
Abstract
Objectives
Outcomes of clinical trials of treatment in patients with Parkinson’s disease (PD) may be influenced by placebo effects. The aim of this study was to determine the factors associated with placebo effects in Parkinson’s disease (PD) for guidance with design of future clinical trials.
Methods
Factors associated with placebo effects in PD were examined in a meta-analysis using a random effects model with pooling of placebo effects on the Unified Parkinson’s Disease Rating Scale part III (UPDRS III) or Movement Disorder Society sponsored revision of UPDRS III (MDS-UPDRS III). The following prespecified variables were included in the analyses: with or without drug at baseline, with or without a placebo run-in phase, with or without motor fluctuation, published year, number of study sites, placebo administration period, age, sex, disease duration, and daily levodopa dose. Publication bias was assessed by visual inspection of funnel plots and adjusted using the trim-and-fill method.
Results
Thirty-eight articles with a total of 4828 subjects satisfied the inclusion criteria. There was a significant placebo effect using UPDRS III or MDS-UPDRS III (SMD = − 0.25; 95% CI − 0.35 to − 0.14; p < 0.001, I2 = 92%). Subgroup and/or multivariate meta-regression analyses revealed that placebo effects were associated with advanced PD (p = 0.04), drug exposure at baseline (p < 0.001), placebo administration period (p < 0.001), and disease duration (p < 0.01).
Conclusions
The results of this study are important as guidance in design of future clinical trials in which the influence of placebo effects is minimized.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Placebos are drugs, devices or treatments that are physically and pharmacologically inert [1]. Placebo effects may be associated with release of molecules such as dopamine [2, 3], endogenous opioids [4, 5], endocannabinoids [6], oxytocin [7], and vasopressin [8], resulting in clinical improvements in many medical conditions. A pronounced placebo effect occurs in Parkinson’s disease (PD) and was seen in 8–9% of subjects assigned to placebo in a 24-week, randomized, double-blind, placebo-controlled clinical trial, with improvement of symptoms in the order of bradykinesia (94%), rigidity (76%), gait balance/midline function (59%), and tremor (47%) [9]. These improvements were induced by dopamine release in the striatum [10], which altered neuronal activity in the basal ganglia and thalamus [2, 11, 12].
Placebo effects are particularly important in PD clinical practice because improvements are common and marked, and affect the results of clinical trials [13]. Research designs and adjustment of placebo-related factors have been proposed to minimize placebo effects and increase the success of clinical trials, but the best approach for diminishing the placebo effect remains unclear. In this vein, previous studies have suggested that the placebo effect could be associated with prior drug exposure, placebo administration period, and the severity/stage of PD [9, 14,15,16,17]; however, a systematic meta-analysis to find a way to minimize placebo effects has not been performed. Here, we identify placebo-related factors and propose a design to control these factors using a meta-analysis of randomized studies.
Methods
Search strategy
The Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) statement was utilized to guide the methodology of the meta-analysis (Supplementary Table 1) [18]. The inclusion criteria were as follows: (1) randomized, placebo-controlled, double-blind, parallel-group design; (2) diagnosis of PD using international consensus criteria including UK Parkinson’s Disease Society Brain Bank criteria [19], Gibb’s criteria [20], Calne’s criteria [21], Gelb’s criteria [22], or Ward and Gibb’s criteria [23]; (3) oral drug proved to be effective for motor symptoms in phase III clinical trials; (4) evaluation of motor symptoms as the primary endpoint; (5) assessment of change in the “on” state using UPDRS III or MDS-UPDRS III from baseline to endpoint; (6) at least 10 subjects reached the endpoint in each group; and (7) written in English. Withdrawal studies in which participants were randomized to continue the investigational drug or placebo after a defined period of the investigational drug administration were excluded. A comprehensive search of three electronic databases (PubMed, Scopus, and Cochrane Library) was conducted on 31st October, 2021. A search of ClinicalTrials.gov of the reference sections of all included articles was also performed. The search terms were (“Parkinson’s disease” OR “Parkinson disease”) AND (“random” OR “randomly” OR “randomized”) AND “placebo”. Two authors (S.H., N.M.) independently evaluated potentially eligible studies identified in the search, after which discrepancies were resolved by mutual agreement between S.H. and N.M.
Data extraction and outcome measures
Data extraction was completed by S.H. and cross-checked by N.M. Intention-to-treat data were used if possible. Extracted data included the publication year, number of study sites, with or without placebo run-in phase, proportion of patients assigned to placebo, treatment period, with or without motor fluctuation, with or without drug treatment at enrollment, age, sex distribution, UPDRS III or MDS-UPDRS III scores in “on” state from baseline to endpoint, disease duration, Hoehn and Yahr stage, levodopa daily dose, rate of withdrawal in the placebo group, and rate of withdrawal due to adverse effects in the placebo group; however, UPDRS III or MDS-UPDRS III scores at the baseline were assessed in the “Without drug at baseline” during the off period. Placebo run-in phase occurs before randomization and all study-eligible subjects are given the placebo treatment [24]. S.H. assessed the risk of bias using the risk of bias tool 2.0 [25] and N.M. cross-checked the result.
Statistical analysis
Means and standard deviations not reported in the original articles were estimated from medians, ranges, and interquartile ranges [26]. Summary statistics were calculated using the DerSimonian and Laird random-effects model [27]. The primary outcome was the change in “on” state for UPDRS III or MDS-UPDRS III scores in the placebo arm. The standardized mean change using change score standardization (SMCC), a type of standardized mean difference (SMD), was used to combine each effect (Hedge’s g). Differences were computed by single group pretest–post-test design, using the following equations: [28, 29]
where yi is the effect size; vi is the variance; Mpre is UPDRS III or MDS-UPDRS part III at baseline; and Mpost is UPDRS III or MDS-UPDRS part III at the endpoint.
Heterogeneity between studies was assessed using Q and I2 statistics, with p < 0.1 or I2 > 50% indicating significant heterogeneity. Subgroup and meta-regression analyses were applied to explore possible sources of heterogeneity. In subgroup analyses, studies were stratified into “Early” or “Advanced” with a cutoff at 50% of recruited patients with motor fluctuations. Studies categorized as “Early” were then classified as “Without drug at baseline” or “With drug at baseline” if participants had not or had received levodopa, a dopamine agonist, amantadine, or a monoamine oxidase B inhibitor at enrollment, respectively. “Advanced” studies were further categorized as “Without a run-in phase” or “With a run-in phase” if a placebo run-in phase was not or was used, respectively.
In the univariate meta-regression analysis, a standard linear mixed effects model was first applied. If this model did not fit the data, a quadratic or cubic polynomial model was used, while in multivariate meta-regression analyses, forced entry was applied to include potential covariates, and a Pearson correlation coefficient > 0.6 was used to check multicollinearity among covariates. The following covariates were prespecified to be included in subgroup and meta-regression analyses: with or without motor fluctuation, with or without drug at baseline, with or without a placebo run-in phase, age, sex distribution, and levodopa daily dose. Publication bias was assessed using visual inspection of funnel plots, and adjusted using the trim-and-fill method [30]. Sensitivity analyses were also performed. p < 0.05 was considered to be significant. All analyses were carried out using the “meta”, “metafor”, “ggplot2”, “regplot”, and “corrplot” packages in the R statistical computing environment ver. 4.0.3 (http://www.r-project.org).
Results
Study characteristics
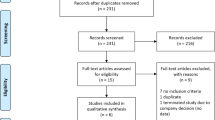
A total of 38 studies [31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68] with 4,828 subjects were included in the meta-analysis (Fig. 1). Of these studies, 21 had a low risk of bias and 17 had a moderate risk (Supplementary Fig. 1). Sixteen studies were in “Early” PD and 22 in “Advanced” PD patients. Of the “Early” studies, 9 were “Without drug at baseline” and 7 were “With drug at baseline”. The “Advanced” studies included 19 “Without a run-in phase” and 3 “With a run-in phase”. The characteristics of all the studies are shown in Table 1. The pooled mean baseline data were: treatment period (range 4–38.6 weeks), study sites (1–129), number of patients administered placebo (20–595), sex distribution (male, 36.4–80.0%), age (59.5–70.2 years), Hoehn and Yahr stage (1.5–3.0), “on” state using UPDRS III or MDS-UPDRS III at baseline (13.9–32.1), levodopa daily dose (0–948 mg/day), and use of the following drugs: entacapone, istradefylline, nebicapone, opicapone, pardoprunox, piribedil, pramipexole, rasagiline, safinamide, tolcapone, tavapadon, and zonisamide.

Flowchart for selection of eligible studies
Placebo effect in patients with Parkinson’s disease
Placebo significantly improved UPDRS III or MDS-UPDRS III in heterogenous studies (SMD = − 0.25, 95% CI − 0.35 to − 0.14, p < 0.001, I2 = 92%; Fig. 2). In subgroup analysis, placebo was not significant in “Early” studies (SMD = − 0.13, 95% CI − 0.30 to 0.04, p = 0.15, I2 = 94%; Fig. 2), but was significant in “Advanced” studies (SMD = − 0.33, 95% CI − 0.41 to − 0.25, p < 0.001, I2 = 70%; Fig. 2). The placebo effect in “Advanced” studies was significantly higher than that in “Early” studies (p = 0.04; Fig. 2). Stratification of the “Early” and “Advanced” studies revealed significant differences in the placebo effect among four subgroups (p < 0.01). In “Early” studies, the placebo effect was not significant in those “Without drug at baseline” (SMD = − 0.01, 95% CI − 0.24 to 0.22, p = 0.94, I2 = 93%), but was significant in those “With drug at baseline” (SMD = − 0.24, 95% CI − 0.33 to − 0.15, p < 0.001, I2 = 39%) (group difference: p = 0.06; Fig. 3). In “Advanced” studies, the placebo effect was significantly higher “With a run-in phase” (SMD = − 0.46, 95% CI − 0.57 to − 0.34, p < 0.001, I2 = 0%) than “Without a run-in phase” (SMD = − 0.31, 95% CI − 0.40 to − 0.22, p < 0.001, I2 = 71%) (group difference: p = 0.04; Fig. 3).

Forest plot showing placebo effects in studies stratified into “Early” and “Advanced” PD. Results on the left indicate improvement with placebo. There was a significant difference in placebo effect between “Early” and “Advanced” studies (p = 0.04)

Forest plot showing placebo effects in studies “Without drug at baseline”, “With drug at baseline”, “Without a run-in phase”, and “With a run-in phase”. “Early” were subdivided to “Without drug at baseline” and “With drug at baseline”. “Advanced” were stratified to “Without a run-in phase” and “With a run-in phase”. There were significant differences between groups (p < 0.01). The placebo effect was not significant in “Without drug at baseline” studies, but was higher in studies “With a run-in phase” than “Without a run-in phase” (p = 0.04). Run-in phase, which was originally expected to attenuate the placebo effect, did not suppress the placebo effect
Meta-regression analysis
The results of univariate and multivariate meta-regression analyses for all studies and for studies “Without drug at baseline” and “Without a run-in phase” are presented in Supplementary Table 2. Bubble plots using univariate meta-regression analysis are shown in Fig. 4. For all studies, univariate meta-regression analyses showed that “Without drug at baseline” (coefficient − 0.33, 95% CI − 0.51 to − 0.16, p < 0.001, R2 = 56.8%), longer placebo administration period (coefficient 0.02, 95% CI 0.01 to 0.03, p < 0.001, R2 = 54.0%), and lower UPDRS III or MDS-UPDRS III scores at baseline (coefficient − 0.03, 95% CI − 0.05 to − 0.01, p < 0.01, R2 = 41.1%) were significantly related to a lower placebo effect (Fig. 4a–c). Disease duration was not significantly associated with a placebo effect using a linear mixed effects model (coefficient − 0.03, 95% CI − 0.05 to 0.00, p = 0.05, R2 = 32.6%; Fig. 4d), but a quadratic polynomial model revealed a significant association (coefficient − 0.16, 95% CI − 0.26 to − 0.07, p < 0.01, R2 = 30.7%; Supplementary Fig. 2). Univariate meta-regression analysis showed that the placebo effect dissipated after 225 days of placebo administration.

Bubble plots for studies “Without drug at baseline” and “Without a run-in phase”. In univariate meta-regression analysis, a lower placebo effect was significantly associated with “Without drug at baseline”, longer placebo administration period, and lower UPDRS III or MDS-UPDRS III scores at baseline. There was no significant association between placebo effect and disease duration. The placebo administration period and disease duration explained heterogeneity in “Without drug at baseline” and “Without a run-in phase” studies, respectively
Multivariate meta-regression analysis revealed that the four significant factors in univariate analysis explained 84% of the variance in estimates across studies (“Without drug at baseline”: coefficient − 0.42, 95% CI − 0.60 to − 0.23, p < 0.001; placebo administration period: coefficient 0.02, 95% CI 0.01 to 0.03, p < 0.001; UPDRS III or MDS-UPDRS III scores at baseline: coefficient − 0.02, 95% CI − 0.03 to 0.00, p = 0.03; disease duration: coefficient 0.04, 95% CI 0.01 to 0.06, p < 0.01; all moderators: p < 0.001, R2 = 83.7%). No significant correlation was found between these covariates.
Meta-regression analyses of studies “Without drug at baseline” and “Without a run-in phase” were performed to explore the cause of heterogeneity. In those “Without drug at baseline”, a longer placebo administration period was a significant predictor of a lower placebo effect and the magnitude of the effect reached zero after 154 days of administration (coefficient 0.03; 95% CI 0.02 to 0.04; p < 0.001; R2 = 83.8%; Fig. 4e). In “Without a run-in phase” studies, a longer disease duration was significantly associated with a lower placebo effect (coefficient 0.09; 95% CI 0.05 to 0.13; p < 0.001; R2 = 72.1%; Fig. 4f).
Publication bias and sensitivity analysis
Visual inspections of funnel plots showed symmetry for “Without a run-in phase” studies, but asymmetry for the other three groups (Supplementary Fig. 3). The trim-and-fill adjusted results were stable for “Without a run-in phase” (SMD = − 0.31, 95% CI − 0.40 to − 0.22, p < 0.001, I2 = 71.1%; Supplementary Fig. 3D) and attenuated the placebo effect in the other three groups (“Without drug at baseline”, SMD = 0.29, 95% CI 0.05 to 0.52, p = 0.02, I2 = 94.8%; “With drug at baseline”, SMD = − 0.19, 95% CI − 0.28 to − 0.09, p < 0.001, I2 = 52.7%; “With a run-in phase”, SMD = − 0.45, 95% CI − 0.55 to − 0.34, p < 0.001, I2 = 0.0%; Supplementary Fig. 3A, B, C). Jackknife sensitivity analysis showed that all results in the meta-analysis were highly reproducible (Supplementary Figs. 4 and 5). These findings indicate the stability of the results.
Discussion
In this meta-analysis, the placebo effect in PD was analyzed separately for all studies and for studies in early and advanced stages of PD, with the following results. (1) There was no placebo effect in studies without use of a drug at baseline. (2) In the analysis in all studies, the placebo effect decreased as the placebo administration period increased, and the effect disappeared after about 7 months. This trend was particularly pronounced for studies without use of a drug at baseline in early-stage PD. (3) The placebo effect was lower in studies of advanced PD without a placebo run-in phase than with a placebo run-in phase. (4) The placebo effect was lower in all studies and in early-stage PD studies with a drug at baseline as disease duration was shorter, and in advanced-stage PD studies as disease duration was longer.
The absence of a placebo effect for studies without a drug at baseline is due to the effect being related to previous drug exposure and its learning effect [14, 69]. Therefore, the first placebo administration does not induce a placebo effect [14, 70]. The current results are consistent with previous reports, although the patients in studies without a drug at baseline were not strictly drug-naive due to inclusion of some patients with a history of prior drug use. The placebo effect may become even smaller if only drug-naive patients are included. Among the studies analyzed, only two reported the number of drug-naive patients [33, 37]. Given that the history of prior drug use affects the placebo effect, the proportion of drug-naive patients should be presented in future clinical trials.
There are multiple lines of evidence that the placebo effect is largely related to expectancy [71, 72]. The expectancy of this effect fades over time. The decrease in the placebo effect with an increased duration of placebo administration and the disappearance of the placebo effect about 230 days after the start of administration are in line with a systematic review that found that placebo-associated improvements occurred throughout a 6-month study [9]. The positive correlation between the placebo effect and duration of placebo administration was strong in studies without a drug at baseline in early stage PD, suggesting that the duration of placebo administration is one of the main causes of heterogeneity in studies without a drug at baseline.
In advanced PD, the placebo effect was enhanced by a placebo run-in phase. Such a run-in phase was originally expected to attenuate the placebo effect [24], but questions have recently been raised concerning placebo suppression by a placebo run-in phase in other diseases [15]. Time and expense spent on clinical trials can be saved if a placebo run-in phase is not required. However, we note that in the analyzed studies this phase was relatively short (1–4 weeks) and a longer period might result in placebo suppression.
The placebo effect was significantly lower with a longer disease duration in studies of advanced PD, but significantly lower with a shorter disease duration in those in early stage PD with a drug at baseline. Thus, the relationship between disease duration and placebo effect differs between early and advanced stages and may be biphasic depending on the disease stage, which was supported by the quadratic polynomial model (Supplementary Fig. 2). The ventral striatum has been linked to the placebo effect. In pain, placebo treatments induce a functional MRI response and dopamine release in the ventral striatum measured by PET. Activation of the ventral striatum during pain is a predictor of high efficacy of opioid analgesia [73]. Moreover, patients with pathological gambling tend to have a higher placebo effect and upregulate release of dopamine in the ventral striatum [74]. PD is characterized by different degeneration in the substantia nigra pars compacta (SNpc) and ventral tegmental area in the midbrain, resulting in different dopamine levels in the efferent dorsal striatum and ventral striatum. SNpc is more affected than the ventral tegmental area in early PD, leading to dominant dopamine depletion of the dorsal striatum. Therefore, the “overdose hypothesis” proposes that dopaminergic therapy overstimulates the ventral striatum [75]. In early PD, this overstimulation becomes stronger as the disease progresses, whereas in advanced PD, the overstimulation is weakened because of degeneration in the ventral tegmental area. In addition, negative experiences such as dyskinesia and visual hallucinations increase with disease duration in advanced PD, which may attenuate the placebo effect. Thus, different disease stage-dependent degeneration may lead to differences in the placebo effect between early and advanced stages.
The placebo effect has been reported to be larger with a higher UPDRS III score at baseline [17], and placebo administration facilitates more dopamine release in patients with severe symptoms than in those with mild symptoms, suggesting that disease severity determines the size of the placebo effect [10]. Using all studies in this analysis, the placebo effect was positively correlated with UPDRS III and MDS-UPDRS III scores at baseline, in agreement with previous reports [17].
There are several limitations to keep in mind when interpreting the results of this study. A meta-analysis demonstrated a placebo effect in sham surgery during the off state [76], whereas the current study only assessed the placebo effect in the on state, due to lack of sufficient studies using UPDRS in the off state. Non-motor symptoms, such as cognition, mood and psychosis, were also not included in this analysis because effects of these factors were not shown in the included studies. Also, despite correction for publication bias using the trim-and-fill method, this bias could not be excluded.
Conclusion
This meta-analysis identified influences of treatment history and disease duration on the placebo effect in PD treatment. The effect was lower for groups with drugs at baseline in early stage PD as disease duration was shorter, and in advanced-stage PD as disease duration was longer. The placebo effect disappeared about 7 months after administration of placebo. A placebo run-in phase failed to attenuate the placebo effect. Based on these findings, we recommend that future randomized controlled clinical trials for patients with PD in the early and advanced stages match treatment history and disease duration between the placebo and active drug groups, and use a follow-up period of at least 7 months. In addition, a placebo run-in phase is not recommended. Knowledge of the factors involved in the placebo effect will both improve the quality of randomized controlled clinical trials and enhance drug efficacy for patients in clinical practice.
Data availability
Data in the manuscript was obtained from original publications.
References
Wager TD, Atlas LY (2015) The neuroscience of placebo effects: connecting context, learning and health. Nat Rev Neurosci 16(7):403–418
Benedetti F, Colloca L, Torre E et al (2004) Placebo-responsive Parkinson patients show decreased activity in single neurons of subthalamic nucleus. Nat Neurosci 7(6):587–588
Lidstone SC, Schulzer M, Dinelle K et al (2010) Effects of expectation on placebo-induced dopamine release in Parkinson disease. Arch Gen Psychiatry 67(8):857–865
Amanzio M, Benedetti F (1999) Neuropharmacological dissection of placebo analgesia: expectation-activated opioid systems versus conditioning-activated specific subsystems. J Neurosci 19(1):484–494
Eippert F, Bingel U, Schoell ED et al (2009) Activation of the opioidergic descending pain control system underlies placebo analgesia. Neuron 63(4):533–543
Benedetti F, Amanzio M, Rosato R, Blanchard C (2011) Nonopioid placebo analgesia is mediated by CB1 cannabinoid receptors. Nat Med 17(10):1228–1230
Kessner S, Sprenger C, Wrobel N, Wiech K, Bingel U (2013) Effect of oxytocin on placebo analgesia: a randomized study. JAMA 310(16):1733–1735
Colloca L, Pine DS, Ernst M, Miller FG, Grillon C (2016) Vasopressin boosts placebo analgesic effects in women: a randomized trial. Biol Psychiatry 79(10):794–802
Goetz CG, Leurgans S, Raman R, Stebbins GT (2000) Objective changes in motor function during placebo treatment in PD. Neurology 54(3):710–714
de la Fuente-Fernández R, Ruth TJ, Sossi V, Schulzer M, Calne DB, Stoessl AJ (2001) Expectation and dopamine release: mechanism of the placebo effect in Parkinson’s disease. Science 293(5532):1164–1166
Benedetti F, Lanotte M, Colloca L, Ducati A, Zibetti M, Lopiano L (2009) Electrophysiological properties of thalamic, subthalamic and nigral neurons during the anti-parkinsonian placebo response. J Physiol 587(Pt 15):3869–3883
Frisaldi E, Carlino E, Lanotte M, Lopiano L, Benedetti F (2014) Characterization of the thalamic-subthalamic circuit involved in the placebo response through single-neuron recording in Parkinson patients. Cortex 60:3–9
Witek N, Stebbins GT, Goetz CG (2018) What influences placebo and nocebo responses in Parkinson’s disease? Mov Disord 33(8):1204–1212
Frisaldi E, Carlino E, Zibetti M et al (2017) The placebo effect on bradykinesia in Parkinson’s disease with and without prior drug conditioning. Mov Disord 32(10):1474–1478
Enck P, Bingel U, Schedlowski M, Rief W (2013) The placebo response in medicine: minimize, maximize or personalize? Nat Rev Drug Discov 12(3):191–204
Benedetti F, Frisaldi E, Carlino E et al (2016) Teaching neurons to respond to placebos. J Physiol 594(19):5647–5660
Shin CW, Hahn S, Park BJ, Kim JM, Park EO, Jeon B (2016) Predictors of the placebo response in clinical trials on Parkinson’s disease: a meta-analysis. Parkinsonism Relat Disord 29:83–89
Moher D, Shamseer L, Clarke M et al (2015) Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst Rev 4(1):1
Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55(3):181–184
Gibb WR, Lees AJ (1988) The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry 51(6):745–752
Calne DB, Snow BJ, Lee C (1992) Criteria for diagnosing Parkinson’s disease. Ann Neurol 32(Suppl):S125-127
Gelb DJ, Oliver E, Gilman S (1999) Diagnostic criteria for Parkinson disease. Arch Neurol 56(1):33–39
Ward CD, Gibb WR (1990) Research diagnostic criteria for Parkinson’s disease. Adv Neurol 53:245–249
Lee S, Walker JR, Jakul L, Sexton K (2004) Does elimination of placebo responders in a placebo run-in increase the treatment effect in randomized clinical trials? A meta-analytic evaluation. Depress Anxiety 19(1):10–19
Sterne JAC, Savovic J, Page MJ et al (2019) RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ (Clin Res Ed) 366:l4898
Wan X, Wang W, Liu J, Tong T (2014) Estimating the sample mean and standard deviation from the sample size, median, range and/or interquartile range. BMC Med Res Methodol 14:135
DerSimonian R, Laird N (1986) Meta-analysis in clinical trials. Control Clin Trials 7(3):177–188
Gibbons RD, Hedeker DR, Davis JM (1993) Estimation of effect size from a series of experiments involving paired comparisons. J Educ Stat 18(3):271–279
Morris SB, DeShon RP (2002) Combining effect size estimates in meta-analysis with repeated measures and independent-groups designs. Psychol Methods 7(1):105–125
Duval S, Tweedie R (2000) Trim and fill: a simple funnel-plot-based method of testing and adjusting for publication bias in meta-analysis. Biometrics 56(2):455–463
Adler CH, Singer C, O’Brien C et al (1998) Randomized, placebo-controlled study of tolcapone in patients with fluctuating Parkinson disease treated with levodopa-carbidopa. Tolcapone Fluctuator Study Group III. Arch Neurol 55(8):1089–1095
Baas H, Beiske AG, Ghika J et al (1997) Catechol-O-methyltransferase inhibition with tolcapone reduces the “wearing off” phenomenon and levodopa requirements in fluctuating parkinsonian patients. J Neurol Neurosurg Psychiatry 63(4):421–428
Bronzova J, Sampaio C, Hauser RA et al (2010) Double-blind study of pardoprunox, a new partial dopamine agonist, in early Parkinson’s disease. Mov Disord 25(6):738–746
Fernandez HH, Greeley DR, Zweig RM et al (2010) Istradefylline as monotherapy for Parkinson disease: results of the 6002-US-051 trial. Parkinsonism Relat Disord 16(1):16–20
Ferreira JJ, Rascol O, Poewe W et al (2010) A double-blind, randomized, placebo and active-controlled study of nebicapone for the treatment of motor fluctuations in Parkinson’s disease. CNS Neurosci Ther 16(6):337–347
Hattori N, Takeda A, Takeda S et al (2018) Efficacy and safety of adjunctive rasagiline in Japanese Parkinson’s disease patients with wearing-off phenomena: a phase 2/3, randomized, double-blind, placebo-controlled, multicenter study. Parkinsonism Relat Disord 53:21–27
Hattori N, Takeda A, Takeda S et al (2018) Rasagiline monotherapy in early Parkinson’s disease: a phase 3, randomized study in Japan. Parkinsonism Relat Disord 60:146–152
Hattori N, Tsuboi Y, Yamamoto A, Sasagawa Y, Nomoto M, Group MES (2020) Efficacy and safety of safinamide as an add-on therapy to l-DOPA for patients with Parkinson’s disease: a randomized, double-blind, placebo-controlled, phase II/III study. Parkinsonism Relat Disord 75:17–23
Hauser RA, Hubble JP, Truong DD (2003) Randomized trial of the adenosine A(2A) receptor antagonist istradefylline in advanced PD. Neurology 61(3):297–303
Hauser RA, Molho E, Shale H, Pedder S, Dorflinger EE (1998) A pilot evaluation of the tolerability, safety, and efficacy of tolcapone alone and in combination with oral selegiline in untreated Parkinson’s disease patients. Tolcapone De Novo Study Group. Mov Disord 13(4):643–647
Hauser RA, Schapira AH, Rascol O et al (2010) Randomized, double-blind, multicenter evaluation of pramipexole extended release once daily in early Parkinson’s disease. Mov Disord 25(15):2542–2549
Hauser RA, Shulman LM, Trugman JM et al (2008) Study of istradefylline in patients with Parkinson’s disease on levodopa with motor fluctuations. Mov Disord 23(15):2177–2185
Hauser RA, Silver D, Choudhry A, Eyal E, Isaacson S (2014) Randomized, controlled trial of rasagiline as an add-on to dopamine agonists in Parkinson’s disease. Mov Disord 29(8):1028–1034
Lees AJ, Ferreira J, Rascol O et al (2017) Opicapone as adjunct to levodopa therapy in patients with Parkinson disease and motor fluctuations: a randomized clinical trial. JAMA Neurol 74(2):197–206
LeWitt PA, Guttman M, Tetrud JW et al (2008) Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson’s disease: a double-blind, randomized, multicenter clinical trial (6002-US-005). Ann Neurol 63(3):295–302
Mizuno Y, Yanagisawa N, Kuno S et al (2003) Randomized, double-blind study of pramipexole with placebo and bromocriptine in advanced Parkinson’s disease. Mov Disord 18(10):1149–1156
Murata M, Hasegawa K, Kanazawa I et al (2015) Zonisamide improves wearing-off in Parkinson’s disease: a randomized, double-blind study. Mov Disord 30(10):1343–1350
Olanow CW, Kieburtz K, Leinonen M et al (2017) A randomized trial of a low-dose Rasagiline and Pramipexole combination (P2B001) in early Parkinson’s disease. Mov Disord 32(5):783–789
Olanow CW, Kieburtz K, Stern M et al (2004) Double-blind, placebo-controlled study of entacapone in levodopa-treated patients with stable Parkinson disease. Arch Neurol 61(10):1563–1568
Olanow CW, Rascol O, Hauser R et al (2009) A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 361(13):1268–1278
Pinter MM, Pogarell O, Oertel WH (1999) Efficacy, safety, and tolerance of the non-ergoline dopamine agonist pramipexole in the treatment of advanced Parkinson’s disease: a double blind, placebo controlled, randomised, multicentre study. J Neurol Neurosurg Psychiatry 66(4):436–441
Poewe W, Rascol O, Barone P et al (2011) Extended-release pramipexole in early Parkinson disease: a 33-week randomized controlled trial. Neurology 77(8):759–766
Pogarell O, Gasser T, van Hilten JJ et al (2002) Pramipexole in patients with Parkinson’s disease and marked drug resistant tremor: a randomised, double blind, placebo controlled multicentre study. J Neurol Neurosurg Psychiatry 72(6):713–720
Pourcher E, Fernandez HH, Stacy M, Mori A, Ballerini R, Chaikin P (2012) Istradefylline for Parkinson’s disease patients experiencing motor fluctuations: results of the KW-6002-US-018 study. Parkinsonism Relat Disord 18(2):178–184
Rajput AH, Martin W, Saint-Hilaire MH, Dorflinger E, Pedder S (1997) Tolcapone improves motor function in parkinsonian patients with the “wearing-off” phenomenon: a double-blind, placebo-controlled, multicenter trial. Neurology 49(4):1066–1071
Rascol O, Bronzova J, Hauser RA et al (2012) Pardoprunox as adjunct therapy to levodopa in patients with Parkinson’s disease experiencing motor fluctuations: results of a double-blind, randomized, placebo-controlled, trial. Parkinsonism Relat Disord 18(4):370–376
Rascol O, Dubois B, Caldas AC et al (2006) Early piribedil monotherapy of Parkinson’s disease: a planned seven-month report of the REGAIN study. Mov Disord 21(12):2110–2115
Rascol O, Fitzer-Attas CJ, Hauser R et al (2011) A double-blind, delayed-start trial of rasagiline in Parkinson’s disease (the ADAGIO study): prespecified and post-hoc analyses of the need for additional therapies, changes in UPDRS scores, and non-motor outcomes. Lancet Neurol 10(5):415–423
Riesenberg R, Werth J, Zhang Y, Duvvuri S, Gray D (2020) PF-06649751 efficacy and safety in early Parkinson’s disease: a randomized, placebo-controlled trial. Ther Adv Neurol Disord 13:1756286420911296
Sampaio C, Bronzova J, Hauser RA et al (2011) Pardoprunox in early Parkinson’s disease: results from 2 large, randomized double-blind trials. Mov Disord 26(8):1464–1476
Schapira AH, Barone P, Hauser RA et al (2011) Extended-release pramipexole in advanced Parkinson disease: a randomized controlled trial. Neurology 77(8):767–774
Schapira AH, Fox SH, Hauser RA et al (2017) Assessment of safety and efficacy of safinamide as a levodopa adjunct in patients with Parkinson disease and motor fluctuations: a randomized clinical trial. JAMA Neurol 74(2):216–224
Schapira AH, McDermott MP, Barone P et al (2013) Pramipexole in patients with early Parkinson’s disease (PROUD): a randomised delayed-start trial. Lancet Neurol 12(8):747–755
Shan DE, Lee SJ, Chao LY, Yeh SI (2001) Gait analysis in advanced Parkinson’s disease—effect of levodopa and tolcapone. Can J Neurol Sci 28(1):70–75
Takeda A, Takahashi R, Tsuboi Y et al (2021) Randomized, controlled study of opicapone in Japanese Parkinson’s patients with motor fluctuations. Mov Disord 36(2):415–423
Zhang L, Zhang Z, Chen Y et al (2013) Efficacy and safety of rasagiline as an adjunct to levodopa treatment in Chinese patients with Parkinson’s disease: a randomized, double-blind, parallel-controlled, multi-centre trial. Int J Neuropsychopharmacol 16(7):1529–1537
Zhang Z, Shao M, Chen S et al (2018) Adjunct rasagiline to treat Parkinson’s disease with motor fluctuations: a randomized, double-blind study in China. Transl Neurodegener 7:14
Zhang Z, Wang J, Chen S et al (2018) Efficacy and safety of rasagiline in Chinese patients with early Parkinson’s disease: a randomized, double-blind, parallel, placebo-controlled, fixed-dose study. Transl Neurodegener 7:32
Quattrone A, Barbagallo G, Cerasa A, Stoessl AJ (2018) Neurobiology of placebo effect in Parkinson’s disease: what we have learned and where we are going. Mov Disord 33(8):1213–1227
Colloca L, Barsky AJ (2020) Placebo and Nocebo effects. N Engl J Med 382(6):554–561
de la Fuente-Fernández R, Phillips AG, Zamburlini M et al (2002) Dopamine release in human ventral striatum and expectation of reward. Behav Brain Res 136(2):359–363
Benedetti F, Pollo A, Lopiano L, Lanotte M, Vighetti S, Rainero I (2003) Conscious expectation and unconscious conditioning in analgesic, motor, and hormonal placebo/nocebo responses. J Neurosci 23(10):4315–4323
Wanigasekera V, Lee MC, Rogers R et al (2012) Baseline reward circuitry activity and trait reward responsiveness predict expression of opioid analgesia in healthy subjects. Proc Natl Acad Sci USA 109(43):17705–17710
Linnet J, Møller A, Peterson E, Gjedde A, Doudet D (2011) Dopamine release in ventral striatum during Iowa Gambling Task performance is associated with increased excitement levels in pathological gambling. Addiction 106(2):383–390
Cools R, Barker RA, Sahakian BJ, Robbins TW (2001) Enhanced or impaired cognitive function in Parkinson’s disease as a function of dopaminergic medication and task demands. Cereb Cortex (New York, NY: 1991) 11(12):1136–1143
Polgar S, Buultjens M, Wijeratne T, Finkelstein DI, Mohamed S, Karimi L (2022) The placebo response in double-blind randomised trials evaluating regenerative therapies for Parkinson’s disease: a systematic review and meta-analysis. J Parkinsons Dis 12(3):759–771
Acknowledgements
This work was supported by JSPS KAKENHI Grant number 23K11880.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
SH has received honoraria from AbbVie Inc., Kyowa Kirin Co. Ltd, and Sumitomo Pharma Co. Ltd. WS has received honoraria from AbbVie Inc., Eisai Co. Ltd, Kyowa Kirin Co. Ltd, Ono Co. Ltd, Sumitomo Pharma Co. Ltd, and Takeda Pharmaceutical Co. Ltd; has received research support from Mitsubishi Tanabe Pharma Co. and JSPS KAKENHI. YI has received honoraria from Mitsubishi Tanabe Pharma Co.; has received research support from Sumitomo Pharma Co. Ltd and Eisai Co. Ltd. Other authors report no competing interests.
Ethical statement
No ethical approval was seeked as only data from previously published studies in which informed consent was obtained were retrieved and analyzed.
Supplementary Information
Below is the link to the electronic supplementary material.
415_2024_12529_MOESM2_ESM.tif
Supplementary file2 Supplementary Fig. S2: Bubble plot using linear regression and quadratic polynomial model. The left figure was applied linear regression model and the right side was calculated by quadratic polynomial model (TIF 159 KB)
415_2024_12529_MOESM3_ESM.tif
Supplementary file3 Supplementary Fig. S3: Funnel plot adjusted using the trim and fill method in each group. Visual inspections of the funnel plots showed symmetry in “Without a run-in phase” studies, but asymmetry in the other three subgroups. The results adjusted using the trim and fill method were stable in “Without a run-in phase” studies and attenuated the placebo effect in the other three subgroups, indicating the stability of the results of the meta-analysis (TIF 534 KB)
415_2024_12529_MOESM4_ESM.tif
Supplementary file4 Supplementary Fig. 4: Sensitivity analysis in all included studies. This analysis showed the robustness of the meta-analysis across all included studies (TIF 424 KB)
415_2024_12529_MOESM5_ESM.tif
Supplementary file5 Supplementary Fig. 5: Sensitivity analyses in subgroups. This analysis showed the robustness of the meta-analysis in each subgroup (TIF 589 KB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Haji, S., Sako, W., Murakami, N. et al. Factors associated with a placebo effect in Parkinson’s disease in clinical trials: a meta-analysis. J Neurol 271, 5825–5837 (2024). https://doi.org/10.1007/s00415-024-12529-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-024-12529-4