
Abstract
Adolescent-onset spastic ataxia is a proposed novel phenotype in compound heterozygous carriers of an intronic mutation (c.1909 + 22G > A) in the POLR3A gene. Here, we present ten new cases of POLR3A-related spastic ataxia and discuss the genetic, clinical and imaging findings. Patients belonged to six pedigrees with hereditary spastic paraplegia or cerebellar ataxia of unknown origin. All affected subjects presented with compound heterozygous variants, comprising c.1909 + 22G > A in combination in each pedigree with one of the following novel mutations (Thr596Met, Tyr665LeufsTer11, Glu198Ter, c.646-687_1185 + 844del). The new mutations segregated with the phenotype in all families. The phenotype combined variable cerebellar ataxia, gait and lower limb spasticity, involvement of central sensory tracts and in some cases also intention tremor. The reportedly characteristic hyperintensity along the superior cerebellar peduncle on MRI was observed in ~ 80% of the cases. Our study extends the clinical and molecular phenotype further supporting the pathogenic role of the c.1909 + 22G4A intronic mutation and identifying four novel causative mutations in POLR3A-related spastic ataxia. Certain characteristic MRI features may be useful to guide genetic diagnosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
POLR3-related disorders comprise a number of clinically overlapping disease entities caused by recessive mutations in POLR3A, POLR3B and POLR1C genes [1]. The best recognized phenotypes consist of an early-childhood-onset hypomyelinating leukodystrophy manifesting with variable combinations of cerebellar ataxia, tremor, spasticity, mental retardation, oligodontia and hypogonadotropic hypogonadism [1,2,3,4,5,6]. Recently, a novel and milder phenotype consisting of adolescent-onset spastic ataxia has been proposed to be specific of an intronic mutation (c.1909 + 22G4A) in the POLR3A gene [7]. In these subjects the typical signs of hypomyelinating leukodystrophy on MRI would be absent and a characteristic hyperintensity along the superior cerebellar peduncle (SCP) in T2 and FLAIR sequences would be observed instead [8,9,10]. Whether or not the reported intronic mutation is specific for this clinical and MRI phenotype is still a matter of debate [11,12,13]. Here, we present ten new cases of POLR3A-related adolescent-onset spastic ataxia, all of them presenting with the c.1909 + 22G > A variant in compound heterozygosity with four novel mutations. Our study extends the clinical and molecular phenotype further supporting the pathogenic role of the c.1909 + 22G4A intronic mutation within the POLR3A gene in this syndrome.
Patients and methods
Subjects
The ten patients reported here belonged to six different pedigrees and were studied at the University Hospital Marqués de Valdecilla in Santander, Spain. The individuals had a previous diagnosis of either cerebellar ataxia or hereditary spastic paraplegia (HSP) of unknown origin. In the setting of clinical practice, a diagnostic multigenic panel was conducted. All patients and relatives signed an informed consent and the study was approved by the local ethical committee.
Genetic analysis
Sequencing was performed using a custom-targeted NGS approach designed to study 187 related ataxia and spastic paraplegia genes using SureSelect Capture Library reagents (Agilent Technologies, Santa Clara, CA, USA) and probes custom designed to capture coding exons, following Agilent protocols and recommendations in combination with CytoScan XON Suite (Applied Biosystems, Waltham, MA, USA). Genetic methodology, primers and PCR conditions are detailed in Supporting information (genetic methodology and Table S1).
MRI imaging
The MRI studies were performed on a 3.0-T scanner (Achieva 3.0T; Philips Healthcare, Best, The Netherlands) with an 8-channel head coil. The protocol included an axial T2-weighted sequence, 3DFLAIR (TR 8000, slice thickness: 0.6 mm), and 3DT1 (slice thickness 1 mm).
Results
Clinical features
The main clinical features of the affected subjects are summarized in Fig. 1 and Videos 1–4. Age at onset of first symptoms ranged from 10 to 31 years. Gait clumsiness or unsteadiness was the presenting symptom in all cases but in subjects from Fam 2 who presented with upper limb tremor. All cases were able to walk independently at the age of examination (mean disease duration 25.8 years), four of them requiring support. A predominantly spastic-type gait was observed in Fam 1 and 3, whereas in subjects from Fam 2 and 4 ataxia predominated over spasticity. Subjects from Fam 5 and 6 showed mixed features. Dysarthria, when present (Fam 2 and 4), was mild. Interestingly, ocular movements were normal in nearly all cases except for Fam 3: II-4, who showed mild gaze-evoked nystagmus. Half of the subjects showed tremor affecting upper limbs, both postural and predominantly kinetic, in one of the subjects also affecting the head. Deep tendon reflexes were conserved in upper limbs and either normal or brisk in lower limbs. All patients showed Babinski sign as well as reduced vibration sense at the lower limbs and Romberg’s sign. Pes cavus was present in three cases and eight also showed genu recurvatum, requiring orthopedic measures in one case (Fam 4: II-3). Distal lower-limb erithrocyanosis was noted in five patients, iris diaphragm sign obtained by pressing on the skin with the fingertip being negative. Two patients from the same pedigree (Fam 4) reported dental abnormalities and another one suffered myopia (Fam 6: II-1). No patient showed clinically overt signs of gonadal dysfunction nor cognitive abnormalities. Electrophysiological studies showed identical findings in all patients consisting of abnormal lower-limb somato-sensory evoked potentials (SEP) and motor evoked potentials, whereas motor and sensory nerve conduction studies were normal.


POLR3A-related spastic ataxia pedigrees. Segregation analysis of the mutations and clinical phenotype. DTR Deep tendón reflexes, UL upper limbs, SCP superior cerebellar peduncle, SEP somatosensory evoked potentials, MEP motor evoked potentials, + present, - absent, + mild, + + moderate, + + + severe, n.a. not assesed
Genetic findings
Genetic diagnosis was obtained for six independent index cases, all of them presenting with compound heterozygous variants identified in the POLR3A gene. PCR and Sanger sequencing confirmed the compound heterozygosity of the identified POLR3A variants and the segregation with the phenotype in all families. The identified variants in POLR3A are summarized in Fig. 1 and Table S2 including five of them not previously described. For index cases for whom NGS only identified an heterozygous variant in POLR3A, analysis with a CytoScan Xon microarray and ChAs allowed to identify a deletion (c.646–687_c.1185 + 844del; NG_029648.1: g.78020702_78023071del; p.Glu216_Lys395del) (Fig. S1) including exons 6, 7 and 8, not previously described. NGS, PCR and Sanger sequencing with specific sequencing primers with 5′ poly(A) and poly(T) were carried out to avoid sequencing errors due to the presence of several homopolymer regions (Fig. S2 and S3). Remarkably, the deleted breakpoint region is surrounded by two identical 21 bp repetitive sequences, suggesting that an unequal crossing over event occurred. This region of the protein codifies partially for an RNA pol N domain (IPR006592 InterPro), which is conserved and characteristic motif for RNA polymerases that bind to DNA for the synthesis of RNA [14].
MR imaging
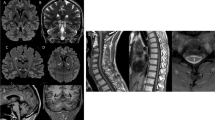
The MRI demonstrated on 3D FLAIR MRI images a striking bilateral hyperintensity along the superior cerebellar peduncles extending from the dentate nucleus up to the midbrain, just below the red nucleus in eight out of nine cases (Fig. 2). In addition, roughly half cases showed mild cervical spinal cord atrophy. No signs of hypomyelinating leukodystrophy were observed on MRI.

MRI findings in affected individuals with POLR3A-realted spastic ataxia. Striking bilateral hyperintensity along the entire superior cerebellar peduncle (SCP) ranging from the cerebellar dentate nucleus (white arrows in a and b) to the midbrain just below the red nucleus (white arrowheads in c, d and e). 3D FLAIR paracoronal view angulated along the course of the SCP (a, b) and standard axial (c), axial T2 (d) and coronal FLAIR (e). Sagital-T1 (f) showing mild but evident atrophy of the spinal cord without cerebellar involvement. a, d, e, f (Fam 2-II:4); b (Fam 3-II:1); c (Fam 2-II:2)
Discussion
Spastic ataxia of adolescent onset is a recently proposed new phenotype observed in compound heterozygous carriers with an intronic mutation (c.1909 + 22G > A) within the POLR3A gene [7,8,9,10,11,12,13, 15]. In this article, we provide ten additional cases that support this association and also report four novel mutations in this gene.
The phenotypic spectrum of hypomyelinating leukodystrophies, caused by recessive mutations in POLR3 genes, comprises a number of clinically overlapping entities [1,2,3,4,5,6]. Recently, Minnerop et al. identified a new intronic mutation in POLR3A gene (c.1909 + 22G > A) in a pedigree with recessive spastic ataxia of unknown origin. From this finding, a large cohort of patients with a diagnosis of HSP or cerebellar ataxia of unknown origin was tested, and POLR3A mutations were identified in ~ 3.1% of cases, 80% of which were compound heterozygous for the intronic c.1909 + 22G > A [7]. The uniform phenotype consisted of a progressive spastic ataxia initiated in adolescence with a high occurrence of tremor and involvement of the central sensory pathways. This phenotype differs from that knowingly attributed to POLR3A-related leukodystrophies because of an later age at onset, absence or scarcity of non-neurological manifestations such as endocrine dysfunction or hypo/oligodontia, absence of the characteristic features of MRI (hypomyelination, cerebellar atrophy and corpus callosum hypoplasia), and the presence of bilateral hyperintensity along the superior cerebellar peduncles in MRI FLAIR sequences [8,9,10]. More recently, Rydning et al. replicated this association in an independent Norwegian cohort [15]. Some authors, however, assert that given the overlap of clinical and imaging characteristics between the different POLR3A-related syndromes, the spastic ataxia phenotype should not be considered as one separate but rather a part of the spectrum [11].
The cases presented here share most of the clinical and MRI characteristics with those reported by Minnerop and Rydning and likewise had received a previous diagnosis of hereditary ataxia or HSP of unknown origin [7, 15]. The mean age at onset was around adolescence in half of the cases, the remaining half beginning at age 30. The phenotype combined variable cerebellar ataxia, gait and lower extremity spasticity, central sensory pathway involvement and, in some cases tremor, predominantly of intention type. It was not as homogeneous, however, as some cases would fall into a clinical diagnosis of HSP and others had received a diagnosis of cerebellar tremor–ataxia syndrome. Our patients mostly lacked non-neurological manifestations. Only 20% presented dentition abnormalities, contrary to Minnerop and Rydning who reported a frequency of 65% and 85%, respectively [7, 15]. Other features such as hypogonadism, myopia, or mental impairment were absent in our series. Peripheral nerve conduction studies showed no abnormalities, similar to the cases reported by Gauquelin, and contrary to the findings of Minnerop and Rydning who reported signs of neuropathy in 30% and 20% of the cases, respectively. Central sensory tracts, however, showed a constant impairment, with abnormal SEP observed in all patients. There is a clinical sign we have observed with unusual frequency and severity, the presence of genu recurvatum. It affected 80% of our cases and in at least one case (Fam 4: II-1, Video 4) at a severe degree. As there is no mention of its incidence in other series, we do not know if it could be a characteristic sign in this entity. The same applies to the presence of distal lower-limb acrocyanosis, observed in half of the patients in our series. Normal iris diaphragm sign points to the absence of sympathetic denervation [16]; in any case, similar findings have been reported in HSP [17].
In accordance with previous series, more than 80% of our patients showed hyperintensity along the SCP on 3D FLAIR MRI; conversely, there were no signs of hypomyelination [7]. Spinal atrophy, or mild cerebellar atrophy, was also an inconstant feature in this series.
Regarding genetics, all the cases were compound heterozygous carriers of the intronic c.1909 + 22G > A variant which has been previously described in spastic ataxia cases [7]. All cases also carried a second genetic variation not previously described that segregated with the phenotype and was predicted to be pathogenic by bioinformatics. Pedigrees 4, 5 and 6 shared a deletion (c.646–687_c.1185 + 844del; NG_029648.1: g.78020702_78023071del p.Glu216_Lys395del) (Supplementary Fig. 3) including exons 6, 7 and 8, while pedigrees 1, 2 and 3 showed single nucleotide changes. Since functional analyses were not carried out in our study, we cannot completely exclude a linkage imbalance with other pathogenic mutations.
Our data support the idea that hereditary spastic ataxia should be considered a distinct phenotype within the spectrum of POLR3A-related disorders. We agree with the authors who propose that the c.1909 + 22G > A mutation would be the main determinant in this phenotype. This mutation has been shown to cause an incomplete loss of the wild allele, which in turn can modulate the phenotype, moving it away from the classical hypomyelination phenotype towards a milder spastic ataxia phenotype. As a nuance, since cases beginning over age 20 are not as uncommon, the term early adolescence would be best omitted when naming this phenotype.
From the perspective of clinical practice, we consider truly useful to classify these cases as POLR3A-related HSP, ataxia or spastic ataxia. In this clinical context, the very characteristic features of MRI can be of great help in guiding genetic diagnosis.
References
Bernard G, Vanderver A (2012) POLR3-related leukodystrophy. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds) GeneReviews® [Internet]. Seattle [WA]. University of Washington, Seattle, pp 1993–2018
Wolf NI, Vanderver A, van Spaendonk RM, Schiffmann R, Brais B, Bugiani M, Sistermans E, Catsman-Berrevoets C, Kros JM, Pinto PS, Pohl D, Tirupathi S, Strømme P, de Grauw T, Fribourg S, Demos M, Pizzino A, Naidu S, Guerrero K, van der Knaap MS, Bernard G, 4H Research Group (2014) Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 83:1898–1905
Bernard G, Chouery E, Putorti ML, Tétreault M, Takanohashi A, Carosso G, Clément I, Boespflug-Tanguy O, Rodriguez D, Delague V, Abou Ghoch J, Jalkh N, Dorboz I, Fribourg S, Teichmann M, Megarbane A, Schiffmann R, Vanderver A, Brais B (2011) Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet 9(89):415–423
Saitsu H, Osaka H, Sasaki M, Takanashi J, Hamada K, Yamashita A, Shibayama H, Shiina M, Kondo Y, Nishiyama K, Tsurusaki Y, Miyake N, Doi H, Ogata K, Inoue K, Matsumoto N (2011) Mutations in POLR3A and POLR3B encoding RNA Polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am J Hum Genet 89:644–651
Potic A, Brais B, Choquet K, Schiffmann R, Bernard G (2012) 4H syndrome with late-onset growth hormone deficiency caused by POLR3A mutations. Arch Neurol 69:920–923
Terao Y, Saitsu H, Segawa M, Kondo Y, Sakamoto K, Matsumoto N, Tsuji S, Nomura Y (2012) Diffuse central hypomyelination presenting as 4H syndrome caused by compound heterozygous mutations in POLR3A encoding the catalytic subunit of polymerase III. J Neurol Sci 320:102–125
Minnerop M, Kurzwelly D, Wagner H, Soehn AS, Reichbauer J, Tao F, Rattay TW, Peitz M, Rehbach K, Giorgetti A, Pyle A, Thiele H, Altmüller J, Timmann D, Karaca I, Lennarz M, Baets J, Hengel H, Synofzik M, Atasu B, Feely S, Kennerson M, Stendel C, Lindig T, Gonzalez MA, Stirnberg R, Sturm M, Roeske S, Jung J, Bauer P, Lohmann E, Herms S, Heilmann-Heimbach S, Nicholson G, Mahanjah M, Sharkia R, Carloni P, Brüstle O, Klopstock T, Mathews KD, Shy ME, de Jonghe P, Chinnery PF, Horvath R, Kohlhase J, Schmitt I, Wolf M, Greschus S, Amunts K, Maier W, Schöls L, Nürnberg P, Zuchner S, Klockgether T, Ramirez A, Schüle R (2017) Hypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia. Brain 140:1561–1578
Daoud H, Tétreault M, Gibson W, Guerrero K, Cohen A, Gburek-Augustat J, Synofzik M, Brais B, Stevens CA, Sanchez-Carpintero R, Goizet C, Naidu S, Vanderver A, Bernard G (2013) Mutations in POLR3A and POLR3B are a major cause of hypomyelinating leukodystrophies with or without dental abnormalities and/or hypogonadotropic hypogonadism. J Med Genet 50:194–197
La Piana R, Cayami FK, Tran LT, Guerrero K, van Spaendonk R, Õunap K, Pajusalu S, Haack T, Wassmer E, Timmann D, Mierzewska H, Poll-Thé BT, Patel C, Cox H, Atik T, Onay H, Ozkınay F, Vanderver A, van der Knaap MS, Wolf NI, Bernard G (2016) Diffuse hypomyelination is not obligate for POLR3-related disorders. Neurology 86:1622–1626
La Piana R, Tonduti D, Gordish Dressman H, Schmidt JL, Murnick J, Brais B, Bernard G, Vanderver A (2014) Brain magnetic resonance imaging (MRI) pattern recognition in Pol III-related leukodystrophies. J Child Neurol 29:214–220
Gauquelin L, Tétreault M, Thiffault I, Farrow E, Miller N, Yoo B, Bareke E, Yoon G, Suchowersky O, Dupré N, Tarnopolsky M, Brais B, Wolf NI, Majewski J, Rouleau GA, Gan-Or Z, Bernard G (2018) POLR3A variants in hereditary spastic paraplegia and ataxia. Brain 141:e1
Minnerop M, Kurzwelly D, Rattay TW, Timmann D, Hengel H, Synofzik M, Stendel C, Horvath R, Schüle R, Ramirez A (2018) Reply: POLR3A variants in hereditary spastic paraplegia and ataxia. Brain 141:e2
Minnerop M, Kurzwelly D, Wagner H, Schüle R, Ramirez A (2019) Reply: Biallelic POLR3A variants confirmed as a frequent cause of hereditary ataxia and spastic paraparesis. Brain 142:e13
Mitchell AL, Attwood TK, Babbitt PC, Blum M, Bork P, Bridge A, Brown SD, Chang HY, El-Gebali S, Fraser MI, Gough J, Haft DR, Huang H, Letunic I, Lopez R, Luciani A, Madeira F, Marchler-Bauer A, Mi H, Natale DA, Necci M, Nuka G, Orengo C, Pandurangan AP, Paysan-Lafosse T, Pesseat S, Potter SC, Qureshi MA, Rawlings ND, Redaschi N, Richardson LJ, Rivoire C, Salazar GA, Sangrador-Vegas A, Sigrist CJA, Sillitoe I, Sutton GG, Thanki N, Thomas PD, Tosatto SCE, Yong SY, Finn RD (2019) InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res 47(1):D351–D360
Rydning SL, Koht J, Sheng Y, Sowa P, Hjorthaug HS, Wedding IM, Erichsen AK, Hovden IA, Backe PH, Tallaksen CME, Vigeland MD, Selmer KK (2019) Biallelic POLR3A variants confirmed as a frequent cause of hereditary ataxia and spastic paraparesis. Brain 142(4):e1
Mumenthaler M, Schliack H (1991) Peripheral nerve lesions. Diagnosis and therapy. Thieme Medical Publishers, Inc, New York
Miranda S, Lévesque H (2017) Acrocyanosis: a common but poorly understood condition. Rev Med Intern 38(4):225–227
Acknowledgements
The research of this work was funded by the Spanish Health Institute Carlos III grants CP08/00027 and CPII14/00029 (to AM-D), and FIS PI14/00136 and PI17/00534 (to AM-D). Antoni Matilla Dueñas was a Miguel Servet Investigator in Neuroscience supported by the Spanish Health Institute Carlos III (ISCIII; CPII14/00029).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
JI received honoraria as speaker from Zambon and financial support for attending conferences from Abbie. KMSC, MCJ, XF, IS, EML, AGG, JLMG, JB and AM report no conflict of interests.
Ethical approval
This study was conducted according to the ethical principles for medical research involving human subjects according to the Declaration of Helsinki.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fam 1-II:2. Segment 1: spastic gait and moderate genu recurvatum. Segment 2: normal ocular movements. Segment 3: absence of tremor or ataxia in upper limbs. Segment 4: lower limb ataxia pyramidal signs and acrocyanosis (M4V 15253 kb)
Fam 2-II:3. Segment 1: ataxic gait and mild genu recurvatum. Segment 2: positive Romberg’s sign. Segment 3: kinetic tremor–ataxia in upper limbs. Segment 4: lower limb ataxia and pyramidal signs (M4V 14863 kb)
Fam 3-II:1. Segment 1: spastic gait and mild genu recurvatum. Segment 2: positive Romberg’s sign. Segment 3: absence of tremor or ataxia in upper limbs. Segment 4: lower limb ataxia and pyramidal signs (M4V 12843 kb)
Fam 4-II:3. Segment 1: ataxic-spastic gait and marked genu recurvatum. Segment 2: cervical dystonic tremor and upper limb action tremor. Segment 3: lower limb ataxia, pyramidal signs and acrocyanosis (M4V 18919 kb)
Rights and permissions
About this article

Cite this article
Infante, J., Serrano-Cárdenas, K.M., Corral‐Juan, M. et al. POLR3A-related spastic ataxia: new mutations and a look into the phenotype. J Neurol 267, 324–330 (2020). https://doi.org/10.1007/s00415-019-09574-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-019-09574-9