
Abstract
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL) represents the most common monogenic cause of adult-onset ischemic stroke and vascular dementia. It is caused by heterozygous missense mutations in the NOTCH3 gene, encoding a transmembrane receptor protein on vascular smooth muscle cells. Classical CADASIL mutations affect conserved cysteine residues of the Notch3 protein. By contrast, the role of non-canonical genetic variation in NOTCH3, in particular of variants causing a hypomorphic Notch3 protein, is subject to an ongoing scientific debate. In this context, we here report a novel NOTCH3 frameshift variant in exon 18 (NM_000435.2: c.2853_2857delTCCCG), causing a frameshift and introducing a premature stop codon, which was detected in a 43-year-old woman and her father. Both carriers of the variant were carefully evaluated, including serial follow-up in the index. Neither clinical nor imaging features provided convincing evidence for a classical CADASIL phenotype, thus reinforcing the concept of hypomorphic NOTCH3 variants most likely not being causative for CADASIL. Our finding, which is discussed in the light of the published literature, has practical implications for interpreting results of NOTCH3 molecular genetic testing as well as patient counseling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The hereditary cerebral small-vessel disease CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy) represents the most common monogenic cause of adult-onset ischemic stroke and vascular dementia [1]. Migraine, in particular migraine with aura, is a typical early disease manifestation, which is present in approximately 40% of patients [2, 3]. Other core clinical features, which occur in subsequent disease stages, include recurrent subcortical ischemic strokes and cognitive deficits, eventually leading to vascular dementia. Classical findings on cranial magnetic resonance imaging (cMRI) include lacunar infarcts, leukoencephalopathy (especially with involvement of the external capsule and the temporal poles), and microbleeds [1, 4].
CADASIL is most commonly caused by heterozygous missense mutations in the NOTCH3 gene [5], which encodes a transmembrane receptor protein expressed on vascular smooth muscle cells and pericytes. CADASIL NOTCH3 mutations affect the extracellular domain of the receptor [6], which contains 34 epidermal growth factor-like repeat (EGFR) domains. Each of these EGFR domains features six conserved cysteine residues, and CADASIL mutations affect one of these cysteine residues, thus causing an uneven number of cysteine residues, i.e., leaving one cysteine unpaired [7]. Diagnosis is established by direct sequencing analysis of NOTCH3 and/or electron microscopic evidence of ultrastructural changes on skin biopsy (so called granular osmiophilic material, GOM) [8].
Beyond classical cysteine-sparing CADASIL mutations, there have been several reports on non-canonical genetic variation in NOTCH3, in particular variants causing a hypomorphic Notch3 protein. The pathophysiological relevance of these findings is subject to an ongoing and unresolved scientific debate. This discussion is closely related to the pathophysiological mechanisms underlying CADASIL, i.e., the question, whether CADASIL is rather caused by a neomorphic effect (i.e., toxic accumulation of mutated protein [9]), the currently prevailing standpoint, or whether a loss-of-function may potentially play a role, as suggested by some experimental findings [10].
In this context, we here report a young female with diverse unspecific neurological complaints, in whom a novel NOTCH3 frameshift variant was detected. We present results of detailed clinical work-up and, in the light of the published literature, discuss the pathophysiological relevance of this genetic finding.
Clinical and genetic findings
The index patient is a now 43-year-old woman. Past medical history revealed Hashimoto’s thyroiditis treated by oral thyroid hormones and episodic migraine without as well as with aura (age at onset: 12 years; attack frequency: 1–2/year), but was otherwise unremarkable, and there were no vascular risk factors.
At the age of 37, she began to experience diverse physical complaints, including back pain, bilateral pressing headache, intermittently persisting for several weeks and described as different from her migraine attacks, dizziness, a “diffuse feeling of being exhausted”, increased daytime sleepiness and bilateral tremor of the hands, especially when she felt nervous. Because of these symptoms, she presented to another hospital at the age of 38, where cMRI revealed “bilateral subcortical T2-hyperintense lesions” (data not shown), while electrophysiology (visually and somatosensory evoked potentials) and CSF analysis were unremarkable. Assuming a tentative diagnosis of “autoimmune inflammatory disorder of the central nervous system”, a 3-day course of intravenous steroids (500 mg methylprednisolone) was administered, followed by oral tapering over 4 weeks.
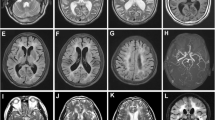
When she presented to our center a few months later for re-evaluation, neurological exam was completely normal. On neuroimaging (Fig. 1), no progression of the non-confluent T2 hyperintense lesions was noted upon manual review, importantly without specific involvement of the temporal poles or the external capsule. Furthermore, there was no evidence of lacunes, microbleeds, or lesions on diffusion-weighted sequences. Repeated electrophysiology, CSF (including oligoclonal bands) and laboratory analysis (including vasculitis parameters) were all within normal limits. Spinal MRI was also normal. Detailed neuropsychological testing was unremarkable except for discrete fluctuations of concentration. In summary, a differential diagnosis of somatoform complaints was considered, as supported by psychosomatic evaluation, and treatment with amitriptyline was initiated.

Representative results of brain imaging in the index patient. On cMRI, T2-weighted sequences revealed a few non-confluent T2 hyperintense lesions (left panel). Of note, there was no specific involvement of the temporal poles (right panel) or the external capsule
The medical team primarily in charge of the patient at that time initiated molecular genetic testing of NOTCH3. This revealed a novel heterozygous variant in exon 18 (NM_000435.2: c.2853_2857delTCCCG). On the protein level, this variant causes a change from prolin to leucin in codon 952 followed by a frameshift, with the introduction of a premature stop codon 9 codons downstream within extracellular EGF-like domain number 24 (p.Pro952Leufs*9). The variant was not detected in various public databases (Human Genome Mutation Database, Leiden Open (source) Variation Database, genome Aggregation Database). Skin biopsy was without evidence of GOM upon electron microscopy.
Family history was negative for symptoms suggestive of CADASIL; in particular, there was no evidence of migraine, recurrent transient ischemic attacks, early strokes, or vascular dementia. The patient’s sister was reported to have had similar changes on cranial MRI and to have committed suicide. No further clinical details were available.
The only other family member, from whom a DNA sample could be obtained, was the index patient’s father. Genetic analysis revealed the presence of the NOTCH3 variant in exon 18. We were able to personally interview and examine him at age 69. In line with his daughter’s earlier reports, there was no history of migraine or recurrent strokes or focal neurological deficits. He had a long-time history as an episodic drinker and had retired 10 years earlier after “knee surgery” (further details unknown). Vascular risk factors included hypertension and a long-term history of smoking. MRI of the brain revealed lacunar lesions, a single microbleed and extensive confluent leukoencephalopathy, yet without involvement of the temporal regions or the external capsule (Fig. 2). On neurological examination, which was without focal deficits, the patient presented with a multifactorial gait disorder, related to polyneuropathy and most likely also to the extensive white matter disease. Neuropsychological testing (Montreal Cognitive Assessment) revealed a score of 21 (out of 30).

Representative results of Brain imaging in the index patient’s father. cMRI revealed lacunar lesions and extensive confluent leukoencephalopathy (left panel), importantly without involvement of the temporal regions (right panel) or the external capsule
We have been following-up on the index patient over a course of more than 5 years with regular telephone interviews and re-evaluations in our outpatient unit. During that interval, the symptoms, which had initially caused her to seek medical advice, had completely remitted, and she was without subjective complaints. There were no strokes or stroke-like episodes, and bedside neuropsychology continued to be normal. Repeated neuroimaging (over a period of more than 5 years) displayed no progression of the T2 hyperintense subcortical lesions upon detailed manual review.
Discussion
We here report a novel non-canonical variant in the NOTCH3 gene, i.e., the gene involved in the hereditary cerebral small-vessel disease CADASIL [1, 5]. For both the index patient and her father, both of whom were heterozygous carriers of the novel allele, we acquired detailed clinical and imaging data, which were neither suggestive nor supportive of a diagnosis of CADASIL. Thus, in other words, our observation provides evidence that the reported novel NOTCH3 variant does not cause a classical CADASIL phenotype.
With respect to the index patient, several lines of evidence argue against a diagnosis of CADASIL: First of all, although clinical presentation of CADASIL is in principle variable, the clinical symptomatology was not suggestive of CADASIL: many of the diverse unspecific complaints can be interpreted as somatoform in origin, and a substantial part of these remitted over time without specific interventions. Neurological status and detailed neuropsychological evaluation were unremarkable. The patient had a history of migraine, but there was no evidence of atypical or complicated aura symptoms, as has been reported in the context of CADASIL [2, 3], and finally, there were no recurrent lacunar syndromes. Second, cranial MRI displayed only a few unspecific non-confluent white matter lesions, which, importantly, did not progress over an extended time course of more than 5 years, although stability of WML has been occasionally observed in CADASIL. Importantly, neither during initial evaluation nor during follow-up MRI, there were any features typical of CADASIL, such as involvement of the temporal lobes or the external capsule [11,12,13,14]. Especially, anterior temporal lobe involvement—although not an obligatory finding—is usually found already early in the course of CADASIL [13] and has been shown to have high sensitivity and specificity [11]. Furthermore, at no time point, any lacunes, microbleeds, or lesions on diffusion-weighted imaging were noted. Finally, skin biopsy was unremarkable, without evidence of GOM, although we are aware of the limited diagnostic sensitivity of this method [11].
Along the same lines, there was no evidence of CADASIL in the index patient’s father: his chief complaint was a 10-year history of gait disturbance, explained by (most likely alcohol-related) polyneuropathy as well as related to extensive white matter disease. Clinical neurological evaluation at the age of 69, except for signs of polyneuropathy, revealed no focal deficits, and there was no history of migraine or recurrent strokes or focal neurological deficits. Bedside neuropsychological testing revealed mild cognitive deficits (MOCA score: 21/30), with some impact on activities of daily living, which, in our interpretation, can be related to a long-standing history as an episodic drinker. cMRI, in addition to some lacunar lesions, revealed a substantial load of leukoencephalopathy (without involvement to the temporal poles or the external capsule), which, in the light of the patient’s extensive vascular risk factor profile (including hypertension, smoking and chronic alcohol abuse), is compatible with a diagnosis of sporadic cerebral small-vessel disease, possibly aggravating the cognitive deficits.
We were not able to obtain DNA samples or retrieve clinical information from any other family members, which is a limitation of our report. Specifically, the lack of genetic, clinical and imaging data on the index patient’s sister is a limitation, in particular given the anecdotal report of WML in her and the fact that she committed suicide, suggesting a possible psychiatric phenotype. However, since she was already deceased at the time when we were first in contact with the index patient, we had no opportunity to obtain more detailed information.
On the other hand, we provide in-depth clinical information on both the index patient and her father, which, as a further strength, includes longitudinal clinical and imaging data for the index, covering a follow-up period of 6 years.
Looking into the literature, a few other authors have reported non-canonical NOTCH3 variants and controversially discussed their pathophysiological relevance: Rutten and colleagues reported two hypomorphic NOTCH3 alleles (one nonsense mutation as well as a large intragenic frameshift deletion) [15]; using a combination of clinical and imaging data as well as skin biopsy, they found no evidence for an association with CADASIL for both variants (although neuroimaging was not available in one individual and RNA analysis was not performed); in line with these findings, NOTCH3 nonsense variants have also been detected in an exome resequencing study of patients with colorectal cancer [16]. On the other hand, Moccia et al. investigated a family with the same NOTCH3 nonsense mutation (p. Arg103X) initially reported by Rutten et al. [17]; importantly, the authors performed mRNA analysis which suggested nonsense-mediated mRNA decay, leaving only the wild-type allele. Based on clinical and imaging data, the authors established a diagnosis of CADASIL in four carriers, while one 27-year-old female had only migraine with aura. Interestingly, skin biopsy in all carriers of the allele was without evidence of GOM. Referring to the seemingly controversial findings in their family and the pedigree reported by Rutten et al., the authors suggest that nontypical genetic variation in NOTCH3 may cause what they call a “CADASIL-like syndrome” with “variable penetrance and/or age at onset” [17].
More specifically, with respect to small frameshift NOTCH3 variants (caused by out-of-frame NOTCH3 deletions), i.e., the same type of mutation as reported here, a few reports have concluded that these may cause CADASIL [18, 19]; however, as put forward by Rutten et al. in the discussion of their manuscript [15], skin biopsy was missing and there were no cosegregation data.
Looking specifically at the variant detected in our family, it would have been potentially interesting to gain more detailed molecular insight, e.g., focussing on the question whether the mutant mRNA is degraded (by nonsense-mediated mRNA decay, NMD) or if it is transcribed into a truncated protein. However, no material for RNA analysis was ascertained upon initial evaluation, and given the spontaneous remission of symptoms in the index, this was also not a feasible option later on.
Summing up, in the light of the controversial data from the literature, how should we interpret our own finding? As outlined above, both patients from our family did not reveal a classical clinical and imaging CADASIL (or CADASIL-like) phenotype, and clinical findings in both individuals can be sufficiently explained by other etiologies. This reinforces the concept of hypomorphic NOTCH3 variants most likely not being causative for CADASIL.
Does this automatically mean that the detected variant is non-pathogenic, i.e., a coincidental finding? This question is more difficult to answer. Especially considering the fact that the variant was not detected in public databases, we cannot exclude a potential association with other phenotypes. However, given the low number of published reports on similar non-canonical NOTCH3 variants, accurate interpretation of these findings obviously remains a substantial challenge. To gain further insight in the pathophysiological relevance of such types of genetic variation in NOTCH3 and to clarify their potential association with neurological and/or psychiatric phenotypes other than CADASIL, prospective assessment of similar genetic findings together with deep phenotyping information is necessary.
References
Chabriat H, Joutel A, Dichgans M et al (2009) CADASIL. The Lancet Neurol 8:643–653. https://doi.org/10.1016/S1474-4422(09)70127-9
Vahedi K, Chabriat H, Levy C et al (2004) Migraine with aura and brain magnetic resonance imaging abnormalities in patients with CADASIL. Arch Neurol 61:1237–1240. https://doi.org/10.1001/archneur.61.8.1237
Guey S, Mawet J, Hervé D et al (2015) Prevalence and characteristics of migraine in CADASIL. Cephalalgia. https://doi.org/10.1177/0333102415620909
Dichgans M, Holtmannspötter M, Herzog J et al (2002) Cerebral microbleeds in CADASIL: a gradient-echo magnetic resonance imaging and autopsy study. Stroke 33:67–71
Joutel A, Corpechot C, Ducros A et al (1996) Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 383:707–710. https://doi.org/10.1038/383707a0
Peters N, Opherk C, Bergmann T et al (2005) Spectrum of mutations in biopsy-proven CADASIL: implications for diagnostic strategies. Arch Neurol 62:1091–1094. https://doi.org/10.1001/archneur.62.7.1091
Duering M, Karpinska A, Rosner S et al (2011) Co-aggregate formation of CADASIL-mutant NOTCH3: a single-particle analysis. Hum Mol Genet 20:3256–3265. https://doi.org/10.1093/hmg/ddr237
Tikka S, Mykkänen K, Ruchoux M-M et al (2009) Congruence between NOTCH3 mutations and GOM in 131 CADASIL patients. Brain 132:933–939. https://doi.org/10.1093/brain/awn364
Joutel A, Andreux F, Gaulis S et al (2000) The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J Clin Invest 105:597–605. https://doi.org/10.1172/JCI8047
Arboleda-Velasquez JF, Manent J, Lee JH et al (2011) Hypomorphic Notch 3 alleles link Notch signaling to ischemic cerebral small-vessel disease. Proc Natl Acad Sci USA 108:E128–E135. https://doi.org/10.1073/pnas.1101964108
Markus HS, Martin RJ, Simpson MA et al (2002) Diagnostic strategies in CADASIL. Neurology 59:1134–1138
Auer DP, Pütz B, Gössl C et al (2001) Differential lesion patterns in CADASIL and sporadic subcortical arteriosclerotic encephalopathy: MR imaging study with statistical parametric group comparison. Radiology 218:443–451. https://doi.org/10.1148/radiology.218.2.r01fe24443
O’Sullivan M, Jarosz JM, Martin RJ et al (2001) MRI hyperintensities of the temporal lobe and external capsule in patients with CADASIL. Neurology 56:628–634
Chabriat H, Joutel A, Dichgans M et al (2009) CADASIL. The Lancet Neurol 8:643–653. https://doi.org/10.1016/S1474-4422(09)70127-9
Rutten JW, Boon EMJ, Liem MK et al (2013) Hypomorphic NOTCH3 alleles do not cause CADASIL in humans. Hum Mutat 34:1486–1489. https://doi.org/10.1002/humu.22432
Smith CG, Naven M, Harris R et al (2013) Exome resequencing identifies potential tumor-suppressor genes that predispose to colorectal cancer. Hum Mutat 34:1026–1034. https://doi.org/10.1002/humu.22333
Moccia M, Mosca L, Erro R et al (2015) Hypomorphic NOTCH3 mutation in an Italian family with CADASIL features. Neurobiol Aging 36(547):e5–e11. https://doi.org/10.1016/j.neurobiolaging.2014.08.021
Dotti MT, De Stefano N, Bianchi S et al (2004) A novel NOTCH3 frameshift deletion and mitochondrial abnormalities in a patient with CADASIL. Arch Neurol 61:942–945. https://doi.org/10.1001/archneur.61.6.942
Weiming F, Yuliang W, Youjie L et al (2013) A novel Notch3 deletion mutation in a Chinese patient with cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). J Clin Neurosci 20:322–323. https://doi.org/10.1016/j.jocn.2012.02.026
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical standards
The study was performed in accordance with the 1964 Declaration of Helsinki and its later amendments. Patients provided their informed consent, including consent to the publication of clinical and genetic findings.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Schubert, V., Bender, B., Kinzel, M. et al. A novel frameshift variant in the CADASIL gene NOTCH3: pathogenic or not?. J Neurol 265, 1338–1342 (2018). https://doi.org/10.1007/s00415-018-8844-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-018-8844-5