
Abstract
Objective
To comprehensively examine the clinical presentation of patients diagnosed with frontotemporal dementia–motor neuron disease (FTD–MND) compared to FTD subtypes. To clarify the heterogeneity of behavioural and language deficits in FTD–MND using a data-driven approach.
Methods
Patients with FTD–MND (n = 31), behavioural variant FTD (n = 119), non-fluent variant primary progressive aphasia (n = 47), semantic variant primary progressive aphasia (n = 42), and controls (n = 127) underwent comprehensive clinical, cognitive and behavioural assessments. Two-step cluster analysis examined patterns of behavioural and language impairment. Voxel-based morphometry and tract-based spatial statistics were used to investigate differences across the subgroups that emerged from cluster analysis.
Results
More than half of FTD–MND patients initially presented with variable combinations of deficits (e.g., mixed behaviour/cognitive, mixed behaviour/cognitive/motor deficits), with 74% of them meeting criteria for FTD–MND within 24 months with a median of 12 months. The frequency and severity of behavioural and language abnormalities in FTD–MND lie between that seen in the three FTD phenotypes. Cluster analysis identified three patterns of behavioural and language impairment in FTD–MND. The three FTD–MND subgroups demonstrated different profiles of white matter tract disruption, but did not differ in age at onset, disease duration or patterns of cortical atrophy.
Conclusions
While highly heterogeneous, in terms of behavioural and language deficits, and disease severity, the presentation of FTD–MND may be distinct to that of FTD. Distinct white matter degeneration patterns may underpin heterogeneous clinical profiles in FTD–MND. FTD presenting with mixed behavioural-language disturbances should be monitored closely for at least 12–24 months for the emergence of MND symptoms/signs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One of the challenges in the study of neurodegenerative diseases is identifying uniform patient groups that can be targeted for trials of potential therapies. Frontotemporal dementia–motor neuron disease (FTD–MND), the syndrome where diagnostic criteria for frontotemporal dementia (FTD) and motor neuron disease (MND) are met concurrently, represents an attractive cohort of patients for the development of therapies aimed at slowing progression of or reversing TAR DNA binding protein 43 kDa (TDP-43) pathology. Firstly, almost all FTD–MND patients have underlying TDP-43 pathology [6], whereas other FTD phenotypes can variably be caused by TDP-43, tau, or ubiquitinated fused in sarcoma (FUS) pathology [8]. Secondly, patients with FTD–MND have a worse prognosis than other FTD phenotypes, with rapid progression over months or a few years, rather many years or decades [17]. Not only does rapid progression highlight an urgent need for effective therapies, but it may facilitate the development of effective treatments, because shorter trial protocols may be sufficient to determine the efficacy of potential treatments.
Clinical, genetic, and pathological investigations have highlighted the links between FTD and MND [7, 12, 27] suggesting that FTD and MND may be part of the same disease spectrum, which includes FTD–MND [6]. However, the relationship between behaviour, language, and motor symptoms in FTD–MND remains incompletely understood. Do all patients initially present with FTD–MND, or do most convert to FTD–MND from either pure FTD or pure MND? Can such a conversion be predicted? Direct comparisons of behaviour and language deficits in FTD–MND to the behaviour and language phenotypes of FTD are scarce. Some studies have classified FTD–MND cases along the lines of existing FTD phenotypes [44], but others [14] including our own [18] have emphasized mixed behaviour and language deficits. Such studies suggest that improved sub-classification of FTD–MND may aid prognostication, readying the stage for trials of TDP-43 therapies [39].
The present study aimed to address the following questions: (1) Is the frequency and severity of behaviour and language disturbances in FTD–MND comparable to those of other FTD phenotypes? (2) Given the clinical heterogeneity of FTD–MND, can subgroups of patients be discerned? (3) Do FTD–MND subgroups differ? (4) Does the pattern of neuroanatomical change drive clinical heterogeneity?
Methods
Participants
Patients diagnosed with FTD–MND, behavioural variant FTD (bvFTD), non-fluent variant primary progressive aphasia (nfvPPA), semantic variant primary progressive aphasia (svPPA), and healthy controls, were included in the study. FTD–MND and FTD patients were recruited from FRONTIER, the frontotemporal dementia research clinic in Sydney Australia. Healthy control participants were recruited from a database of volunteers. The diagnosis of FTD was made according to the current consensus criteria and further classified into the established subtypes: bvFTD, nfvPPA, and svPPA [13, 29, 35]; only patients with probable or definite bvFTD were included in this study. MND was diagnosed in accordance with the El Escorial and Awaji criteria [5, 11]. FTD–MND was diagnosed when both FTD and MND diagnostic criteria were fulfilled [45]. FTD–MND patients were either diagnosed with FTD–MND at the initial assessment, or at a later review assessment. For those patients who developed FTD–MND after presenting with FTD or MND, only data acquired after the fulfillment of the FTD–MND diagnosis were included in the study. Data extracted from the clinical file included: age at symptom onset, initial mode of presentation, and ‘Time to FTD–MND’, defined as the time from first symptom onset to established FTD–MND diagnosis. The majority of FTD–MND patients met criteria for the dual diagnosis within 24 months. In contrast, 56% of bvFTD, 52% of nfvPPA and 83% of svPPA had a disease duration longer than 36 months. All FTD patients were monitored for the development of FTD–MND.
Patients with the right temporal variant of FTD, or possible (as opposed to probable or definite) bvFTD, as well as patients with extensive cerebrovascular disease, significant head injury, prior history of mental illness, movement disorder, alcohol and other drug abuse were excluded from the present study. The study was approved by the ethics committees of the University of New South Wales and South Eastern Sydney Local Health District. In accordance with the Declaration of Helsinki, written informed consent was obtained from all participants, or their carers.
Clinical assessments
Clinical features, including comprehensive behavioural changes, neuropsychiatric symptoms and language impairments, were assessed by experienced behavioural neurologists (JRB and JRH) using a consistent approach [16]. These features were reported by carers and recorded as present or absent for behavioural disturbances, including loss of insight, insidious change in personality/social behaviour, disinhibition, apathy, loss of sympathy/empathy, stereotyped behaviours and hyperorality/dietary changes. Neuropsychiatric features including aggression, agitation, delusions, hallucinations, mood disturbance, anxiety and mental rigidity were recorded. In addition, the presence or absence of dysarthria—including flaccid (i.e., moist, indistinct) and spastic (i.e., harsh, strained voice)—motor speech apraxia, slowed speech rate, phonological errors, syntax errors, anomia, word finding difficulties, impaired word/sentence comprehension, impaired word/sentence repetition, surface dyslexia, and dysgraphia were systematically assessed. Aphasic features, such as motor speech apraxia (i.e., effortful, distorted, and impaired ability of coordinating phonemes on single word production) or phonological errors (i.e., incorrectly selecting phonemes on single word production) [31] were distinguished from dysarthria (i.e., selecting phoneme correctly, but indistinct, moist, or breathy production due to lower motor neuron weakness, or harsh, effortful, strangled speech due to upper neuron weakness).
Cognitive assessments
The Addenbrooke’s Cognitive Examination, either the second (ACE-R) [25] or third (ACE-III) [19] iteration, was used as a measure of general cognition. To ensure a uniform dataset, ACE-R scores were transformed to ACE-III scores using recently validated algorithms [42]. Disease severity was measured using the Frontotemporal Dementia Rating Scale (FRS) [26].
Digit span forwards and backwards tasks assessed basic attention and working memory, respectively [15, 23, 43]. The Trail making test parts A and B measured processing speed and divided attention [4]. Trails B-A time and letter fluency (F, A, S) were used as measures of executive function [3]. The Rey Auditory Verbal Learning Test (RAVLT) [38] and Rey Complex Figure (RCF) recall were used to assess verbal and visuospatial memory [33].
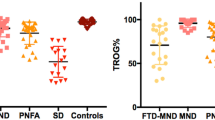
Behavioural disturbances were measured using the Cambridge Behavioural Inventory-Revised (CBI-R) [47]. The Sydney Language Battery (SYDBAT) was used to assess expressive and receptive language skills, and used as a measure of confrontational naming and semantic knowledge [37]. The single word repetition subtask was not included in the final analysis to avoid potential confounding effects of dysarthria, resulting in a revised SYDBAT total score of 0–90 points. The Test for Reception Of Grammar (TROG) examined syntactic comprehension ability [10]. To avoid fatigue affecting performance in FTD–MND patients, the TROG was abbreviated to 2 instead of 4 sentences across 20 blocks, resulting in a revised TROG total score of 0–40 points. Both SYDBAT and TROG can be completed without the need for participants to produce speech, so performance was unlikely to be affected by dysarthria.
Statistical analyses
Data analyses were performed using IBM SPSS Statistics 24. One-way analysis of variance (ANOVA) with post hoc Sidak tests (normally distributed data) and Kruskal–Wallis with post hoc Mann–Whitney U tests (non-normally distributed data) were applied. Chi-square tests were employed for categorical variables. A p value < 0.05 was considered statistically significant for group comparisons. Bonferroni correction was used for multiple comparisons. The corrected p value was based on the number of multiple comparisons, with a corrected p value < 0.005 (for a comparison number n = 10) or a corrected p value < 0.0083 (number of comparisons n = 8). To avoid confounding effects of disease duration on test performance of disease groups, multivariate analysis of covariance (MANCOVA) was employed for tasks with total as well as sub-scores (i.e., ACE-III, CBI-R, RAVLT and SYDBAT), and analysis of covariance (ANCOVA) was applied for the remainder of cognitive assessments.
To explore the distribution of behaviour and language disturbances of FTD–MND, and to avoid reducing statistical power when using FTD–MND group alone, a two-step cluster analysis was conducted using bvFTD, FTD–MND, nfvPPA and svPPA patients. Furthermore, to avoid lowering the quality of the clustering solution and to improve the discrimination based on behaviour and language performance in cluster analysis, a limited number of variables sensitive to deficits encountered in FTD phenotypes were selected: (1) a combined behavioural score of CBI-R subdomains reported to be sensitive to changes in bvFTD [48] (i.e., abnormal behaviours, eating habits, motivation, stereotypic and motor behaviours); (2) SYDBAT naming was chosen given the demonstration of confrontational naming impairments in FTD–MND [21, 22]. Euclidean distance was used for distance measure and Bayes Information Criterion (BIC) was for clustering criterion [28]. To validate stability, multiple steps were conducted: (1) examination of the predictor importance and the score of silhouette measure of cohesion and the separation were used to assess cluster solution quality [36]; (2) the resulting cluster number was used in a hierarchical cluster analysis using the average-linkage method with squared Euclidean distance [49] to verify whether cluster membership changed from one algorithm to another for each case. Using these approaches, a stable cluster model is evident when more than 80% of cases do not change cluster membership [36].
Imaging acquisition
MRI acquired after FTD–MND criteria being fulfilled were included in imaging analyses. Only scans of sufficient quality for voxel-based morphometry (VBM) and tract-based spatial statistics (TBSS) analyses were included (n = 20). Age- and sex-matched healthy controls (n = 20) were included for comparison.
Three-dimensional, structural T1-weighted and diffusion-weighted images were obtained on a 3 T Philips MRI scanner with an eight-channel head coil. Acquisition sequences for T1-weighted images were as follows: coronal orientation, matrix 256 × 256, 200 slices, 1 mm3, echo time/inversion time = 2.6/5.8 ms, flip angle α = 8°. Diffusion-weighted acquisition: 32 gradient directions, repetition time/echo time/inversion time: 8400/68/90 ms, b value = 1000 s/mm2, 55 2.5 mm horizontal slices, end resolution: 2.5 × 2.5 × 2.5 mm3, 96 × 96 matrix; repeated 2 times.
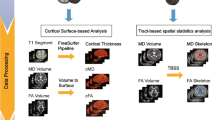
Voxel-based morphometry (VBM) analysis
The FMRIB FSL software package (https://fsl.fmrib.ox.ac.uk/fsl/fslwiki/) was used for voxel-based morphometry (VBM) analysis. Brain extraction [40] was followed by grey matter (GM) and white matter (WM) segmentation. Spatial intensity variations (bias field or radio-frequency inhomogeneities) were then corrected [50]. GM volumes were concatenated and averaged to Montreal Neurological Institute standard space (MNI152) [2]. All grey matter images were registered nonlinearly after a study-specific template being created. The Jacobian of the warp field was used for modulation of each voxel. An isotropic Gaussian kernel (sigma = 3 mm) was used for smoothing the normalised and modulated grey matter images. Patterns of grey matter intensity decrease were explored using a whole-brain general linear model (GLM). Non-parametric permutation-based testing was then conducted with 5000 permutations per contrast [30].
Diffusion tensor imaging (DTI) analysis
The acquired two DTI sequences were averaged for each subject, and then visually inspected for field inhomogeneity distortions. Images were subsequently corrected for eddy current and motion distortions. Diffusion tensor models were fitted for each voxel through the FDT toolbox of the FMRIB software library (http://fsl.fmrib.ox.ac.uk/fsl/fslwiki/FDT), creating maps of three eigenvalues (λ1, λ2, λ3) which were used for calculating fractional anisotropy (FA).
Tract-based Spatial Statistics (TBSS) [41] from FSL were applied to conduct a skeleton-based analysis of white matter FA. FA maps for each participant were co-registered using non-linear registration (FNIRT) [2] to the MNI standard space using the FMRIB58_FA template. Then, FA images were averaged to generate a group mean FA image, followed by a skeletonized algorithm [41] to define a group template of maximum FA lines. FA values for each subject were projected onto this group template skeleton. Clusters were also statistically tested using permutation-based non-parametric testing.
Age and disease duration (defining disease duration as zero in controls) were used as nuisance variables for comparisons across FTD–MND subgroups derived from cluster analysis both in VBM and TBSS analyses. Significant clusters were reported using a threshold-free cluster enhancement method with Family-Wise Error (FWE) corrected at p < 0.05.
Results
Participant demographics
In total, 366 participants including 31 FTD–MND, 119 bvFTD, 47 nfvPPA, 42 svPPA and 127 controls were recruited. Table 1 displays the demographic, clinical, and neuropsychological profiles of study samples. Age at symptom onset in FTD–MND did not differ from that in other patient groups. The svPPA group had longer disease duration at diagnosis than FTD–MND (p < 0.001) and nfvPPA (p < 0.001). Overall, 72 patients were deceased at the time of data analysis (29 bvFTD, 21 FTD–MND, 13 nfvPPA, 6 svPPA). Survival duration (from symptom onset) was significantly shorter in FTD–MND relative to bvFTD (p < 0.001), nfvPPA (p = 0.003), and svPPA (p = 0.001).
Clinical features at presentation
Behavioural changes and language difficulties were highly prevalent in FTD–MND patients, with apathy found in 83.3% and language symptoms in 92.3% of patients at presentation (Table 2). Compared to bvFTD, patients with FTD–MND had lower rates of personality change/social behaviour disturbance (p = 0.002) and disinhibition (p < 0.001). No differences were detected between bvFTD and FTD–MND in terms of psychiatric symptoms. Compared to nfvPPA, the FTD–MND group exhibited lower rates of slowed speech rate, motor speech apraxia, syntax errors and impaired word/sentence repetition (all p values < 0.005). Relative to svPPA, the FTD–MND group had lower rates of word finding difficulties, anomia, impaired word comprehension, and surface dyslexia (all p values < 0.001) (Table 2).
Initial diagnosis/symptoms and ‘time to FTD–MND’ in FTD–MND
As shown in Fig. 1a, the initial presentation of FTD–MND was highly variable. Of 31 cases, 22.6% presented with concurrent FTD and ALS features, while the majority had either pure behavioural (32.3%), mixed behavioural and language (29%), or pure language disturbances (9.7%). One patient presented (3.2%) with memory impairment. Only one patient (3.2%) presented with pure ALS, potentially reflecting recruitment bias (see “Discussion”).

Summary of the differences between FTD–MND and the three FTD phenotypes, and the heterogeneity in FTD–MND. a Summary of the differences between FTD–MND and FTD: most FTD–MND cases started with behavioural or variable combination of deficits then developed ALS, and met diagnostic criteria for FTD–MND within 24 months. While three FTD phenotypes started with either predominant behavioural changes or dominant language disturbances. FTD–MND and FTD three subtypes had comparable age at symptoms onset, but FTD–MND presented with much shorter survival duration (from symptoms onset) than bvFTD, nfvPPA and svPPA. FTD–MND was not simply bvFTD plus ALS. Most FTD–MND had a mixture of behavioural and language features, and the language was a hybrid of nfvPPA and svPPA. The severity of behavioural and language deficits in FTD–MND lie between the FTD three phenotypes after controlling the disease duration. b The ‘time to FTD–MND’ in FTD–MND: time length (months) from symptoms onset to diagnosis of FTD–MND. 74% of FTD–MND patients met diagnosis of FTD–MND within 24 months. c The distribution of all patients into three re-classified subgroups from cluster analysis. d The initial diagnosis/symptoms and ‘time to FTD–MND’ in FTD–MND subgroups. The initial mode of presentation and median ‘time to FTD–MND’ did not differ across the three FTD–MND subgroups. a, b, d Behaviour = diagnosis of bvFTD or pure behavioural symptoms before the onset of ALS symptoms and/or signs; language = diagnosis of nfvPPA or mixed (semantic and syntactic) language disturbances; mixed BC = diagnosis of FTD-mixed (bvFTD combined nfvPPA and/or svPPA) or had co-occurred behavioural and cognitive symptoms (including language features); mixed B/C-M = simultaneous onset of FTD and ALS features, either with diagnosis of FTD–MND or with mixed behavioural/cognitive-motor deficits; Others = one with diagnosis of ALS, one case with memory impaired. c Mild mixed subgroup = mild mixture of behaviour-language impairments; moderate-severe mixed subgroup = moderate behaviour-severe language impairments; behaviour dominant subgroup = dominantly behavioural disturbances
The ‘time to FTD–MND’, defined as the time from symptom onset to diagnosis of FTD–MND (see “Methods”) was variable with a median onset of 12 months. 74% of patients met criteria for FTD–MND within 24 months and all but two within 48 months (Fig. 1b). One patient, who had an initial diagnosis of nfvPPA, developed ALS after 60 months, and another patient who had an initial diagnosis of bvFTD developed ALS after 132 months.
Neuropsychological profile
The results of neuropsychological testing are presented in Table 1. In short, all patient groups demonstrated deficits on tests of executive function and most aspects of memory compared to controls (all post hoc p < 0.005). FTD–MND patients did not differ from nfvPPA patients on any cognitive domain but displayed disproportionate executive deficits relative to svPPA and bvFTD patients (all post hoc p ≤ 0.004) (Table 1).
Behavioural and language disturbances
Compared to controls, all patient groups demonstrated wide ranging behavioural disturbances, as reflected in CBI-R responses (Table 3). After controlling for disease duration, FTD–MND patients exhibited less disinhibition and apathy than bvFTD (both post hoc p ≤ 0.005) and more severe impairments of everyday skills and self-care than svPPA (both post hoc p ≤ 0.003). FTD–MND patients demonstrated greater overall levels of behavioural disturbance than nfvPPA (CBI-R total: p < 0.001; CBI-R subdomains: all p ≤ 0.005).
All patient groups had multimodal language impairments relative to controls, reflected in performance on the SYDBAT and TROG (all post hoc p < 0.001). Comparison of patient groups revealed that FTD–MND patients were more impaired than bvFTD on SYDBAT naming (p = 0.002) and more impaired than nfvPPA in SYDBAT single word comprehension and semantic association (both p ≤ 0.005), but less impaired relative to svPPA on SYDBAT naming, single word comprehension and total (all p < 0.005). No difference was identified across disease groups on TROG performance (Table 3). Summary of the differences between FTD–MND and the three FTD phenotypes were illustrated in Fig. 1a.
Cluster analysis
All patients
In this cluster solution, the quality of the three-cluster model was ‘good’, as determined by the algorithm: silhouette measure of cohesion and separation score > 0.5 [36]. The predictor importance, indicating an overview of variables’ overall importance [36], for the SYDBAT naming and behavioural scores were 1.0 and 0.75, respectively. Additionally, the cluster membership did not change for 80% of cases. Together, these validation approaches suggest a high degree of model stability.
Cluster analysis of all clinically defined patient groups identified three subgroups, labeled as ‘mild mixed’, ‘moderate-severe mixed’ (i.e., moderate behaviour-severe language) and ‘behaviour dominant’. The three subgroups derived from cluster analysis largely mapped onto established FTD phenotypes, with 83.3% of nfvPPA, 84.6% of svPPA, and 58% of bvFTD allocated to the ‘mild mixed’, ‘moderate-severe mixed’ and ‘behaviour dominant’ subgroups, respectively (Fig. 1c). Interestingly, FTD–MND patients were spread across the three classifications (37.9%, 37.9% and 24.2% of FTD–MND allocated to ‘mild mixed’, ‘moderate-severe mixed’ and ‘behaviour dominant’ subgroups, respectively) (Fig. 1c, Supplementary Table 1).
FTD–MND patients only
The three FTD–MND subgroups arising from cluster analysis were matched on age of onset and disease duration. Relative to the ‘mild mixed’ subgroup, the ‘moderate-severe mixed’ subgroup had more severe cognitive, behavioural and language disturbances, reflected by the ACE-III, CBI-R, SYDBAT and TROG (all p < 0.05), while the ‘behaviour dominant’ subgroup showed greater behavioural disturbances on CBI-R total/subdomains (all p < 0.05) (Supplementary Table 2), indicating that FTD–MND patients can be meaningfully sub-grouped according to the pattern and severity of cognitive and behavioural changes. The initial mode of presentation and median ‘time to FTD–MND’ (Fig. 1d) did not differ across the three FTD–MND subgroups (Supplementary Table 2).
Imaging profiles in FTD–MND subgroups
Grey matter atrophy
Compared to controls, all three FTD–MND subgroups showed frontal and anterior temporal atrophy. The ‘mild mixed’ and ‘moderate-severe mixed’ subgroups also demonstrated atrophy of the left peri-insular regions, the middle/inferior frontal gyrus, and the precentral/postcentral gyrus. No significant differences were detected across FTD–MND subgroups (Fig. 2a, Table 4).

Neuroimaging profiles in FTD–MND subgroups. Three FTD–MND subgroups showed cortical atrophy of frontal orbital cortex, frontal pole, anterior temporal gyrus bilaterally. No significant difference in white matter was detected in FTD–MND ‘mild mixed’ subgroup relative to controls. The FTD–MND ‘moderate-severe mixed’ subgroup had bilateral white matter changed in comparison with FTD-MND ‘mild mixed’ subgroup. a, b Clusters are overlaid on the Montreal Neurological Institute standard brain and FMRIB58_FA template. Age and disease duration were included as covariates in the analyses. R right, L left. a Colored voxels show cortical regions that were significant in voxel-based morphometry analyses at p < 0.05 FWE corrected. b Colored voxels show white matter tracts that were significant in tract-based spatial statistics at p < 0.05 FWE corrected
White matter profiles
Relative to controls, analysis of FA revealed changes in white matter projections underlying the bilateral prefrontal cortex, anterior temporal lobe and peri-insular regions in the ‘moderate-severe mixed’ and ‘behaviour dominant’ subgroups. Specifically, the uncinate fasciculus, forceps minor, inferior fronto-occipital fasciculus, and corticospinal tract were abnormal compared to controls (Table 5). No significant difference in FA was detected in the ‘mild mixed’ subgroup compared to controls. Relative to the ‘mild mixed’ subgroup, the ‘moderate-severe mixed’ subgroup showed decreased FA in the bilateral corticospinal tracts, inferior fronto-occipital fasciculus, superior longitudinal fasciculus, cingulum (cingulate gyrus) and forceps minor (Fig. 2b, Table 5).
Discussion
The present study is the first to systematically characterize the presentation and heterogeneity of clinical and neuroimaging features in FTD–MND. Most FTD–MND patients presented with variable combinations of deficits in behaviour, language, or motor function. Language dysfunction was multimodal, with syntactic comprehension and semantic deficits. If absent at presentation, ALS typically developed within 24 months. Three FTD–MND subgroups, which differed in severity of behavioural and language disturbances, were identified through cluster analysis. Multimodal imaging analyses showed the grey matter and distinct white matter tract abnormalities may underlie clinical heterogeneity in FTD–MND.
In the current study, more than half of FTD–MND patients started with a variable combination of deficits rather than a clearly defined syndrome. Most FTD–MND (74.2%) developed FTD features before motor deficits, but others developed them with (22.6%) or even after (3.2%) motor deficits. The number of the cases who presented with ALS then developed FTD was less than previously reported [1, 20], potentially due to recruitment bias. However, our clinic shares expertise and resources with a co-located ALS research clinic, with frequent cross referral, so the risk of recruitment bias is partially mitigated. In line with a previous study, the present study results suggest that a combination of bvFTD and nfvPPA features may indicate an increased risk of developing FTD–MND. On the other hand, the risk FTD–MND in patients presenting with FTD may decrease when disease duration exceeds 24 months [46]. The frequency of behavioural changes and language deficits in FTD–MND were found to lie between those seen in the behaviour and language forms of FTD. For example, disinhibition was less frequent in FTD–MND than in bvFTD, while apathy and delusions were more frequent in FTD–MND than in nfvPPA and svPPA. In contrast, motor speech apraxia and syntax errors were less frequent in FTD–MND than in nfvPPA, and anomia and surface dyslexia were less frequent than in svPPA. All documented language deficits were more frequent in FTD–MND than in bvFTD. These findings are broadly consistent with previous reports showing overlap between behaviour and language impairments in patients with frontotemporal lobar degeneration (FTLD) [14] pathology, but extends this understanding by demonstrating that behavioural and language deficits coexist in FTD–MND, quite unlike other FTD phenotypes. In addition, the severity of behavioural and language disturbances in FTD–MND lie between that seen in FTD phenotypes. The pattern of cortical atrophy across FTD–MND and FTD three phenotypes had been studied widely, with previous studies reporting less atrophy in the superior frontal and paracingulate gyri in FTD–MND relative to bvFTD [24] and less atrophy in Broca’s and Wernicke’s areas in FTD–MND compared to FTD language subtypes [32]. As such, intermediate degrees of atrophy in these regions may explain intermediate behaviour/language impairments in FTD–MND. Furthermore, survival duration was shorter in FTD–MND compared to FTD subtypes, despite a similar age at onset. Collectively, these results suggest that FTD–MND represents a distinct, but variable, clinical syndrome.
We employed a data-driven approach to determine if clinical heterogeneity in FTD–MND could be explained by discernible disease subgroups, defined by the pattern and severity of behavioural and language deficits. This approach revealed three distinct subgroups, which largely mapped FTD phenotypes. Interestingly, FTD–MND patients were fairly evenly distributed across the subgroups, rather than demonstrating a uniform pattern of deficits. Furthermore, the three FTD–MND subgroups did not differ in age of symptom onset or disease duration. These findings reinforce the clinical heterogeneity of FTD–MND.
To date, few studies have attempted to define distinct subgroups of FTD–MND subgroups, likely reflecting issues with sample sizes or failure of distinguishing FTD–MND from ALS. One previous study classified FTD–MND patients as behaviour- or language-dominant [9] based on the early/dominant clinical features, and showed that language-dominant FTD–MND patients were more likely to be associated with bulbar-onset ALS and shorter survival [9]. Another study classified FTD–MND patients as ‘long-term’ survivors or ‘typical’ survivors, and reported that longer survival was associated with a longer ‘time to FTD–MND’ (i.e., symptoms to FTD–MND) [20]. While the issue of meaningful subgrouping of FTD–MND may be unresolved, the presence of marked heterogeneity in mode of presentation and clinical features is incontrovertible and certainly worthy of future investigation.
After controlling for age and disease duration, multimodal imaging analysis revealed that all three FTD–MND subgroups had atrophy of bilateral orbitofrontal, frontopolar, and anterior temporal lobes. The pattern of cortical atrophy appears to be similar in FTD–MND to that seen in FTD [6, 21, 22, 34], but the degree of white matter tract involvement differed. Marked white matter disruption was present in the ‘moderate-severe mixed’ and ‘behaviour dominant’ FTD–MND subgroups, with particular involvement of the corticospinal tract and tracts underlying frontal and temporal lobes. White matter was relatively intact in the ‘mild mixed’ FTD–MND subgroup, when compared to controls or the ‘moderate-severe mixed’ FTD–MND subgroup. As such, differential white matter tract disruption may underpin clinical heterogeneity in FTD–MND.
The following potential limitations need to be considered. In particular, detailed information on motor involvement and genetics were not available making it difficult to establish links between motor involvement and survival, and difficult to clarify associations between genetic variability and clinical features. One strength of the present study is that patients were recruited from an FTD research clinic, meaning that detailed information on behaviour and cognition was available for a comparatively large group of FTD–MND patients, as well as a large cohort of patients with well-defined FTD phenotypes. On the other hand, a limitation of the present study is that patients who develop FTD–MND after presenting with ALS may be under-represented. FTD–MND is a rare disease, and although the cohort recruited for the present study was one of the largest assembled, sub-classification was challenging. Our sub-classification of FTD–MND patients appears to differ from that outlined in previous studies, probably reflecting different methodologies. Importantly, we used a data-driven definition of subgroups, rather than an arbitrary clinical definition. Future studies employing larger samples and longitudinal assessments will be essential to extend on these findings and further clarify the natural history and progression of FTD–MND.
Conclusion
Clinical features are highly heterogeneous in FTD–MND, in terms of range (i.e., of behaviour/language deficits) and severity. Grey matter atrophy and distinct white matter tract abnormalities may underpin the heterogeneous clinical profiles in FTD–MND. The mode of presentation of FTD–MND appears distinct to that of established FTD phenotypes, potentially arguing against the concept of an FTD–MND disease continuum.
Data availability
Anonymized data not published within the article will be shared on reasonable request from a qualified investigator for replication purposes.
References
Ahmed RM, Devenney EM, Strikwerda-Brown C, Hodges JR, Piguet O, Kiernan MC (2020) Phenotypic variability in ALS–FTD and effect on survival. Neurology 94:e2005–e2013
Andersson JL, Jenkinson M, Smith S (2007) Non-linear registration, aka spatial normalisation FMRIB technical report TR07JA2. FMRIB Anal Group Univ Oxf 2:1–21
Benton AL, Hamsher KD (1983) Multilingual aphasia examination: manual of instructions. In: Department of Neurology and Psychology, University of Iowa, Iowa City
Bowie CR, Harvey PD (2006) Administration and interpretation of the Trail Making Test. Nat Protoc 1:2277–2281
Brooks BR, Miller RG, Swash M, Munsat TL (2000) El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 1:293–299
Burrell JR, Halliday GM, Kril JJ, Ittner LM, Götz J, Kiernan MC, Hodges JR (2016) The frontotemporal dementia–motor neuron disease continuum. Lancet 388:919–931
Burrell JR, Kiernan MC, Vucic S, Hodges JR (2011) Motor neuron dysfunction in frontotemporal dementia. Brain 134:2582
Chare L, Hodges JR, Leyton CE, McGinley C, Tan RH, Kril JJ, Halliday GM (2014) New criteria for frontotemporal dementia syndromes: clinical and pathological diagnostic implications. J Neurol Neurosurg Psychiatry 85:865–870
Coon E, Sorenson E, Whitwell J, Knopman D, Josephs K (2011) Predicting survival in frontotemporal dementia with motor neuron disease. Neurology 76:1886–1892
Croot K, Hodges JR, Patterson K (1999) Evidence for impaired sentence comprehension in early Alzheimer’s disease. J Int Neuropsychol Soc 5:393–404
de Carvalho M, Dengler R, Eisen A, England JD, Kaji R, Kimura J, Mills K, Mitsumoto H, Nodera H, Shefner J, Swash M (2008) Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol 119:497–503
Elamin M, Bede P, Byrne S, Jordan N, Gallagher L, Wynne B, O’Brien C, Phukan J, Lynch C, Pender N (2013) Cognitive changes predict functional decline in ALS a population-based longitudinal study. Neurology 80:1590–1597
Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, Manes F (2011) Classification of primary progressive aphasia and its variants. Neurology 76:1006–1014
Harris JM, Jones M, Gall C, Richardson AM, Neary D, du Plessis D, Pal P, Mann DM, Snowden JS, Thompson JC (2016) Co-occurrence of language and behavioural change in frontotemporal lobar degeneration. Dementia Geriatr Cogn Disord Extra 6:205–213
Hodges JR (2007) Cognitive assessment for clinicians. Oxford University Press, Oxford
Hodges JR (2017) Cognitive assessment for clinicians, 3rd edn. Oxford University Press, Oxford
Hodges JR, Davies R, Xuereb J, Kril J, Halliday G (2003) Survival in frontotemporal dementia. Neurology 61:349–354
Hsieh S, Caga J, Leslie FV, Shibata M, Daveson N, Foxe D, Ramsey E, Lillo P, Ahmed RM, Devenney E (2016) Cognitive and behavioral symptoms in ALSFTD: detection, differentiation, and progression. J Geriatr Psychiatry Neurol 29:3–10
Hsieh S, Schubert S, Hoon C, Mioshi E, Hodges JR (2013) Validation of the Addenbrooke’s Cognitive Examination III in frontotemporal dementia and Alzheimer’s disease. Dement Geriatr Cogn Disord 36:242–250
Hu WT, Seelaar H, Josephs KA, Knopman DS, Boeve BF, Sorenson EJ, McCluskey L, Elman L, Schelhaas HJ, Parisi JE (2009) Survival profiles of patients with frontotemporal dementia and motor neuron disease. Arch Neurol 66:1359–1364
Kamminga J, Leslie FV, Hsieh S, Caga J, Mioshi E, Hornberger M, Ballard KJ, Kiernan MC, Hodges JR, Burrell JR (2016) Syntactic comprehension deficits across the FTD–ALS continuum. Neurobiol Aging 41:11–18
Leslie FV, Hsieh S, Caga J, Savage SA, Mioshi E, Hornberger M, Kiernan MC, Hodges JR, Burrell JR (2015) Semantic deficits in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 16:46–53
Lezak M, Howieson D, Bigler E, Tranel D (2012) Neuropsychological assessment, 5th edn. Oxford University Press, New York
Lillo P, Mioshi E, Burrell JR, Kiernan MC, Hodges JR, Hornberger M (2012) Grey and white matter changes across the amyotrophic lateral sclerosis-frontotemporal dementia continuum. PLoS ONE 7:e43993
Mioshi E, Dawson K, Mitchell J, Arnold R, Hodges JR (2006) The Addenbrooke’s Cognitive Examination Revised (ACE-R): a brief cognitive test battery for dementia screening. Int J Geriatr Psychiatry 21:1078–1085
Mioshi E, Hsieh S, Savage S, Hornberger M, Hodges JR (2010) Clinical staging and disease progression in frontotemporal dementia. Neurology 74:1591–1597
Montuschi A, Iazzolino B, Calvo A, Moglia C, Lopiano L, Restagno G, Brunetti M, Ossola I, Presti AL, Cammarosano S (2015) Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J Neurol Neurosurg Psychiatry 86:168–173
Mooi E, Sarstedt M (2011) Cluster analysis. A concise guide to market research. Springer, Berlin
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51:1546–1554
Nichols TE, Holmes AP (2002) Nonparametric permutation tests for functional neuroimaging: a primer with examples. Hum Brain Mapp 15:1–25
Ogar J, Slama H, Dronkers N, Amici S, Luisa Gorno-Tempini M (2005) Apraxia of speech: an overview. Neurocase 11:427–432
Omer T, Finegan E, Hutchinson S, Doherty M, Vajda A, McLaughlin RL, Pender N, Hardiman O, Bede P (2017) Neuroimaging patterns along the ALS–FTD spectrum: a multiparametric imaging study. Amyotroph Lateral Scler Frontotemporal Degener 18:611–623
Osterrieth PA (1944) Le test de copie d'une figure complexe; contribution a l'etude de la perception et de la memoire. Archives de psychologie
Piguet O, Hornberger M, Mioshi E, Hodges JR (2011) Behavioural-variant frontotemporal dementia: diagnosis, clinical staging, and management. Lancet Neurol 10:162–172
Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, Van Swieten JC, Seelaar H, Dopper EG, Onyike CU (2011) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134:2456–2477
Sarstedt M, Mooi E (2014) Cluster analysis. A concise guide to market research. Springer, Heidelberg
Savage S, Hsieh S, Leslie F, Foxe D, Piguet O, Hodges JR (2013) Distinguishing subtypes in primary progressive aphasia: application of the Sydney language battery. Dement Geriatr Cogn Disord 35:208–218
Schmidt M (1996) Rey auditory verbal learning test: a handbook. Western Psychological Services, Los Angeles
Scotter EL, Chen H-J, Shaw CE (2015) TDP-43 proteinopathy and ALS: insights into disease mechanisms and therapeutic targets. Neurotherapeutics 12:352–363
Smith SM (2002) Fast robust automated brain extraction. Hum Brain Mapp 17:143–155
Smith SM, Jenkinson M, Johansen-Berg H, Rueckert D, Nichols TE, Mackay CE, Watkins KE, Ciccarelli O, Cader MZ, Matthews PM (2006) Tract-based spatial statistics: voxelwise analysis of multi-subject diffusion data. Neuroimage 31:1487–1505
So M, Foxe D, Kumfor F, Murray C, Hsieh S, Savage G, Ahmed RM, Burrell JR, Hodges JR, Irish M, Piguet O (2018) Addenbrooke’s Cognitive Examination III: psychometric characteristics and relations to functional ability in dementia. J Int Neuropsychol Soc JINS 24:1–10
Strauss E, Sherman EM, Spreen O (2006) A compendium of neuropsychological tests: administration, norms, and commentary. American Chemical Society, Washington, DC
Strong MJ, Abrahams S, Goldstein LH, Woolley S, Mclaughlin P, Snowden J, Mioshi E, Roberts-South A, Benatar M, HortobáGyi T (2017) Amyotrophic lateral sclerosis-frontotemporal spectrum disorder (ALS–FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener 18:153–174
Strong MJ, Abrahams S, Goldstein LH, Woolley S, Mclaughlin P, Snowden J, Mioshi E, Roberts-South A, Benatar M, HortobáGyi T (2017) Amyotrophic lateral sclerosis-frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener 18(3–4):153–174. https://doi.org/10.1080/21678421.2016.1267768
Van Langenhove T, Piguet O, Burrell JR, Leyton C, Foxe D, Abela M, Bartley L, Kim WS, Jary E, Huang Y (2017) Predicting development of amyotrophic lateral sclerosis in frontotemporal dementia. J Alzheimer’s Dis 58:163–170
Wear HJ, Wedderburn CJ, Mioshi E, Williams-Gray CH, Mason SL, Barker RA, Hodges JR (2008) The Cambridge behavioural inventory revised. Dement Neuropsychol 2:102–107
Wedderburn C, Wear H, Brown J, Mason SJ, Barker RA, Hodges J, Williams-Gray C (2008) The utility of the Cambridge Behavioural Inventory in neurodegenerative disease. J Neurol Neurosurg Psychiatry 79:500–503
Yim O, Ramdeen KT (2015) Hierarchical cluster analysis: comparison of three linkage measures and application to psychological data. Quant Methods Psychol 11:8–21
Zhang Y, Brady M, Smith S (2001) Segmentation of brain MR images through a hidden Markov random field model and the expectation-maximization algorithm. IEEE Trans Med Imaging 20:45–57
Acknowledgements
We are grateful to the research participants and their families.
Funding
This work was supported by funding to Forefront, a collaborative research group dedicated to the study of frontotemporal dementia and motor neuron disease, from the National Health and Medical Research Council of Australia (NHMRC) program Grant (#1037746) and the Australian Research Council Centre of Excellence in Cognition and its Disorders Memory Node (#CE110001021). In addition, JRB was supported by a NHMRC Early Career Fellowship (#1072451). MI is supported by an ARC Future Fellowship (FT160100096). OP is supported by a NHMRC Senior Research Fellowship (APP1103258).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors report no competing interests.
Ethical approval
The study was approved by the ethics committees of the University of New South Wales and South Eastern Sydney Local Health District. In accordance with the Declaration of Helsinki, written informed consent was obtained from all participants, or their carers.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Long, Z., Irish, M., Foxe, D. et al. Heterogeneity of behavioural and language deficits in FTD–MND. J Neurol 268, 2876–2889 (2021). https://doi.org/10.1007/s00415-021-10451-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-021-10451-7