
Abstract
Pathogenic mutations in the OPA1 gene can be associated with Autosomal Dominant Optic Atrophy (ADOA). In approximately 20 % of patients with OPA1 mutations, a more complex neurodegenerative disorder with extraocular manifestations, known as ADOA Plus, can arise. 12 members of a multigenerational family were assessed clinically and screened for a genetic mutation in OPA1. Eight family members displayed manifestations consistent with ADOA Plus and four did not. Affected members of the oldest available generation displayed the most severe phenotype, which included severe optic atrophy, deafness, ptosis, ophthalmoplegia, proximal myopathy, neuropathy and ataxia. The next generation was less severely affected but several members displayed manifestations only after the fifth decade. Genetic analysis revealed a heterozygous variant in the OPA1 gene (c.1053T>A, p.Asp351Glu) that segregated with disease. The affected family members described here exhibited visual loss later than is typical for OPA1-related disease, as well as later onset of other neurological abnormalities in the fifth or sixth decades of life that progressed to severe neurological disability by the seventh decade. These findings expand the clinical spectrum of OPA1-related disease associated with a novel OPA1 mutation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Optic Atrophy 1 (OPA1) is a dynamin-related protein of the large GTPase superfamily that locates to the inner mitochondrial membrane and is integral for mitochondrial structure and function [1–3]. Mutations in the OPA1 gene have largely been associated with autosomal dominant optic atrophy (ADOA; OMIM 165500), which is typified by atrophy of the retinal ganglion cells [4, 5]. This neuro-ophthalmic degeneration typically manifests with early onset (first decade) insidious bilateral visual loss (defects in colour vision and paracentral scotomas) and an absence of extraocular symptoms. For a subset of OPA1 mutations (~20 %), ADOA is accompanied by sensorineural hearing loss in the first decade, with subsequent onset of peripheral manifestations such as progressive external ophthalmoplegia, ptosis, myopathy, peripheral neuropathy and ataxia, leading to a syndromic disease sub-group known as ADOA Plus (OMIM 125250) [1, 6]. To date, over 300 mutations in OPA1 have been identified and associated with ADOA and ADOA Plus (http://mitodyn.org/home.php?select_db=OPA1) [7]. The majority of variants associated with ADOA Plus are missense mutations confined to the GTPase domain of OPA1, which is highly conserved across the dynamin superfamily and requisite for molecular function. We describe a novel heterozygous mutation in OPA1 that is predicted to disrupt the GTPase domain of OPA1 and is associated with phenotypically variable ADOA Plus.
Clinical background
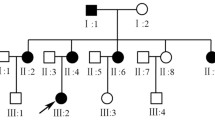
12 members of the affected family were clinically examined (Fig. 1a); six females and two males were found to be symptomatic (Table 1; Fig. 1a). Family members underwent a detailed neurological examination including brain magnetic resonance imaging (MRI), nerve conduction studies (NCS), electromyography (EMG), electrocardiography (ECG), optical coherence tomography (OCT) and auditory testing. The family pedigree presentation was indicative of an autosomal dominant mode of inheritance (Fig. 1a).

(a) Multigenerational family pedigree showing patients available for genetic testing (black outline symbol) and unavailable for genetic testing (grey outline symbol). A novel heterozygous OPA1 mutation (c.1053T>A; half coloured symbols) was found to segregate with disease symptoms (asterisk) that are consistent with autosomal dominant optic atrophy Plus. Circles female, squares male, arrows proband. (b) Sequencing electropherogram showing wild-type sequence (upper trace) and representative novel heterozygous OPA1 c.1053T>A mutation (lower trace) from exon 11. (c) The novel mutation abolishes a restriction enzyme site for MboI (enzyme cut sites indicated by black vertical lines, site abolished by mutation indicated by cross) and creates a banding pattern on agarose gel electrophoresis that is indicative of mutation carriers (upper 242 bp band unique to mutation carriers). Colours of fragments from the upper diagram are shown next to the respective digest fragment on the agarose gel. −C = negative control, NTC = no-template control. 100 bp ladder shown. (d) The novel nucleotide mutation is predicted to cause a p.Asp351Glu amino acid change to an evolutionarily conserved residue (red outline) located within the G-box 1 motif (blue outline) of the GTPase domain of OPA1. (e) Comparison of G-box 1 motifs from other dynamin-related protein superfamily members shows that the aspartic acid at position 351 of OPA1 (red box) is not conserved in the consensus motif of related proteins, suggesting a structural rather than functional role for this amino acid in OPA1. Upper sequences relate to mitochondria-associated proteins and the lower sequences relate to other cellular proteins from the dynamin superfamily
The proband (II.9) developed her first clinical symptom of bilateral visual loss in her fourth decade, which gradually progressed to legal blindness by her seventh decade. Hearing loss was first noticed in her fifth decade, with an ocular myopathy developing around the same time (Table 1). Further neurological problems including sensorimotor neuropathy, mild proximal myopathy and ataxia only became evident when the patient was in her seventh decade. At the time of examination, she demonstrated severe visual loss, with bilateral visual acuity of 6/60. The patient had moderate ptosis and a moderate to severe generalised ophthalmoplegia. Upper and lower limb examination revealed mild proximal weakness and sensory disturbance in a ‘glove and stocking’ distribution. Deep tendon reflexes were absent and her gait was unsteady and mildly wide based.
Within the same generation, there were three other similarly affected family members (II.1, II.4 and II.11). Two of these patients developed visual loss from the fifth decade, with the third experiencing childhood onset visual loss (Table 1). Other neurological deficits progressed from the fifth decade onwards (Fig. 2).
The third generation was found to be less affected, which is likely to represent an age/expressivity correlation. These patients generally developed mild visual and hearing loss in their fourth decade (Table 1). However, one patient (III.20) had childhood onset visual loss and subsequent ptosis.
All affected patients had optic nerve head pallor with an increased cup to disc ratio, regardless of visual acuity (Table 1).
Clinical investigation
The visual acuity in the second generation ranged from 6/7.5 to count fingers acuity, as compared with that of the third generation, which ranged from 6/6 to 6/12. OCT of the optic nerve was performed on all family members, with affected patients showing markedly reduced average retinal nerve fibre layer thickness, even when visual loss was mild (Table 1). These measurements ranged from 45 to 70 μm, and were all below the third centile when compared to age- and sex-matched controls. There was little difference between generations. Affected patients generally showed an increased cup to disc ratio on OCT, ranging from 0.56 to 0.86, when compared to non-affected family members and normal population values.
Five of the eight affected patients underwent MRI of the brain, with all patients showing optic nerve atrophy. There were no other relevant abnormal findings. Six of the eight affected patients had NCS and EMG. Two patients had evidence of an axonal neuropathy on NCS, and four patients had features of myopathy on EMG. None of the patients had elevated serum lactate, pyruvate or creatine kinase. Additionally, cardiological investigations were normal for all patients examined.
Genetic investigation
Initial genetic screening of the OPA1 gene in II.4 and II.9 (Fig. 1a) was outsourced to Centogene (Rostock, Germany). A heterozygous nucleotide transversion (c.1053T>A; numbering according to NM_130837) (Fig. 1b) was screened for in available family members (Fig. 1a) by Sanger sequencing of a 420 base pair amplicon encompassing OPA1 exon 11. The mutation abolishes an MboI restriction digest site creating a banding pattern unique to mutation carriers on restriction digestion agarose gel electrophoresis (Fig. 1c). The sequencing and restriction digest results confirmed segregation of the mutation with disease phenotypes (Fig. 1a, c).
The nucleotide change is predicted to alter the amino acid sequence by substituting Glutamic acid for a highly conserved Aspartic acid (p.Asp351Glu; protein numbering according to UniProt entry E5KLJ5) located within the G-box 1 motif of the OPA1 GTPase domain (Fig. 1d) [8]. In silico mutation prediction using Mutation Taster, PolyPhen 2 and SIFT Human Protein indicated the p.Asp351Glu mutation would be ‘disease causing’, ‘probably damaging’ and ‘damaging’, respectively [9–11]. Although the Aspartic acid at p.351 is completely evolutionarily conserved in OPA1 across species (Fig. 1d), the affected residue is variable within homologous G-box 1 motifs of other dynamin-related proteins from Homo sapiens (Fig. 1e). This is indicative of a highly important structural role for Aspartic acid at this position in OPA1, where it likely stabilises the functional G-box 1 motif, which is known to be responsible for binding to and coordinating the GTP molecule for subsequent hydrolysis, a requisite for OPA1 molecular function.

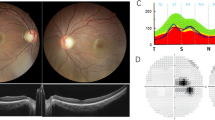
(a) Patient II.4 developed symptoms in his first decade of life, later progressing to ptosis, progressive external ophthalmoplegia, hearing loss and ataxia. (b) Fundus photograph of affected patient demonstrating optic nerve head pallor and an increased cup to disc ratio
Discussion
We report the identification of a novel heterozygous OPA1 mutation that is predicted to affect the GTPase domain of OPA1 and manifests as ADOA Plus. The variable and often delayed onset phenotypic manifestation and progression of ADOA Plus in the large pedigree described here is at odds with the generally observed temporal phenotypic spectrum [6]. The family also provides an example of the phenotypic heterogeneity and potential neurological complications of OPA1 mutations.
As multiple tissues are affected in the patients described here, it is likely that the mutation exerts a dominant negative effect on the wild-type allelic product. This would be consistent with the majority of other ADOA Plus mutations and with dominant negative influences observed in GTPase-deficient dynamins [12]. Furthermore, disruption of the G-box1 motif (Fig. 1d) has been shown to disrupt GTPase activity required for proper OPA1 function [13, 14]. Due to the complete conservation of Aspartic acid at the affected position in OPA1 across species (Fig. 1d) and the precise spatial requirements for GTP (and Mg2+) binding, coordination and subsequent hydrolysis by the GTPase domain, the predicted amino acid change appears sufficient to disrupt the structure of the OPA1 GTPase domain.
The clinical features of the family described are relatively unique, in being particularly late onset for the majority of those affected. The largest collection of ADOA Plus patients, described by Yu-Wai-Man et al., displayed visual loss in the first and second decades followed by hearing loss in the second and third decades and other neurological abnormalities from the third decade onwards [6]. In our cohort, only two of the eight patients had visual loss in the first decade of life. The remaining six did not become visually symptomatic until they were in their fourth or fifth decades (Table 1). Whilst there have been cases of late-onset visual loss in OPA1 patients, including a patient in her seventh decade, this observation is unusual and divergent from the large majority of ADOA/ADOA Plus cases [15]. In the late-onset patients described herein, hearing loss often occurred concurrently with visual loss. Notably, some features including neuropathy and gait ataxia did not manifest until patients were in their seventh decade. The basis of the atypical phenotypic presentation in this family is unclear. Other genetic and environmental factors may play a role in modulating the disease progression and next-generation sequencing may provide relevant information.
OCT was markedly abnormal in all symptomatic patients and may be useful as a screening tool in pre-symptomatic/early stage patients who have not yet developed decreased visual acuity and where genetic testing is not available. With increasing evidence that OPA1 mutations can lead to a multisystemic disorder of variable onset, with neurological complications potentially arising decades after the onset of visual loss, ADOA patients may benefit from regular neurological screening by way of clinical examination and diagnostic investigations.
References
Amati-Bonneau P, Valentino ML, Reynier P et al (2008) OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 131(Pt 2):338–351
Elachouri G, Vidoni S, Zanna C et al (2011) OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res 21(1):12–20
Olichon A, Baricault L, Gas N et al (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278(10):7743–7746
Alexander C, Votruba M, Pesch UE et al (2000) OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26(2):211–215
Delettre C, Lenaers G, Griffoin JM et al (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26(2):207–210
Yu-Wai-Man P, Griffiths PG, Gorman GS et al (2010) Multi-system neurological disease is common in patients with OPA1 mutations. Brain. 133(Pt 3):771–786
Ferre M, Amati-Bonneau P, Tourmen Y, Malthiery Y, Reynier P (2005) eOPA1: an online database for OPA1 mutations. Hum Mutat 25(5):423–428
Liesa M, Palacin M, Zorzano A (2009) Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89(3):799–845
Adzhubei IA, Schmidt S, Peshkin L et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4(7):1073–1081
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11(4):361–362
Marks B, Stowell MH, Vallis Y et al (2001) GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature 410(6825):231–235
Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM (2004) Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem 279(18):18792–18798
Patten DA, Wong J, Khacho M et al (2014) OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J 33(22):2676–2691
Lenaers G, Hamel C, Delettre C et al (2012) Dominant optic atrophy. Orphanet J Rare Dis. 7:46
Acknowledgments
The authors wish to acknowledge the clinical assistance of Dr N. Gerbis. KEA is a NHMRC postgraduate scholar (#1074763). RLD is a NHMRC early career research fellow (#1037797). CMS is a NHMRC practitioner fellow (#1008433).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflicts of interest.
Ethical standards
The authors hereby declare that the research documented in the submitted manuscript has been carried out in accordance with ethical standards laid down in the 1964 declaration of Helsinki and was approved by the ethics committee of the Northern Sydney Local Health District.
Additional information
K. E. Ahmad and R. L. Davis contributed equally to this work.
Rights and permissions
About this article

Cite this article
Ahmad, K.E., Davis, R.L. & Sue, C.M. A novel OPA1 mutation causing variable age of onset autosomal dominant optic atrophy plus in an Australian family. J Neurol 262, 2323–2328 (2015). https://doi.org/10.1007/s00415-015-7849-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-015-7849-6