow). Neuron-like cell is dual-positive pointed (white arrow) (g). A differentiated cell, pointed (white arrow), demonstrates photoreceptor-like cell morphology, displaying a cell body and segment region in red and synaptic region in green (h). A differentiated cell, (red arrow), shows synaptic region in yellow, demonstrating dual positivity to recoverin and tubulin-beta 3 (H). Scale bar = 50 μm
mRNA expression changes in activated microglia compared to inactivated microglia
IFN-γ/LPS-activated microglia displayed typical amoeboid morphology (Fig. 4a and b). We further analysed changes in mRNA expression in three independent experiments when microglia are activated and found, in comparison to non-activated microglia, the following: enhanced expressions of CXCL-10 (27,612-fold, p < 0.05), IL-27 (1,243-fold, p < 0.05), IL-1β (74-fold, p > 0.05), IL-6 (62-fold, p < 0.05), TNF-alpha (38-fold, p < 0.05), IRF1 (59-fold, p > 0.05), and SOCS1 (11-fold, p > 0.05), and reduced expression of CD68 (p > 0.05), CD11b (p > 0.05), and AIF1 (Iba1, p > 0.05) (Fig. 4c).

Gene expression changes in activated microglia in comparison to non-activated microglia. Microglia cultures developed using pooled CD11b+ cells from eyes of three donors (two female and one male; 77, 71 and 68 years of age respectively). Activated microglial cells, 24 h after being activated using lipopolysaccharide and interferon-γ display amoeboid cell morphology (b) in contrast to non-activated microglia control a, scale bar = 50 μm. c shows the fold changes in expression of target genes relative to the internal control gene GAPDH between the two groups. Data obtained in three independent experiments using CD11b+ cells from seven donors are presented as mean + SD. * p < 0.05. GAPDH: glyceraldehyde 3-phosphate dehydrogenase; CD68: cluster of differentiation molecule 68; CD11b: cluster of differentiation molecule 11B; CD200: cluster of differentiation 200; CD200R: CD200 receptor; IL-27: interleukin-27; IL-27RA: IL-27 receptor; IL-6: interleukin-6; IL-6R: IL-6 receptor; IL-10: interleukin 10; IL-10RA: IL-10 receptor; CXCL10: C-X-C motif chemokine 10; IL-1b: interleukin-1 beta; TNF: tumour necrosis factor alpha; AIF1:allograft inflammatory factor 1; IRF1: interferon regulatory transcription factor 1; IRF8: interferon regulatory transcription factor 8; SOCS1: suppressor of cytokine signaling 1; SOCS3: suppressor of cytokine signaling 3
Activated microglia conditioned media suppressed retinal progenitor cell proliferation
Microglia produce various cytokines and proteins, which are neurotrophic or cytotoxic in physiological and pathological conditions. To continue our study on the roles of activated microglia on neurosphere development [20], we modified our experimental protocol by quantitatively analysing neurosphere size and performing BrdU incorporation. Delayed neurosphere growth was seen in neurosphere cultures in the presence of conditioned medium from activated microglia (Fig. 5a-d, g). This result was supported by our observation of a 48 % reduction of BrdU positive cells in neurosphere cultures in presence of conditioned medium from activated microglia, compared to those in presence of conditioned medium from non-activated microglia control (2.28 versus 4.42 % respectively) (Fig. 5e and f). This result was compatible with the reduced mRNA expression for nestin (NES) and antigen MI-67 (MKI67) in neurosphere cultures in presence of conditioned medium from activated microglia (Fig. 5h).

Impact of soluble factors secreted by activated microglia on retinal progenitor proliferation. Neurospheres and microglia cultures were generated using pooled CD133+ cells and pooled CD11b+ cells respectively, isolated from donors’ eyes. Neurospheres of 30 μm or more were cultured in medium with (a and c) or without (b and d) conditioned medium from activated microglia. Neurosphere cultures were photographed at day 0 (a and c) and day 10 (b and d), scale bar = 50 μm. Graph g shows average neurosphere sizes in the two experimental conditions. Data are presented in square micrometres as mean + SD; ** p < 0.01. Graphs e and f show BrdU incorporation data of neurospheres in medium with (f) or without (e) conditioned medium from activated microglia. Figures in the graphs represent percentages of BrdU positive cells. Graph h shows the fold changes in expression of target genes relative to the internal control gene GAPDH between the two groups. Data obtained in four independent experiments using CD133+ and CD11b+cells from nine donors are presented as mean + SD. GAPDH: glyceraldehyde 3-phosphate dehydrogenase; PAX6: Paired box protein Pax-6; NES: nestin; PCNA: proliferating cell nuclear antigen; RCVRN: recoverin; TUBB3: tubulin-beta 3; MKI67: antigen KI-67; RHD: rhodopsin; CD133: cluster of differentiation 133
Activated microglia influenced retinal progenitor migration and differentiation
Retinal progenitor cells exhibited rapid differentiation towards morphologically neuron-like cells in co-culture with microglial cells (Fig. 3) using co-culture medium without additional supplement of neurotropic factors N2 and B27, which are essential for an efficient differentiation of human stem cells toward retinal photoreceptor cells [28]. This data indicates that microglial cells support retinal progenitor cell differentiation toward morphologically neuron-like cells. During degeneration, microglia alter morphology into an activated ‘amoeboid’ form. We hypothesised that activated microglial cells will regulate retinal progenitor cell differentiation compared to non-activated microglial cells. This hypothesis was upheld by observation of delayed demonstration of formation and migration of neuron-like cell morphology in the co-culture with activated microglial cells (Fig. 6b and d) compared to the co-culture with non-activated microglial cells (Fig. 6a and c). This notion was partially supported by the observation of reduced mRNA expression of recoverin (RCVRN) and rhodopsin (RHD) in the co-cultures with activated microglial cells, in comparison to the co-cultures with non-activated microglial cells (Fig. 6e). Statistical analysis of combining individual experiments failed to achieve significance between the two groups, which is likely, given the profound gene alterations in each experiment, to be due to inherent donor variance in material and the levels of gene change observed in independent experiments.

Impact of activated microglia on the differentiation of retinal progenitor cells (neurospheres). Neurospheres were co-cultured with activated microglia or control for 14 days. b and d show cell morphology and immune reactivity of the co-cultures using activated microglia; a and c show data of the controls. Recoverin was stained in red, tubulin-beta 3 in green, and nucleus in blue, scale bar = 50 μm. Graph e shows the fold changes in expression of target genes relative to the internal control gene GAPDH between the two groups. Data obtained in three independent experiments using CD133+ cells and CD11b+ cells from seven donors are presented as mean + SD. GAPDH: glyceraldehyde 3-phosphate dehydrogenase; PAX6: paired box protein Pax-6; NES: nestin; PCNA: proliferating cell nuclear antigen; RCVRN: recoverin; TUBB3: tubulin-beta 3; MKI67: antigen KI-67; RHD: rhodopsin
Discussion
A pathological role of microglia in neurodegenerative diseases, including Alzheimer’s disease and macular degeneration, has been recognized. However, the precise role of microglia during outer retinal degeneration or retinal repair and ultimately microglial influence on retinal stem cell transplant integration [14] remains to be elucidated. A recent study suggests that retinal stem cells isolated from adult mouse retina have the potential of producing functional photoreceptor cells [29], while the finding of retinal stem cells in pigmented ciliary bodies in adult animals remains debatable [30–32]. Here, as with our previous work, we demonstrate that cells with retinal stem/progenitor cell behaviour (CD133+) can be isolated from the adult human retina and form neurospheres in vitro. Retinal progenitor-derived neurospheres differentiated into morphologically neuron-like cells in a new co-culture system with microglia. We also provide direct evidence of human microglia influence on retinal progenitor cells proliferation and differentiation.
Neurospheres were propagated following CD133+ cell isolation, and when developed these neurospheres displayed 61 % CD133 positivity, and 4.42 % cells were BrdU positive, indicating low level but active proliferation. The low proliferative potential found in adult retinal progenitor cells is similar to that found in adult mouse neural stem cells [33], in which 5.27–2.22 % of Prom-1+ (CD133+) mouse cells are BrdU-positive. This data suggests that CD133+cells represent a slowly dividing neural stem and progenitor cell subtype. Whilst CD133 may not be a good marker for isolating retinal stem/progenitor cells for stem cell transplantation, it allowed us to develop an in-vitro disease modelling, as a surrogate to isolate cells with proliferative capacity using its status as recognized neural stem cell marker. Adding conditioned medium from activated microglia in neurosphere cultures led to a reduced neurosphere growth and fewer BrdU positive cells. The reduced retinal progenitor cell proliferation (reduced neurosphere size) can be understood, as we found reduced mRNA expression of nestin (NES) and antigen KI-67 (MKI67) (Fig. 5h). The data supports the conclusion that reduced nestin expression represents a reduced cell capacity to differentiate, and KI-67 reduction reflects reduced proliferation capacity. Future work could consider further phenotyping of neural stem cells such as with CD15 and CD81, to assess whether stem cell properties were maintained when differentiation was affected. Using carboxyfluorescein succinimidyl ester (CFSE) would illuminate cell fate with respect to cells undergoing differentiation.
Adult human retinal progenitor cells generated morphologically neuron-like cells that expressed photoreceptor cells marker recoverin or/and neuron marker tubulin-beta 3 (Fig. 3g and h). Recoverin is expressed in photoreceptors and bipolar neural cells in the developing and mature mammalian retina [34, 35]. Tubulin-beta 3, a constituent of neuronal microtubules, has been frequently used as a marker for the neuronal lineage in developmental biology, as well as a marker for ganglion cells in the retina. We observed tubulin-beta 3 expression in both microglia and differentiated neuron-like cells from neurospheres by immunofluorescence when in co-culture (Fig. 3g and h). The mechanism of tubulin-beta 3 expression in adult human retinal microglia remains unknown. Given that the expression of tubulin-beta 3 is found in early born lineage of retinal neurons, not being related to cell morphology or cell function, but rather to the cell lineage [36], one is led to infer whether yolk sac adult human retinal microglia have the potential to generate retinal neurons [37]. However, the expression does not rely on cell contact, as enhanced mRNA expression of tubulin-beta 3 was observed in the retinal progenitor cells (neurospheres) that were cultured with activated microglia supernatant compared to control (Fig. 5h ). These results are consistent with a previous report that shows a significant increase in the number of tubulin-beta 3-expressing neuronal phenotype in a co-culture of optic nerve cells with activated microglia compared to a co-culture using inactivated microglia. Their data suggests that activated microglia increase neurogenesis through secretion of protease-serine 2 [38]. Understanding the mechanisms underlying the roles of activated microglia via soluble factors or through cell–cell interaction opens the potential of new therapeutic strategies for facilitating retinal repair.
Various cell sources have been used to develop stem cell therapies for retinal degeneration, including embryonic stem cells [4] and induced pluripotent stem (iPS) cells [5], either of which have the ability to differentiate into any cell type in the body and provide unlimited supply of human cells for transplantation and disease modeling. Optimization of transplanted cell integration is essential for the success of a stem cell therapy using any cell source, since barriers exist in the degenerative retina for the transplanted photoreceptor precursor migration and integration into the host outer nuclear layer (ONL), in which both Mϋller and microglial cells are involved [39]. In-vitro and in-vivo animal studies demonstrate that macrophage/microglia can benefit repairing neurodegeneration [16–18]. We co-cultured retinal progenitor cells (neurospheres) with non-activated microglia in a medium without additional N2 or B27, which are essential to induce human pluripotent stem cell differentiation toward retinal photoreceptors in vitro [28]. This approach led to a vigorous retinal progenitor cell migration and rapid differentiation toward morphologically neuron-like cells (Fig. 3) within only 10 to 14 days, though less time was required for inducing human pluripotent stem cell differentiation toward retinal photoreceptor cells [28]. Our notion was that non-activated microglia provide support through substrate formation and cytokines and neurotrophic factors to regulate retinal progenitor cells differentiation. This was in part confirmed when cells isolated from different donors were used in the co-culture model; microglia, depending on activation status, regulated the migration, proliferation, and differentiation of retinal progenitor cells. Our data supported the thesis that manipulating the recipient retinal environment can improve transplanted photoreceptor integration and survival [40].
Several obstacles, including microglia, are supposed to prevent stem cell therapy from successful implementation in patients with retinal degenerative disorders [41]. This includes a physical barrier to migration [11] as well as, for example, induction of photoreceptor apoptosis [42]. Using the co-culture model described here, we demonstrate an inhibition through activated microglia on retinal progenitor migration and differentiation (Fig. 6), as well as an inhibition of gene expression of microglia-secreted mediators influencing retinal progenitor proliferation (Fig. 5). Those observations can be explained, in part, by the observations of enhanced expression of proinflammatory cytokines including CXCL10, IL-6, IL-27, and TNF-alpha from activated microglia (Fig. 4c). Together with our previous study demonstrating inhibition through microglia-secreted IL-6 on neurosphere generation from retinal progenitor cell cultures [20], there remains compelling evidence that activation of microglia and soluble factors will directly regulate progenitor cell behaviour [12, 14]. Enhanced mRNA expression of IL-27 in activated microglia (Fig. 4e) implicates a role in photoreceptor survival, as photoreceptors constitutively express IL-27 receptor. Endogenous production of IL-27 and IL-10 by retinal cells suppresses intraocular inflammation, wherein SOCS proteins induced by IL-27 during uveitis may protect the neuroretina from pro-inflammatory cytokines and apoptosis [43]. Encouraging cell replacement, IL-27 regulates proliferation and differentiation of human hematopoietic stem/progenitor cells [44], although to date no reports on IL-27 influence on retinal progenitor cells have been elucidated. Further to our observations, enhanced expression of IP-10 (CXCL10) in the retinal pigment epithelial (RPE) cells of eyes with early age-related macular degeneration (AMD), geographic atrophy, or choroidal neovascularization has been reported [45], and expression of various apoptotic factors in RPE cells in AMD is up-regulated by TNF-alpha [46]. Whether, therefore, neutralization of TNF-alpha activity (TNF-alpha inhibitor) as tested in animal models and in clinical trial in AMD shows benefit toward retinal degeneration or indeed promoting retinal repair has not been pursued [47].
In summary, these data further support the view that adult human retina retains retinal progenitor cell potential with capacity to proliferate and differentiate into recoverin-expressing morphologically neuron-like cells. Adult human microglia support autologous and heterologous retinal progenitor cell differentiation into photoreceptor-like cells in vitro. Activation of microglia leads to suppression of retinal progenitor cell proliferation and capacity to differentiate toward photoreceptor-like cells. Modulating microglia activity, therefore, is an approach for promoting retinal repair and as an adjunct benefiting stem cell transplant integration into the retina.
References
Ali RR (2012) Gene therapy for retinal dystrophies: twenty years in the making. Hum Gene Ther 23(4):337–339
Dalkara D, Byrne LC, Klimczak RR, Visel M, Yin L, Merigan WH, Flannery JG, Schaffer DV (2013) In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med 5(189):189ra176
MacLaren RE, Groppe M, Barnard AR, Cottriall CL, Tolmachova T, Seymour L, Clark KR, During MJ, Cremers FP, Black GC, Lotery AJ, Downes SM, Webster AR, Seabra MC (2014) Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet 383(9923):1129–1137
Schwartz SD, Hubschman JP, Heilwell G, Franco-Cardenas V, Pan CK, Ostrick RM, Mickunas E, Gay R, Klimanskaya I, Lanza R (2012) Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet 379(9817):713–720
Song P, Inagaki Y, Sugawara Y, Kokudo N (2013) Perspectives on human clinical trials of therapies using iPS cells in Japan: reaching the forefront of stem-cell therapies. Biosci Trends 7(3):157–158
Huang Y, Enzmann V, Ildstad ST (2011) Stem cell-based therapeutic applications in retinal degenerative diseases. Stem Cell Rev 7(2):434–445
Klassen HJ, Ng TF, Kurimoto Y, Kirov I, Shatos M, Coffey P, Young MJ (2004) Multipotent retinal progenitors express developmental markers, differentiate into retinal neurons, and preserve light-mediated behavior. Invest Ophthalmol Vis Sci 45(11):4167–4173
MacLaren RE, Pearson RA, MacNeil A, Douglas RH, Salt TE, Akimoto M, Swaroop A, Sowden JC, Ali RR (2006) Retinal repair by transplantation of photoreceptor precursors. Nature 444(7116):203–207
Liu Y, Yang X, Utheim TP, Guo C, Xiao M, Liu Y, Yin Z, Ma J (2013) Correlation of cytokine levels and microglial cell infiltration during retinal degeneration in RCS rats. PLoS One 8(12):e82061
Roque RS, Imperial CJ, Caldwell RB (1996) Microglial cells invade the outer retina as photoreceptors degenerate in Royal College of Surgeons rats. Invest Ophthalmol Vis Sci 37(1):196–203
Singhal S, Lawrence JM, Bhatia B, Ellis JS, Kwan AS, Macneil A, Luthert PJ, Fawcett JW, Perez MT, Khaw PT, Limb GA (2008) Chondroitin sulfate proteoglycans and microglia prevent migration and integration of grafted Muller stem cells into degenerating retina. Stem Cells 26(4):1074–1082
Dick AD (2009) Influence of microglia on retinal progenitor cell turnover and cell replacement. Eye (Lond) 23(10):1939–1945
Karlstetter M, Ebert S, Langmann T (2010) Microglia in the healthy and degenerating retina: insights from novel mouse models. Immunobiology 215(9–10):685–691
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8(1):57–69
Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312(5778):1389–1392
Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH (2006) Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 103(43):16021–16026
Corti S, Locatelli F, Donadoni C, Guglieri M, Papadimitriou D, Strazzer S, Del Bo R, Comi GP (2004) Wild-type bone marrow cells ameliorate the phenotype of SOD1-G93A ALS mice and contribute to CNS, heart and skeletal muscle tissues. Brain 127(Pt 11):2518–2532
London A, Itskovich E, Benhar I, Kalchenko V, Mack M, Jung S, Schwartz M (2011) Neuroprotection and progenitor cell renewal in the injured adult murine retina requires healing monocyte-derived macrophages. J Exp Med 208(1):23–39
Carter DA, Dick AD, Mayer EJ (2009) CD133+ adult human retinal cells remain undifferentiated in leukaemia inhibitory factor (LIF). BMC Ophthalmol 9:1
Balasubramaniam B, Carter DA, Mayer EJ, Dick AD (2009) Microglia derived IL-6 suppresses neurosphere generation from adult human retinal cell suspensions. Exp Eye Res 89(5):757–766
Chaichana KL, Guerrero-Cazares H, Capilla-Gonzalez V, Zamora-Berridi G, Achanta P, Gonzalez-Perez O, Jallo GI, Garcia-Verdugo JM, Quinones-Hinojosa A (2009) Intra-operatively obtained human tissue: protocols and techniques for the study of neural stem cells. J Neurosci Methods 180(1):116–125
Carter DA, Mayer EJ, Dick AD (2007) The effect of postmortem time, donor age and sex on the generation of neurospheres from adult human retina. Br J Ophthalmol 91(9):1216–1218
Nakazawa T, Hisatomi T, Nakazawa C, Noda K, Maruyama K, She H, Matsubara A, Miyahara S, Nakao S, Yin Y, Benowitz L, Hafezi-Moghadam A, Miller JW (2007) Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc Natl Acad Sci U S A 104(7):2425–2430
Carter DA, Balasubramaniam B, Dick AD (2013) Functional analysis of retinal microglia and their effects on progenitors. Methods Mol Biol 935:271–283
Yaddanapudi K, De Miranda J, Hornig M, Lipkin WI (2011) Toll-like receptor 3 regulates neural stem cell proliferation by modulating the Sonic Hedgehog pathway. PLoS One 6(10):e26766
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 25(4):402–408
Melief J, Koning N, Schuurman KG, Van De Garde MD, Smolders J, Hoek RM, Van Eijk M, Hamann J, Huitinga I (2012) Phenotyping primary human microglia: tight regulation of LPS responsiveness. Glia 60(10):1506–1517
Mellough CB, Sernagor E, Moreno-Gimeno I, Steel DH, Lako M (2012) Efficient stage-specific differentiation of human pluripotent stem cells toward retinal photoreceptor cells. Stem Cells 30(4):673–686
Li T, Lewallen M, Chen S, Yu W, Zhang N, Xie T (2013) Multipotent stem cells isolated from the adult mouse retina are capable of producing functional photoreceptor cells. Cell Res 23(6):788–802
Ahmad I, Tang L, Pham H (2000) Identification of neural progenitors in the adult mammalian eye. Biochem Biophys Res Commun 270(2):517–521
Froen R, Johnsen EO, Nicolaissen B, Facsko A, Petrovski G, Moe MC (2013) Does the adult human ciliary body epithelium contain “true” retinal stem cells? Biomed Res Int 2013:531579
Tropepe V, Coles BL, Chiasson BJ, Horsford DJ, Elia AJ, McInnes RR, van der Kooy D (2000) Retinal stem cells in the adult mammalian eye. Science 287(5460):2032–2036
Obermair FJ, Fiorelli R, Schroeter A, Beyeler S, Blatti C, Zoerner B, Thallmair M (2010) A novel classification of quiescent and transit amplifying adult neural stem cells by surface and metabolic markers permits a defined simultaneous isolation. Stem Cell Res 5(2):131–143
Gunhan E, van der List D, Chalupa LM (2003) Ectopic photoreceptors and cone bipolar cells in the developing and mature retina. J Neurosci 23(4):1383–1389
Milam AH, Dacey DM, Dizhoor AM (1993) Recoverin immunoreactivity in mammalian cone bipolar cells. Vis Neurosci 10(1):1–12
Sharma RK, Netland PA (2007) Early born lineage of retinal neurons express class III beta-tubulin isotype. Brain Res 1176:11–17
Katsumoto A, Lu H, Miranda AS, Ransohoff RM (2014) Ontogeny and functions of central nervous system macrophages. J Immunol 193(6):2615–2621
Nikolakopoulou AM, Dutta R, Chen Z, Miller RH, Trapp BD (2013) Activated microglia enhance neurogenesis via trypsinogen secretion. Proc Natl Acad Sci U S A 110(21):8714–8719
West EL, Pearson RA, MacLaren RE, Sowden JC, Ali RR (2009) Cell transplantation strategies for retinal repair. Prog Brain Res 175:3–21
West EL, Pearson RA, Duran Y, Gonzalez-Cordero A, MacLaren RE, Smith AJ, Sowden JC, Ali RR (2012) Manipulation of the recipient retinal environment by ectopic expression of neurotrophic growth factors can improve transplanted photoreceptor integration and survival. Cell Transplant 21(5):871–887
Bhatia B, Singhal S, Jayaram H, Khaw PT, Limb GA (2010) Adult retinal stem cells revisited. Open Ophthalmol J 4:30–38
Roque RS, Rosales AA, Jingjing L, Agarwal N, Al-Ubaidi MR (1999) Retina-derived microglial cells induce photoreceptor cell death in vitro. Brain Res 836(1–2):110–119
Lee YS, Amadi-Obi A, Yu CR, Egwuagu CE (2011) Retinal cells suppress intraocular inflammation (uveitis) through production of interleukin-27 and interleukin-10. Immunology 132(4):492–502
Seita J, Asakawa M, Ooehara J, Takayanagi S, Morita Y, Watanabe N, Fujita K, Kudo M, Mizuguchi J, Ema H, Nakauchi H, Yoshimoto T (2008) Interleukin-27 directly induces differentiation in hematopoietic stem cells. Blood 111(4):1903–1912
Mo FM, Proia AD, Johnson WH, Cyr D, Lashkari K (2010) Interferon gamma-inducible protein-10 (IP-10) and eotaxin as biomarkers in age-related macular degeneration. Invest Ophthalmol Vis Sci 51(8):4226–4236
Yang P, McKay BS, Allen JB, Jaffe GJ (2004) Effect of NF-kappa B inhibition on TNF-alpha-induced apoptosis in human RPE cells. Invest Ophthalmol Vis Sci 45(7):2438–2446
Theodossiadis PG, Liarakos VS, Sfikakis PP, Vergados IA, Theodossiadis GP (2009) Intravitreal administration of the anti-tumor necrosis factor agent infliximab for neovascular age-related macular degeneration. Am J Ophthalmol 147(5):825–830, 830 e821
Acknowledgments
We thank Dr. Lindsay B. Nicholson for advice on data analysis and manuscript writing. The work was supported through Guide Dogs for the Blind Association, UK (OR2009-02e). A.D.D. is partially supported through funding. This work was partly supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology. The views expressed are those of the author (s) (A.D.D.) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Disclosure of potential conflicts of interest
The authors indicate no potential conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
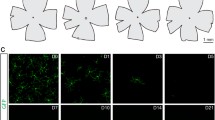
Supplement Fig. 1
Co-expression of Iba-1 in CD11b+ cells by immunofluorescence. CD11b+ cells isolated from donor eye were further cultured for 10 days. Cells were then fixed in paraformaldehyde (4 %), permeabilized in Triton-100, and then stained using mouse-anti-human mAb, Iba-1 (Abcam; 1:100) overnight at 4 °C, and detected following 2 h incubation with FITC-labelled bovine anti-mouse IgG (Santa Cruz; 1:200) at room temperature, mounted and counterstained with DAPI (Vector Laboratories). Photos show phase image (a), DAPI image (b), FITC image (c) and merged image (d); scale bar = 50 μm (DOCX 2195 kb)
Supplement Table 1
(DOCX 18 kb)
Rights and permissions
About this article

Cite this article
Xu, Y., Balasubramaniam, B., Copland, D.A. et al. Activated adult microglia influence retinal progenitor cell proliferation and differentiation toward recoverin-expressing neuron-like cells in a co-culture model. Graefes Arch Clin Exp Ophthalmol 253, 1085–1096 (2015). https://doi.org/10.1007/s00417-015-2961-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-015-2961-y