
Abstract
Mutations of myelin protein zero gene (MPZ) are found in 5% of Charcot–Marie–Tooth patients. In 2004, Shy et al. identified two main phenotypes associated with them: an early-onset subtype with mainly demyelinating features and a late-onset subgroup with prominent axonal impairment. We evaluated whether novel MPZ mutations described in literature during the last 14 years could still fit with this classification. We collected and revised reports of 69 novel MPZ mutations. Almost 90% of them could be alternatively classified as responsible for: (a) an early-onset phenotype, with first limitations starting before 3 years (2.5 ± 0.50 years), motor milestones delays, frequently severe course and upper limb MNCVs below 15 m/s; (b) late-onset neuropathy, with mean age of onset of 42.8 ± 1.5 years and mean upper limbs motor nerve conduction velocities (MNCVs) of 47.2 ± 1.4 m/s; (c) a phenotype more similar to typical CMT1A neuropathy, with onset during the 2nd decade, MNCV in the range of 15–30 m/s and slowly progressive course. The present work confirms that P0-related neuropathies may be separated into two main distinct phenotypes, while a third, relatively small, group comprehend patients carrying MPZ mutations and a childhood-onset disease, substantiating the subdivision into three groups proposed by Sanmaneechai et al. (Brain 138:3180–3192, 2015). Interestingly, during the last years, an increasing number of novel MPZ mutations causing a late-onset phenotype has been described, highlighting the clinical relevance of late-onset P0 neuropathies. Since the family history for neuropathy is often uncertain, due to the late disease onset, the number of patients carrying this genotype is probably underestimated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Charcot–Marie–Tooth disease (CMT), with a prevalence of 9.37–20.1/100.000 [1], is one of the most common hereditary disorders of the nervous system. The clinical hallmarks of the disease are distal weakness, muscle wasting and sensory loss; nevertheless, the clinical phenotype can be quite variable in terms of age of onset, disease progression and severity. Based on nerve conduction velocity, CMT can be distinguished into two main forms: demyelinating (CMT1), characterized by upper limb motor nerve conduction velocities (MNCVs) below 38 m/s, and axonal (CMT2), with preserved or mildly slowed nerve conduction velocities in the upper limbs (> 38 m/s). From a molecular point of view, CMT is a complex disorder, with more than 1000 putative mutations in 80 different disease-associated genes [2]. Mutations in myelin protein zero gene (MPZ), encoding for P0, are found in 4.1% of all CMT patients [3, 4], and were initially described as responsible for CMT1B; subsequently, mutations in this gene have been found also in cases of congenital hypomyelinating neuropathy/Dejerine Sottas disease and CMT2. To analyse the phenotypes associated with MPZ mutations, Shy et al. [5] evaluated patients seen in their clinic and re-evaluated clinical data from 64 cases of CMT1B reported in literature until 2004. They found that most patients could be classified as having either an early-onset or a late-onset neuropathy, the former presenting with onset of the disease during the 1st decade of life and with upper extremity MNCVs lower than 15 m/s, the latter with an age of onset over 20 years of age and with upper extremity MNCVs faster than 30 m/s. In a more recent work examining clinical severity of patients from Inherited Neuropathy Consortium [6], 47 MPZ mutations, of whom 15 were novel and 6 were described in literature after 2004, were classified as responsible for three phenotypes with onset in infancy, childhood and adulthood, respectively.
In the present study, we collected literature data related to novel MPZ mutations described between 2005 and 2018 to assess whether the distinction between early- and late-onset neuropathy can still be considered a landmark and to provide further insight into the disease mechanisms.
Methods
Literature evaluation
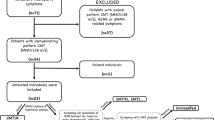
Reports about novel MPZ mutations described between 2005 and 2018 were collected from a search on PubMed and on the online database http://molgen-www.uia.ac.be/CMTMutations/. We used the following search terms: “myelin protein zero”, “Charcot–Marie–Tooth disease”, and “novel MPZ mutation”. Reports of novel MPZ mutations not providing adequate clinical and neurophysiological data, i.e., a complete description of the clinical presentation and an upper limb neurophysiological study were excluded (Fig. 1). Mutations described by more than one author and associated with more than one phenotype were included in each of the described groups. MPZ mutations found in patients with a negative or not available family history were included if segregation analysis had been performed or if the mutation was absent in unrelated healthy controls. For each mutation, clinical (age of onset, number of affected family members, severity and additional clinical features) and neurophysiological data (upper and lower limbs motor conduction velocities and compound motor action potentials) were derived from the considered study for all the available patients. Mutations were classified as responsible for early-, childhood–adolescence- or late-onset neuropathy according to age of onset of the neuropathy (≤ 5 years, 6–20 years; ≥ 21 years), defined by the age at which the proband or another family member started to manifest clinical and/or neurophysiological signs; for each group, average values and standard error were calculated for the considered variables. The severity of the disease was classified based on CMTNS, when reported, or on clinical aspects described in the report.

Flowchart of the inclusion process for the review
Results
Between 2005 and 2018, 76 novel MPZ mutations in 203 patients have been reported in the literature [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51] as responsible for Dejerine–Sottas disease, congenital hypomyelinating neuropathy, CMT1B or CMT2 neuropathies. We excluded three mutations described in five patients for insufficient clinical and neurophysiological data and one report about one variant of unknown significance (Figs. 1, 2; Supplementary Table 4); three reports [21, 29, 47] on MPZ copy number variation in 14 patients were considered separately (Supplementary Table 5). Reports about 69 novel MPZ mutations in 180 patients contained adequate clinical and neurophysiological data, thus allowing us to derive the phenotype, including a group of 15 novel MPZ mutations in 18 patients newly described in 2015 [6] and whose phenotype had already been classified has responsible for early-onset, CMT1B or late-onset neuropathy (Fig. 1).

Venn diagram showing mutations included and excluded from the review
Overall, 60 out of 69 mutations (87%) in 144 patients could be unambiguously classified, according to clinical and neurophysiological parameters, as responsible for an early-onset neuropathy (35 patients—24%; 23 mutations—38%), a late-onset neuropathy (74 patients—51%; 27 mutations—45%) or a childhood-onset phenotype (36 patients—25%; 10 mutations—17%). Six (9%) other mutations in 18 patients (10%) have been described in more than one report and associated by the authors to more than one phenotype and were therefore considered in each of the described groups; eventually, 3 further MPZ mutations (4%) in 14 patients (7%) were associated with an atypical phenotype [14, 20, 48] (Fig. 2).
From the descriptions of MPZ mutation in the literature, excluding those already classified in 2015 [6], we can derive as follows (Table 1, Fig. 3).

(updated from Sanmaneechai et al. [6])
Mutations in the MPZ gene associated with inherited neuropathies. Adhesive interface, fourfold interface and head-to-head interface, marked with colour to the border of circle, refer to amino acid residues deemed essential for cis and trans adhesion between adjacent myelin wraps. Both the numbering systems for MPZ mutations, including or not the 29 amino acid leader peptide cleaved before insertion in the myelin sheath, are reported. Mutations demonstrated by adding the letters that represent amino acid change, arrows that represent frameshift mutation and lines that represent nonsense mutation. Mutations causing early-onset phenotype are filled or noted with red colour, while those causing childhood-onset phenotype are in orange, and those causing late-onset phenotype are in purple
Twenty-six patients with 15 different novel mutations could be classified as having an early-onset neuropathy (Table 2). Among them, 17 patients with eight different mutations were described by the authors as having DSS, five patients with four different mutations were described as affected by CHN, and four patients with three different mutations received a diagnosis of CMT1. The mean age at onset was 2.5 ± 0.50 years (median 2 years; range 0–9), with eight cases where the neuropathy started within the 2nd year of life and seven patients presenting at birth as floppy babies. Family history was negative in the nine cases (60%) and six families (40%) with at least two affected members were described. A severe clinical course, defined by a CMTNS > 20 was associated with 13 mutations in 19 cases (76%); 2 mutations in 6 cases were responsible for a mild to severe neuropathy. The phenotype was complicated by the occurrence of kyphoscoliosis in six patients (24% of patients, 33% of all mutations). Cranial nerves involvement was reported for several patients with early-onset neuropathy. In particular, five patients carrying the His81Gln [13] mutation and one patient carrying the Ile30Thr mutation [19] showed Adie’s pupils, while two patients carrying the Gly137Gly mutation [50] had sensory-neural hearing loss and ptosis as adjunctive features. Neurophysiology was consistent with a severe hypo- or demyelinating neuropathy with secondary axonal degeneration. All but three cases presented with MNCV of the median and ulnar nerves below 15 m/s, with mean MNCV of the median nerve of 7.4 ± 1.2 m/s; three patients showed absence of sensory and motor nerve responses both in upper and lower limbs. When obtainable, conduction velocities from lower limbs were similar to those obtained from the upper limbs. Compound muscle action potentials (CMAPs) and sensory nerve action potentials (SNAPs) were reduced, with mean CMAP = 1.8 ± 0.4 mV.
Seventy-six patients carrying 27 novel mutations could be classified as affected by a late-onset neuropathy (Table 3). In most cases, symptoms ensued after the 4th decade of life, and in several cases they started during the 6th or 7th decade (median 43 years; range 15–75; mean age of onset of 42.8 ± 1.5 years). Family history was negative in 33% of the cases. Emerging features of late-onset P0 neuropathy were consistent with a sensory-motor neuropathy with prominent involvement of lower limbs and causing a relatively low disability, with 56 patients (78%) having a slowly progressive, mild neuropathy (CMTNS < 10). Nevertheless, cases of rapidly progressive symptoms, appearing late in life but quickly conducting to severe disability and ambulation aid requirement have been described for Arg36Gly, Pro70Ser and His81Leu mutations [15, 27, 29]. Moreover, neuropathic pain was a prominent feature in several cases of late-onset P0 neuropathies, as in the case of Arg36Trp [11] and Arg36Gly [15]: in both cases, patients started to complain of spontaneous burning pain and tingling of hands and feet, and showed neurophysiologic features of an axonal sensory-motor neuropathy in all limbs. Additional clinical findings included slow or absent pupillary light reflexes in one patient carrying the Phe95Leu mutation [38], and sensory-neural hearing impairment in five patients harbouring the Phe95Leu, Pro105Thr or Arg106Cys mutations [23, 33, 37].
Electrophysiological analysis was consistent with an axonal neuropathy, with mean MNCV from ulnar nerve of 47.2 ± 1.4 m/s, and normal CMAP amplitudes from the upper limbs. Lower limbs neuronography, when recorded, was consistent with an axonal neuropathy (mean CMAP of common peroneal nerve = 2 ± 0.5 mV).
Forty-four patients with 14 different mutations presented a childhood–adolescence-onset phenotype that fits neither with an early- nor with a late-onset neuropathy, resembling rather CMT1A (Table 4). Usually patients showed a normal motor development and started to complain of sensory-motor symptoms during the 1st or 2nd decade of life, with a mean age of neuropathy onset of 15.1 ± 1.1 years (median 15 years; range 6–40). Nine cases were familiar (64%) and five sporadic (36%). The clinical course of disease within this group was mild in 82% of the cases, with a mean age of 39.1 ± 3.3 years at first neurological visit, usually 2 decades after first signs or symptoms’ onset. Only eight patients with six mutations experienced a neuropathy of moderate severity (CMTNS 10–20). Cranial nerves involvement was never reported and patients had no additional features other than the polyneuropathy. Neurophysiological examination was consistent with a demyelinating polyneuropathy with MNCV approaching the so-called intermediate range, a mean ulnar nerve MNCV of 31.2 ± 2.6 m/s, and usually preserved upper limbs CMAP (6.6 ± 0.6 mV).
Eventually, four mutations were associated with a phenotype slightly different from the classical ones (Table 5): Gly103Gly synonymous mutation [14] has been described in two families with variable age of onset, neuropathic pain and predominant sensory involvement; Arg67Pro [20] has been described in a family with a compound genotype and phenotype of periodic palsy and myotonia, Leu48Pro [49] has been reported in two families with high intrafamilial variability in terms of age of onset, electrophysiological findings and severity of the disease. Moreover, Tyr145 fs has been found by two different groups, in one case in a family with classical CMT2 phenotype [32], in the other [31] in three patients from one family with onset during the 4th decade of life and electrodiagnostic and pathologic findings of HNPP. In all the atypical cases, the identified mutation segregated with the disease in the families.
In addition to the classical small intragenic mutations, cases of MPZ copy number variation have recently been described (Supplementary Table 5). Hoyer [21] first described a family carrying an extra copy of the MPZ gene from exon 1 to exon 6, associated with a demyelinating neuropathy with onset within the 1st decade of life and slowly progressive course, while Maeda [30] described a family carrying MPZ triplication, affected by a demyelinating sensory-motor neuropathy with pupillary abnormalities and high intrafamilial heterogeneity in terms of age of onset and disease severity. Recently, Speevak [48] reported another case of MPZ duplication in a child affected by motor hypo-demyelinating neuropathy with onset during the 1st year of life and a copy number variation of a region including MPZ and succinate dehydrogenase complex subunit C (SDHC) genes.
Six patients among all those included in our study, six carrying late-onset variants (Arg36Trp, Cys50Gly, Ser54 fs, Asp224Tyr, Arg36Gly, Val136Gly) [11, 12, 15, 36, 41, 43] initially received a diagnosis of inflammatory neuropathy, given an acute/subacute fluctuating clinical course eventually associated with pain as a prominent feature, markedly increased CSF protein concentration, and diffuse hypertrophy and slight gadolinium enhancement of the cauda equina roots at MRI. Moreover, co-occurrence with multiple sclerosis or asymptomatic CNS myelin abnormalities has been described for the Asp224Tyr [17] and Val136Gly [41] mutations, respectively.
Discussion
In this study, we aimed to define the spectrum of MPZ-associated phenotypes [5, 6] collecting and reviewing literature data regarding novel MPZ mutations described during the last 14 years (2005–2018), to better characterize the clinical features associated with them.
We identified 76 novel MPZ mutations associated with CMT in the recent literature. We excluded one VUS and other three mutations (5%) from the current work because of inadequate clinical data and considered separately three MPZ copy number variations (5%). Four mutations resulted in an atypical phenotype and six were described by more than one author as responsible for distinct phenotypes. The remaining 60 mutations, including 15 newly reported in a recent paper [6] analysing the clinical records of patients carrying MPZ mutations recruited by the Inherited Neuropathy Consortium in a natural history study, corresponded to either early demyelinating, a childhood demyelinating or the late axonal phenotype.
In more detail, one quarter of the cases presented an early-onset phenotype, manifesting during motor development and characterized by very slow MNCV, while one half of the patients with novel MPZ mutations had an adult-onset axonal neuropathy with normal or minimally impaired MNCV. Interestingly, another quarter of the cases evaluated in this study had a distinct phenotype, presenting with motor and sensory symptoms usually during the 2nd decade of life and showing a slowly progressive course, with MNCV in the lower limit of the intermediate range. Such a phenotype, definable as childhood–adolescence onset, has been also highlighted, with a similar percentage, by Sanmaneechai et al. [6].
Myelin protein zero is the most abundant protein of the peripheral compact myelin, where it maintains the cohesion between adjacent myelin wraps through homophilic interactions [52]. As P0 is a prominent myelin protein, the mechanisms whereby MPZ mutations act in the generation of disorders alternatively affecting myelin or axons remain to be elucidated.
In the last years, different pathomechanisms for MPZ mutations have been dissected, particularly for mutations causing an early-onset neuropathy, ranging from endoplasmic reticulum retention and activation of the canonical unfolded protein response [53,54,55,56] to altered trafficking of the protein to non-myelin plasma membranes and altered radial axonal sorting during the early phases of myelination [57]. Other proposed mechanisms include the disruption of the intercellular adhesion properties with a structural packing defect [53] and mis-glycosylation of P0 with either loss of the native glycosylation site, the Asp122, frequently associated to a late-onset axonal neuropathy, or the gain of a new glycol site, resulting in an hyperglycosylated P0 variant, observed for mutations causing a severe demyelinating neuropathy [54, 58].
Advances have been achieved by the evidence that P0 not only localises in the compact myelin but also at the paranode and node of Ranvier, where it interacts with neurofascin 155 (NF155) and neurofascin 186 (NF186), participating in the maintenance of the nodal structure [59] and that Asp6Tyr, Asp32Gly, and His52Tyr mutations, responsible for late-onset CMT, display homophilic adhesion properties comparable to wild-type P0, but are unable to interact with components of the paranodal and nodal complexes.
CMT2 accounts for 30% of all CMT patients [60]. From our results, in the last decade, there was an increasing amount of novel MPZ mutations associated with late-onset axonal neuropathies rather than early-onset hypo- or demyelinating neuropathies. If we consider that while severe early-onset P0 variants often occur de novo and remain limited to a restricted number of cases, late-onset mutations are frequently familial and involve a larger number of subjects and recessive transmission, usually more common in severe early-onset diseases, has been described as associated with a late-onset phenotype in a patient carrying Val136Gly mutation, located in the same residue of Val136Glu but associated with heterozygous DSS and the prevalence of late-onset MPZ mutations, which has long been underestimated, is likely to be higher than expected, as growing awareness is increasing diagnostic rate and patients without overt family history may still be misdiagnosed.
Our data also include cases with presenting or superimposed features of an acquired inflammatory neuropathy. Given that the association between hereditary and inflammatory neuropathies is not yet clarified, and that neurophysiological and laboratory findings once considered typical for acquired inflammatory neuropathies have been described in various forms of CMT, genetic causes, including MPZ mutations, should be considered in the workup of patients with features of inflammatory neuropathy, especially if not all diagnostic criteria for inflammatory neuropathies are fulfilled and clinical response to immunosuppressive treatment is poor.
Molecular diagnosis of CMT can be achieved in 60–80% of cases [3, 56, 60, 61]. While up to 98% [56] of patients affected by CMT1 eventually receive a molecular diagnosis, this is still frequently challenging for patients suffering from late-onset CMT2, with the rate of diagnosis ranging between 25 and 63% using a gene-by-gene approach.
During the last years, the landscape of mutations responsible for CMT2 has expanded, ranging between the more common GJB1 heterozygous mutations and autosomal dominant GDAP1 mutations, and the incorporation of next-generation sequencing in the diagnostic practice has allowed the discovery of novel CMT genes, especially causing late-onset CMT2 [62], including HARS [63], MARS [64], MME [65] MORC2 [66] and NEFH [67].
The increasing amount of new genes associated with CMT2 and by the fact that other genetically determined neuropathies such as transthyretin-related amyloidosis, a systemic disease with involvement of peripheral nerves, can also manifest after the 6th decade, it is conceivable that an increasing number of late-onset neuropathies, previously misdiagnosed as acquired or age related, have a genetic cause.
In conclusion, our data, focusing on P0-related neuropathies heterogeneity, confirms the existence of three distinct phenotypes deriving from MPZ mutations, and highlights the clinical relevance of late-onset P0 neuropathies as well as the need to increase our knowledge about their underlying molecular mechanisms.
References
Barreto LC, Oliveira FS, Nunes PS et al (2016) Epidemiologic study of Charcot–Marie–Tooth disease: a systematic review. Neuroepidemiology 46:157–165
Timmerman V, Strickland AV, Zuchner S (2014) Genetics of Charcot–Marie–Tooth (CMT) disease within the frame of the human genome project success. Genes (Basel) 5(1):13–32
Fridman V, Bundy B, Reilly MM et al (2015) CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry 86(8):873–878
Pareyson D, Saveri P, Pisciotta C (2017) New developments in Charcot–Marie–Tooth neuropathy and related diseases. Curr Opin Neurol 30(5):471–480
Shy ME, Jani A, Krajewski K et al (2004) Phenotypic clustering in MPZ mutations. Brain 127(Pt 2):371–384
Sanmaneechai O, Feely S, Scherer SS et al (2015) Genotype–phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene. Brain 138:3180–3192
Banchs I, Casasnovas C, Alberti A et al (2009) Diagnosis of Charcot–Marie–Tooth disease. J Biomed Biotechnol 2009:985415
Bienfait HM, Faber CG, Baas F et al (2006) Late onset axonal Charcot–Marie–Tooth phenotype caused by a novel myelin protein zero mutation. J Neurol Neurosurg Psychiatry 77(4):534–537
Braathen GJ, Sand JC, Russell MB (2010) Two novel missense mutations in the myelin protein zero gene causes Charcot–Marie–Tooth type 2 and Dejerine–Sottas syndrome. BMC Res Notes 3:99
Brozkova D, Mazanec R, Haberlova J, Sakmaryova I, Seeman P (2010) Clinical and in silico evidence for and against pathogenicity of 11 new mutations in the MPZ gene. Clin Genet 78(1):81–87
Burns TM, Phillips LH, Dimberg EL, Vaught BK, Klein CJ (2006) Novel myelin protein zero mutation (Arg36Trp) in a patient with acute onset painful neuropathy. Neuromuscul Disord 16(5):308–310
Chavada G, Rao DG, Martindale J, Hadjivassiliou M (2012) A novel MPZ gene mutation in exon 2 causing late-onset demyelinating Charcot–Marie–Tooth disease. J Clin Neuromusc Dis 13(4):206–208
Choi BO, Kim SB, Kanwal S et al (2011) MPZ mutation in an early-onset Charcot–Marie–Tooth disease type 1B family by genome-wide linkage analysis. Int J Mol Med 28(3):389–396
Corrado L, Magri S, Bagarotti A et al (2016) A novel synonymous mutation in the MPZ gene causing an aberrant splicing pattern and Charcot–Marie–Tooth disease type 1B. Neuromusc Disord 26:516–520
Dacci P, Taroni F, Bella ED et al (2012) Myelin protein zero Arg36Gly mutation with very late onset and rapidly progressive painful neuropathy. J Peripher Nerv Syst 17(4):422–425
Duan X, Gu W, Hao Y et al (2016) A novel Asp121Asn mutation of myelin protein zero is associated with late-onset axonal Charcot–Marie–Tooth disease, hearing loss and pupil abnormalities. Front Aging Neurosci 8:222 (eCollection)
Fabrizi GM, Pellegrini M, Angiari C et al (2006) Gene dosage sensitivity of a novel mutation in the intracellular domain of P0 associated with Charcot–Marie–Tooth disease type 1B. Neuromuscul Disord 16(3):183–187
Finsterer J, Miltenberger G, Rauschka H, Janecke A (2006) Novel C59T leader peptide mutation in the MPZ gene associated with late-onset, axonal, sensorimotor polyneuropathy. Eur J Neurol 13(10):1149–1152
Floroskufi P, Panas M, Karadima G, Vassilopoulos D (2007) New mutation of the MPZ gene in a family with the Dejerine–Sottas disease phenotype. Muscle Nerve 35(5):667–669
Hisama FM (2005) Familial periodic paralysis and Charcot–Marie–Tooth disease in a 7-generation family. Arch Neurol 62(1):135–138
Hoyer H, Braathen GJ, Eek AK, Skjelbred CF, Russell MB (2011) Charcot–Marie–Tooth caused by a copy number variation in myelin protein zero. Eur J Med Genet 54(6):e580–e583
Iida M, Koike H, Ando T et al (2012) A novel MPZ mutation in Charcot–Marie–Tooth disease type 1B with focally folded myelin and multiple entrapment neuropathies. Neuromuscul Disord 22(2):166–169
Kabzinska D, Korwin-Piotrowska T, Drechsler H, Drac H, Hausmanowa-Petrusewicz I, Kochanski A (2007) Late-onset Charcot–Marie–Tooth type 2 disease with hearing impairment associated with a novel Pro105Thr mutation in the MPZ gene. Am J Med Genet Part A 143A(18):2196–2199
Keckarevic-Markovic M, Milic-Rasic V, Mladenovic J et al (2009) Mutational analysis of GJB1, MPZ, PMP22, EGR2, and LITAF/SIMPLE in Serbian Charcot–Marie–Tooth patients. J Peripher Nerv Syst 14(2):125–136
Kleffner I, Schirmacher A, Gess B, Boentert M, Young P (2010) Four novel mutations of the myelin protein zero gene presenting as a mild and late-onset polyneuropathy. J Neurol 257(11):1864–1868
Lin KP, Soong BW, Chang MH et al (2012) Clinical and cellular characterization of two novel MPZ mutations, p.I135 M and p.Q187PfsX63. Clin Neurol Neurosurg 114(2):124–129
Laurà M, Milani M, Morbin M et al (2007) Rapid progression of late onset axonal Charcot–Marie–Tooth disease associated with a novel MPZ mutation in the extracellular domain. J Neurol Neurosurg Psychiatry 78(11):1263–1266
Lee YC, Yu CT, Lin KP et al (2008) MPZ mutation G123S characterization: evidence for a complex pathogenesis in CMT disease. Neurology 70(4):273–277
Liu L, Li X, Zi X et al (2013) Two novel MPZ mutations in Chinese CMT patients. J Peripher Nerv Syst 18(3):256–260
Maeda MH, Mitsui J, Soong BW et al (2012) Increased gene dosage of myelin protein zero causes Charcot–Marie–Tooth disease. Ann Neurol 71(1):84–92
Magot A, Latour P, Mussini JM et al (2008) A new MPZ mutation associated with a mild CMT1 phenotype presenting with recurrent nerve compression. Muscle Nerve 38(2):1055–1059
Mandich P, Fossa P, Capponi S et al (2009) Clinical features and molecular modelling of novel MPZ mutations in demyelinating and axonal neuropathies. Eur J Hum Genet 17(9):1129–1134
Marttila M, Rautenstrauss B, Huehne K, Laitinen V, Majamaa K, Karppa M (2012) A novel mutation of myelin protein zero associated with late-onset predominantly axonal Charcot–Marie–Tooth disease. J Neurol 259(8):1585–1589
McMillan HJ, Santagata S, Shapiro F et al (2010) Novel MPZ mutations and congenital hypomyelinating neuropathy. Neuromuscular Disorders 20(11):725
Miltenberger-Miltenyi G, Schwarzbraun T, Loscher WN et al (2009) Identification and in silico analysis of 14 novel GJB1, MPZ and PMP22 gene mutations. Eur J Hum Genet 17(9):1154–1159
Nishiyama S, Sugeno N, Tateyama M, Aoki M (2013) Late-onset Charcot–Marie–Tooth disease type 1B due to a novel mutation in the extracellular disulfide bridge of MPZ gene. Clin Neurol Neurosurg 115(2):208–209
O’Connor G, McNamara P, Bradley D, Connolly S, Langan Y, Redmond J (2012) Late-onset CMT phenotype caused by a novel mutation in the MPZ gene. Eur J Neurol 19(7):e65–e66
Piazza S, Baldinotti F, Fogli A et al (2010) A new truncating MPZ mutation associated with a very mild CMT1 B phenotype. Neuromuscul Disord 20(12):817–819
Prada V, Capponi S, Ursino G et al (2015) Sural nerve biopsy and functional studies support the pathogenic role of a novel MPZ mutation. Neuropathology 35(3):254–259
Ramirez JD, Barnes PR, Mills KR, Bennett DL (2012) Intermediate Charcot–Marie–Tooth disease due to a novel Trp101Stop myelin protein zero mutation associated with debilitating neuropathic pain. Pain 153(8):1763–1768
Reyes-Marin K, Jimenez-Pancho J, Pozo L et al (2011) A novel myelin protein zero (V136G) homozygous mutation causing late onset demyelinating polyneuropathy with brain white matter lesions. Clin Neurol Neurosurg 113(3):243–244
Sabet A, Li J, Ghandour K et al (2006) Skin biopsies demonstrate MPZ splicing abnormalities in Charcot–Marie–Tooth neuropathy 1B. Neurology 67(7):1141–1146
Schneider-Gold C, Kotting J, Epplen JT et al (2010) Unusual Charcot–Marie–Tooth phenotype due to a mutation within the intracellular domain of myelin protein zero. Muscle Nerve 41(4):550–554
Sevilla T, Lupo V, Sivera R et al (2011) Congenital hypomyelinating neuropathy due to a novel MPZ mutation. J Peripher Nerv Syst 16(4):347–352
Shimizu H, Oka N, Kawarai T et al (2010) Late-onset CMT2 associated with a novel missense mutation in the cytoplasmic domain of the MPZ gene. Clin Neurol Neurosurg 112(9):798–800
Simpson BS, Rajabally YA (2010) Charcot–Marie–Tooth disease due to novel myelin protein zero mutation presenting as late-onset remitting sensory neuropathy. J Clin Neuromusc Dis 11(4):187–190
Smit LS, Roofthooft D, Van Ruissen F, Baas F, van Doorn PA (2008) Congenital hypomyelinating neuropathy, a long term follow-up study in an affected family. Neuromuscul Disord 18(1):59–62
Speekav MD, Farrel SA (2013) Charcot–Marie–Tooth 1B caused by expansion of a familial myelin protein zero (MPZ) gene duplication. Eur J Med Genet 56:566–569
Szabo A, Zuchner S, Siska E, Mechler F, Molnar MJ (2005) Marked phenotypic variation in a family with a new myelin protein zero mutation. Neuromuscul Disord 15(11):760–763
Taioli F, Cabrini I, Cavallaro T, Simonati A, Testi S, Fabrizi GM (2011) Dejerine–Sottas syndrome with a silent nucleotide change of myelin protein zero gene. J Peripher Nerv Syst 16(1):59–64
Zschuntzsch J, Dibaj P, Pilgram S, Kotting J, Gerding WM, Neusch C (2009) Severe demyelinating hypertrophic polyneuropathy caused by a de novo frameshift mutation within the intracellular domain of myelin protein zero (MPZ/P0). J Neurol Sci 281(1–2):113–115
Shapiro L, Doyle JP, Hensley P et al (1996) Crystal structure of the extracellular domain from P0, the major structural protein of peripheral nerve myelin. Neuron 1(7):435–449
Wrabetz L, D’Antonio M, Pennuto M et al (2006) Different intracellular pathomechanisms produce diverse Myelin Protein Zero neuropathies in transgenic mice. J Neurosci 26(8):2358–2368
Grandis M, Vigo T, Passalacqua M et al (2008) Different cellular and molecular mechanisms for early and late-onset myelin protein zero mutations. Hum Mol Genet 17(13):1877–1889
Pennuto M, Tinelli E, Malaguti M et al (2008) Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot–Marie–Tooth 1B mice. Neuron 57(3):393–405
Saporta ASD, Sottile SL, Miller LJ, Feely SME, Siskind CE, Shy ME (2011) Charcot–Marie–Tooth disease subtypes and genetic testing strategies. Ann Neurol 69(1):22–33
Fratta P, Ornaghi F, Dati G et al (2019) A nonsense mutation in Myelin Protein Zero causes congenital hypomyelination neuropathy through altered P0 membrane targeting and gain of abnormal function. Hum Mol Genet 28(1):124–132
Prada V, Passalacqua M, Bono M et al (2012) Gain of glycosylation: a new pathomechanism of myelin protein zero mutations. Ann Neurol 71(3):427–431
Brügger V, Engler S, Pereira JA et al (2015) HDAC1/2-dependent P0 expression maintains paranodal and nodal integrity independently of myelin stability through interactions with neurofascins. PLOS Biol 13(9):e1002258
Murphy SM, Laura M, Fawcett K et al (2012) Charcot–Marie–Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry 83(7):706–710
Sivera R, Sevilla T, Vílchez J et al (2013) Charcot–Marie–Tooth disease: genetic and clinical spectrum in a Spanish clinical series. Neurology 81(18):1617–1625
Rossor AM, Polke JM, Houlden H et al (2013) Clinical implications of genetic advances in Charcot–Marie–Tooth disease. Nat Rev Neurol 9(10):562–571
Brodzkova S, Deconinck T, Griffin LB et al (2015) Loss of function mutations in HARS cause a spectrum of inherited peripheral neuropathies. Brain 138(Pt8):2161–2172
Gonzalez M, McLaughlin H, Houlden H et al (2013) Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J Neurol Neurosurg Psychiatry 84(11):1247–1249
Lupo V, Frasquet M, Sánchez-Monteagudo A et al (2018) Characterising the phenotype and mode of inheritance of patients with inherited peripheral neuropathies carrying MME mutations. J Med Genet 55(12):814–823. https://doi.org/10.1136/jmedgenet-2018-105650 (Epub 2018 Nov 10)
Sevilla T, Lupo V, Martínez-Rubio D et al (2016) Mutations in the MORC2 gene cause axonal Charcot–Marie–Tooth disease. Brain 139(Pt 1):62–72
Rebelo AP, Abrams AJ, Cottenie E et al (2016) Cryptic amyloidogenic elements in the 3’ UTRs of neurofilament genes trigger axonal neuropathy. Am J Hum Genet 98(4):597–614
He J, Guo L, Xu G et al (2018) Clinical and genetic investigation in Chinese patients with demyelinating Charcot-Marie-Tooth disease. J Peripher Nerv Syst 23(4):216–226
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
None of the authors have any conflict of interest regarding this work.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study formal consent is not required.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Callegari, I., Gemelli, C., Geroldi, A. et al. Mutation update for myelin protein zero-related neuropathies and the increasing role of variants causing a late-onset phenotype. J Neurol 266, 2629–2645 (2019). https://doi.org/10.1007/s00415-019-09453-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-019-09453-3