
Abstract
Introduction
Ubiquitin C-terminal hydrolase L1 (UCHL1) has been associated with a severe, complex autosomal recessive spastic paraplegia (HSP79) [1] [2] [3] [4]. More recently, UCHL1 loss of function (LoF) variants have been associated to an autosomal dominant disease characterized by late-onset spastic ataxia, neuropathy, and frequent optic atrophy [5].
Methods
Routine clinical care whole-genome (WGS) and exome (ES) sequencing.
Results
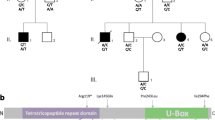
We present three families with autosomal dominant UCHL1-related disorder. The clinical phenotype mainly associated optic atrophy, mixed cerebellar and sensory ataxia, and possible hearing loss. We delineated two major phenotypes, even within the same family: (1) juvenile severe optic atrophy followed by a later-onset ataxia, or (2) late-onset ataxia with asymptomatic or mild optic atrophy. The families harboured three novel heterozygous variants in UCHL1: two loss of function (p.Lys115AsnfsTer40; c.171_174 + 7del11), and one missense (p.Asp176Asn) involving the catalytic site of the protein and potentially altering the adjacent splice site.
Discussion
We confirm the existence of dominantly inherited UCHL1 pathogenic variants. We describe a considerable intrafamilial phenotypic variability, with two main phenotypes. Optic atrophy was consistently present, but with varying degrees of severity. Neither delayed motor or intellectual development, nor dysmorphic features were part of the dominant phenotype in comparison with the autosomal recessive form. The molecular mechanism appears to be haploinsufficiency. UCHL1 monoallelic variants should therefore be considered in any case of early-onset optic atrophy or in late-onset complex ataxic syndrome with asymptomatic optic atrophy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ubiquitin C-terminal hydrolase L1 (UCHL1) has been associated with an autosomal recessive (AR) spastic paraplegia (HSP79) characterized by an early-onset cerebellar ataxia, spastic paraplegia, intellectual deficiency, dysmorphism, and optic atrophy. This is a rare and severe disease with only four families described up to now [1,2,3,4]. More recently, UCHL1 heterozygous loss of function (LoF) variants have been associated to an autosomal dominant (AD) disease characterized by late-onset spastic ataxia, neuropathy, and frequent optic atrophy [5]. We present three independent families (7 subjects) harbouring three novel heterozygous variants in UCHL1, confirming the occurrence of an AD form, clinically different from the recessive one, and showing a great intrafamilial phenotypic variability.
Methods
The variations in the UCHL1 gene were found after genetic investigation in the context of clinical care. Family 1 was analysed by Whole-Genome Sequencing (WGS) using Illumina NovaSeq technology, in the context of the French National Genomic Program (PFMG 2025, AURAGEN platform). Family 2 was analysed by exome sequencing (ES) by Illumina NextSeq technology (https://www.illumina.com/products/by-type/clinical-research-products/trusight-one.html). Sequence alignment and variant calling were performed following the Broad Institute’s Genome Analysis Toolkit (GATK4) best practices. Family 3 was analysed by WGS using Illumina DNA PCR-Free Library Prep and Illumina NovaSeq 6000 platform (Illumina, Inc.). Alignment and variant calling were performed using Illumina DRAGEN Bio.IT platform (Illumina Inc.). Clinical sequence interpretation was performed following the guidelines from the American College of Medical Genetics [6].
This study was approved by the institutional review boards of the Montpellier University Medical Centre (Number: IRB-MTP_2021_11_202100959) and was conducted in accordance with the principles of the Declaration of Helsinki. All the presented individuals gave their consent for the genetic analysis, for sharing data in the context of the research about their disease and for the publication of anonymous data.
Results
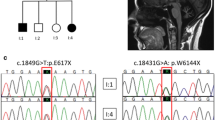
Family 1 (Fig. 1): the proband is a 71 year-old man presenting at the age of 61 years with late-onset ataxia, due to both cerebellar involvement and mild sensory axonal peripheral neuropathy. He had normal visual acuity (20/20 Right Eye; 20/25 left eye) but ophthalmological examination found bilateral temporal disc pallor, decreased nerve fibre layer thickness, and bilateral mild alteration of the temporal upper visual field. Cerebral Magnetic Resonance Imaging (MRI) showed mild hemispheric cerebellar atrophy (Fig. 2). He also had late-onset progressive bilateral and moderate down sloping hearing loss (hearing thresholds were 15 dB above age and sex normative thresholds (ISO7024), requiring hearing aids since the age of 70 years. The personal history revealed noise trauma exposure in young adulthood (hunting). Tonal and vocal audiometry were concordant. Auditory evoked brainstem potentials (BAEP) (at the age of 72 years) found a prolonged central conduction time on the left and non-reproducible responses on the right for a 70 dB click stimulus. The absence of reproducible response at 70 dB on right ear was concordant with subjective elevated thresholds but prolonged central conduction time on left ear could suggest auditory neuropathy, possibly related to his genetic disease. Neurological examination found moderate ataxia (Scale for the Assessment and Rating of Ataxia – SARA: 13/40) with gait ataxia, mild dysarthria, saccadic pursuit, positive Romberg sign, and decreased deep sensation from the lower limbs.

The pedigree of the three families with dominant UCHL1-related disorder and the previously reported UCHL1 mutations. a the pedigree of the three families with the corresponding mutations; the arrow indicates the index case; stars indicate tested family members, with affected presented with closed symbols and unaffected with opened ones. We represented in deep grey the patients with late-onset ataxia, in light grey the patients with juvenile-onset visual loss due optic atrophy, and in black the affected patients with ataxia and optic atrophy for which the phenotype is not better detailed. b the UCHL1 gene (upper) and Uchl1 protein (lower) with mutations described in this report (in purple), previously described recessive mutations (in black), and previously described dominant mutations (in red)

Cerebral MRI of patients with dominant UCHL1-related disease.a bilateral optic nerve atrophy without cerebellar atrophy (subject III.2, family 1); b very mild hemispheric cerebellar atrophy at the age of 71 years (subject II.2, family 1); c no cerebellar atrophy at the age of 40 years (subject III.1, family 2)
The complete work-out for genetic and acquired late-onset cerebellar ataxia was negative, including plasma alpha-fetoprotein, vitamin E, ceruloplasmin, lactate, albumin, protein isoelectrofocusing and urinary organic acids analysis; genetic analyses for Friedreich ataxia, SCA1, SCA2, SCA3, SCA6 and SCA7, CANVAS, FXTAS-fragile X tremor ataxia syndrome) were also negative. A DATSCAN was normal. His brother died at 68 years because of a neurodegenerative disease with ataxia and optic atrophy, with an onset at about 45 years. His son presented at 15 years with severe optic atrophy, reduced central visual acuity (20/200) (Fig. 3), and optic nerve atrophy at cerebral MRI (Fig. 2). He denied any other problems but a neurological examination at 36 years showed a mild cerebellar ataxia (SARA = 4/40). Because of juvenile-onset isolated optic atrophy, he was genetically explored and POLG1 and OPA1 were negative, as well as mtDNA sequencing (notably for Leber hereditary optic neuropathy); biotinidase activity was also normal and a muscle biopsy was unremarkable. WGS studies identified in both the father and his son a heterozygous deletion (NM_004181.5: c.345delA) in UCHL1, predicted to cause a frameshift followed by a premature stop codon (p.(Lys115AsnfsTer40)) and a truncation of half of the UCHL1 protein. This variant is absent in the general population according to the Genome Aggregation Database (gnomADv4). The variant was classified as pathogenic. No other likely pathogenic variations of UCHL1 gene were identified in this family.

Ophthalmological findings a Slit-lamp examination and Optical Coherence Tomography (OCT) (subject III.2, family 2, at 52 years): the optic disc involvement is very mild compared to the macular OCT slice (b), where the maculo-papillary bundle is clearly altered, and compared to the clear reduction of the ganglion fibres on the Retinal Nerve fibre layer (RNFL) (c); d Visual field analysis showing reduced central visual sensibility (Subject III.2, family 1, at the age of 30 years)
Family 2 (Fig. 1): the proband is a 52 year-old-man presenting with problems in the colour vision and fluctuating visual acuity (20/25 to 20/20) since his infancy. At the age of 17 years, the visual field showed a central scotoma, initially in the right eye and then bilaterally. Since the age of 30 years, he progressively developed reduced visual acuity, followed by mixed sensory and cerebellar ataxia (SARA = 8/40). Ophthalmological examination found reduced visual acuity (20/125), optic disc atrophy, and reduced nerve fibre layer thickness. Interestingly, at the ocular fundus the optic disc was only mildly abnormal, whereas optical coherence tomography (OTC) showed marked reduced retinal nerve fibre layer (RNFL) thickness, particularly involving the maculo-papillary bundle (Fig. 3). The cerebral MRI showed severe bilateral optic nerve atrophy, without cerebellar atrophy (Fig. 2). He had no auditory complain (hearing loss or tinnitus). However, in the context of visual and neurological deficiencies, audio-vestibular assessment was performed by the age of 40: audiometry was normal (tonal and vocal, in both quiet and noisy environment); auditory brainstem auditory evoked potential (BAEP) measurements showed normal central conduction time but elevated thresholds (reproducible wave 5 was obtained for 70 dB click stimulus and louder); moreover, distorted otoacoustic emissions were present at 2 et 4 kHz in right ear and at 2 kHz in left ear. These results were in favour of an auditory neuropathy, possibly related to his genetic disorder, without clinical impact. Audiological assessment remained stable 15 years later. Vestibular assessment revealed a left partial deficit in caloric testing, a right directional preponderance (2.5 s) and an alteration of ocular fixation; rotary tests were normal, without any directional preponderance. His brother had optic atrophy with preserved visual acuity. His mother had late-onset (at about 60 years) cerebellar ataxia and optic atrophy. She also complained about late-onset auditory problems but a detailed ORL examination was not available. The initial complete work-out for dominant optic atrophies was negative. ES analysis identified a heterozygous deletion (NM_004181.5: c.171_174 + 7del11) in UCHL1 in both the patient and his affected mother. Segregation analyses of the deletion in the family revealed that his affected brother also carried the deletion while his unaffected maternal aunt did not, which confirms perfect segregation with the disease. The 11 nucleotide deletion removes all of the exon 3 donor splice site as well as the last four nucleotides of the exon. Skipping of exon 3 as a result of donor splice site loss is predicted to result in an in-frame deletion of 43 amino acids (p.(Val16_Gln58del43)) involving 2 of the 6 highly conserved beta-strands of the UCHL1 protein. This variant is absent in the general population (gnomADv4). The variant was classified as pathogenic. No other likely pathogenic variations of UCHL1 gene were identified in this family.
Family 3: The proband presented at the age of 17 years with progressive visual loss due optic atrophy. The ophthalmological examination found reduced visual acuity (about 20/200 bilaterally), optic disc atrophy, and reduced nerve fibre layer thickness. At the age of 37 years, he began to present cerebellar ataxia; the cerebral MRI found a mild cerebellar atrophy. Genetic analysis for ataxias due to CAG expansions (SCA 1, 2, 3, 6, 7, SCA17, DRPLA) and for Friedreich ataxia were negative, as well as a panel of 137 genes responsible for cerebellar ataxia, 37 genes responsible for nuclear mitochondrial disorder (including OPA1) and mitochondrial DNA analysis for Leber hereditary optic neuropathy. He had a family history of dominant cerebellar ataxia spanning four generations (Fig. 1, Family 3). A detailed clinical description was not always available because some family members died many years ago. However, his mother was also affected, presenting cerebellar ataxia since the age of 56 years, as well as his grandfather, presenting with cerebellar ataxia since the age of 50 years. His maternal aunt (now 79 years old) was also affected and presented with mixed sensory and cerebellar ataxia since the age of about 55 years; she had no visual complain but detailed ophthalmological examination has never been performed. WGS analysis identified a heterozygous missense change (NM_004181.5: c.526G > A; NP_004172.2: p.Asp176Asn) in UCHL1 in both the proband and his maternal aunt. His mother was not tested but she was symptomatic and obligate carrier of the same variant. Thus, the variant co-segregates with the phenotype in the family (PP1), with a concordant clinical phenotype (PP4). The p.Asp176Asn variant is absent from the GnomAD v4 database (PM2). Previous mutagenesis studies demonstrated that this variant is deleterious on UCHL1 protein function (PS3) because it impairs ubiquitin binding [7] and virtually eliminates UCHL1 monoubiquitination [8]. Aspartate 176 is conserved in all eukaryotes (PP3, Supplementary Fig. 1). The variant was therefore classified as likely pathogenic (PS3 PM2 PP1 PP3) based on ACMG criteria [6]. In addition, the c.526G > A change is located at the last position of exon 7 and may alter the exon 7 donor splice site (MaxEnt score 7.1 → 2.8, SPIP prediction 98% damaging, SpliceAI Acceptor Loss 0.51, Pangolin Splice Loss 0.76). Unfortunately, protein or mRNA expression studies were not available.
A summary of the demographic, clinical, and radiological features of the three families is presented in Table 1.
Discussion
We present three independent families (7 subjects) harbouring three novel heterozygous variants UCHL1. These observations confirm the existence of AD UCHL1-related disorders, which are remarkably different from the AR UCHL1-related disease, since neither delayed motor or intellectual development, nor dysmorphic features were part of the phenotype. The possible presence of a predominant ataxia and of isolated optic atrophy in AD UCHL1-related disease was already reported in the original paper [5], but with our observation we are able to further precise these phenotypes. In our families, differently from the previously published AD UCLH1-related patients, spasticity was minimal or absent. Moreover, we show a large intrafamilial phenotypic variability: from juvenile-onset severe optic atrophy (followed by mild cerebellar ataxia more than 15 years after disease onset) to late-onset ataxia with asymptomatic optic atrophy.
Optic atrophy was constantly present, if searched through detailed ophthalmological examination, but with different severities. In details, optic atrophy was quite particular, showing a relatively mildly pale optic disc (even in patients with severely reduced visual acuity) but a severe reduction of the retinal nerve fibre layer, and particularly of the interpapillomacular bundle (Fig. 3).
Interestingly, hearing assessment in the proband of family 2 showed a sub-clinical auditory neuropathy, with electrophysiological signs but no functional impact during a 15 years’ follow-up. The proband of family 1 also had electrophysiological signs of auditory neuropathy but also a bilateral sensorineural hearing loss; the aetiology of this hearing loss remained uncertain and may be multifactorial (age related, noise trauma induced). Although our data were not sufficient to affirm the presence of hearing phenotype in UCHL1-related disease, we suggest that a cochleo-vestibular assessment should be performed in patients with UCHL1 variants to detect a possible disease-related hearing involvement.
The exact pathophysiological mechanism leading to AR and AD UCHL1-related disease is not yet completely elucidated [5]. A loss of function mechanism has been considered for the AR forms. However, it seems that AR disease are mainly related to missense mutations, in-frame deletions, or in-frame splicing variants, probably leading to some residual functional protein. These findings globally suggest that the UCHL1 variants found in the AR form could be hypomorphic mutations. Instead, up to now, AD UCLH1-related disorders were related to predicted loss of function variants suggesting a pathogenic mechanism by haploinsufficiency. The only exception is the c.526G > A p.Asp176Asn variant present in Family 3. However, this variant could still lead to haploinsufficiency, through either altered splicing or alteration of catalytic site activity. Finally, a neurotoxic gain of function mechanism cannot be ruled out for this variant, since UCHL1 has a role in deubiquitination of other proteins and ubiquitin homeostasis, and its absence or dysfunction could cause an accumulation of ubiquitinated proteins, which is one of the hallmarks of neurodegenerative diseases [9].
In conclusion, we confirm the existence of an autosomal dominant form of UCLH1-related disorder, characterized by less severe neurological disorders and two main phenotypes, even in the same family: (1) early-onset optic atrophy followed by mild sensory and cerebellar ataxia; (2) late-onset ataxia with mild or asymptomatic optic atrophy. A subtle hearing loss may also be associated with the disease. The variants associated with AD-UCHL1 disease are mainly, if not all, predicted loss of function. We suggest that UCHL1 heterozygous variants should be considered in patients with early-onset optic atrophy or in late-onset complex ataxic syndrome with mild or asymptomatic optic atrophy.
References
Das Bhowmik A, Patil SJ, Deshpande DV et al (2018) Novel splice-site variant of UCHL1 in an Indian family with autosomal recessive spastic paraplegia-79. J Hum Genet 63:927–933. https://doi.org/10.1038/s10038-018-0463-6
Bilguvar K, Tyagi NK, Ozkara C et al (2013) Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc Natl Acad Sci U S A 110:3489–3494. https://doi.org/10.1073/pnas.1222732110
Rydning SL, Backe PH, Sousa MML et al (2017) Novel UCHL1 mutations reveal new insights into ubiquitin processing. Hum Mol Genet 26:1217–1218. https://doi.org/10.1093/hmg/ddx072
McMacken G, Lochmüller H, Bansagi B et al (2020) Behr syndrome and hypertrophic cardiomyopathy in a family with a novel UCHL1 deletion. J Neurol 267:3643–3649. https://doi.org/10.1007/s00415-020-10059-3
Park J, Tucci A, Cipriani V et al (2023) Heterozygous UCHL1 loss-of-function variants cause a neurodegenerative disorder with spasticity, ataxia, neuropathy, and optic atrophy. Genet Med Off J Am Coll Med Genet. https://doi.org/10.1016/j.gim.2023.100961
Richards S, Aziz N, Bale S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Larsen CN, Price JS, Wilkinson KD (1996) Substrate binding and catalysis by ubiquitin C-terminal hydrolases: identification of two active site residues. Biochemistry 35:6735–6744. https://doi.org/10.1021/bi960099f
Meray RK, Lansbury PT (2007) Reversible monoubiquitination regulates the Parkinson disease-associated ubiquitin hydrolase UCH-L1. J Biol Chem 282:10567–10575. https://doi.org/10.1074/jbc.M611153200
Mi Z, Graham SH (2023) Role of UCHL1 in the pathogenesis of neurodegenerative diseases and brain injury. Ageing Res Rev 86:101856. https://doi.org/10.1016/j.arr.2023.101856
Acknowledgements
This research was made possible through access to the data generated by the France Genomic Medicine Program 2025 (PFMG 2025). The authors would also like to thank the patients and their families for accepting to share clinical data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
CM received honorary travel to attend scientific meetings (Nutricia and Vitaflo) and honorary for consultant activity (Biogen, Reata Pharmaceuticals, Medesis Pharma); FR, CV, CB, SF, QH, YN, NL, GT, MB, CH, MK, and IM have nothing to disclose. On behalf of all authors, the corresponding author states that there is no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Marelli, C., Ramond, F., Vignal, C. et al. Phenotypic variability related to dominant UCHL1 mutations: about three families with optic atrophy and ataxia. J Neurol 271, 6038–6044 (2024). https://doi.org/10.1007/s00415-024-12574-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-024-12574-z