
Abstract
As an important aspect of epigenetics, DNA methylation has been proven to be suitable for forensic DNA analysis. By detecting changes in DNA methylation, it is desirable to construct a model of age patterns associated with it to infer the age of the individual. The hTERT gene methylation is closely related to tumors, but there are few reports on the relationship between hTERT gene promoter methylation and age. In this study, we utilized the methylation-specific polymerase chain reaction and real-time PCR (relative quantification and absolute quantification) approach to explore the connection between hTERT DNA methylation and age prediction. We fit three models for age prediction based on methylation assay for 90 blood samples from donors aged 1–79 years old. Among them, the model of absolute quantification of real-time enabled the age prediction with R2 = 0.9634. We verified the linear regression model with a validation set of 30 blood samples where prediction average error was 4.29 years. Generally, this reliable method improves the DNA methylation analysis of forensic samples.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
At present, the age inference of forensic individuals is based on the theory of forensic anthropology. The age could be inferred by measuring the bones, teeth, and other specimens or by using the morphological markers of imaging data such as X-ray films. However, the obtained age-inferred results are influenced by subjective factors as the appraisers, further, whether bones or teeth required for analysis are intact may trigger fluctuated results [1]. It is not possible to estimate the age of biological samples without morphological features left on site. For another, the use of biological macromolecular markers for age inference is currently attractive. Case in point is that some age-related molecular markers, such as telomere length, telomerase activity, T cell DNA rearrangement, glycosylation end products, and mtDNA mutations, have been identified, due to the accuracy and duplication of age-inferred results [2].
Studies have shown that epigenetics, especially DNA methylation, has a significant correlation with age [3]. DNA methylation is affected by the environment, resulting in a change in the degree of DNA modification. The expression of the regulated gene under different environmental conditions is more responsive to the age of the individual. Its application to forensic practice is a seminal complement to existing DNA analysis methods. DNA methylation is the earliest and most common mammalian genome modification pathway, which refers to the transfer of methyl group of S-adenosylmethionine to the cytosine ring in DNA molecules catalyzed by DNA methyltransferase. A carbon atom that produces 5-methylcytosine (5mC) [4]. DNA methylation plays an important role in maintaining the stability of a genomic genetic material and regulating gene expression, and is closely related to the occurrence of tumors, genetic diseases, autoimmune diseases, and aging. From a forensic point of view, epigenetics provides a richer set of information beyond the DNA sequence for the continuous improvement of DNA identification technology. There is a certain correlation between DNA methylation and age. Some studies have shown that the overall level of DNA methylation in humans decreases with age [5], especially, the ability of DNA to maintain methylation is reduced during cell differentiation. However, other studies have suggested that while the overall methylation level of the genome is reduced, there are also some phenomena of increased methylation levels of genes, such as the level of DNA methylation in central tissues with the accretion of age [6]. The determination of possible alterations in DNA methylation patterns could aid various forensic investigations, such as differentiating monozygotic twins, identifying the tissue source, or determining the age of tissue donors [7].
According to the known DNA methylation assays, they can be divided into methylation analysis method based on restriction endonuclease, methylation analysis method based on bisulfite, and column method. At present, a methylation-sensitive restriction endonuclease (MSRE), a methylated CpG-binding protein (MeCP), and a sulfite conversion (bisulfite) have been established. Among these series of DNA methylation assays, the most common methods based on the principle of sulfite conversion, including methylation-specific PCR (MSP), methylation fluorescence (MethyLight), high-resolution melting curve (HRM), Sanger clone sequencing (BSP), pyrosequencing, and gene chip method. Following multivariate, linear regression and ANN analysis, Vidaki et al. [8] identified an epigenetic aging signature based on the methylation status of a total of 16 CpG sites. To allow for reliable age predictions, a next-generation sequencing protocol based on Illumina’s MiSeq1 platform was developed and optimized using commercially available DNA methylation standards. Hannum et al. [9] have shown that genome-wide methylation patterns represent a strong and reproducible biomarker of biological aging rate. DNA methylation in 5 CpG sites located in ELOVL2, C1orf132, TRIM59, KLF14, and FHL2 has been analyzed to enable estimation of human chronological age with high accuracy [10]. Furthermore, DNA methylation in ELOVL2 and C1orf132 has been proved to correctly predict chronological age of individuals from three disease groups (early onset Alzheimer’s disease, late onset Alzheimer’s disease, and Graves’ disease) [11]. Human telomerase reverse transcriptase (hTERT) is a pivotal protease involved in tumorigenesis and development. The methylation status of its promoter region affects its expression [12]. The hTERT gene promoter contains a large number of CpG islands, with a GC-rich sequence, suggesting that hTERT expression may be regulated by methylation. In tumor cells and tissues, most of the hTERT promoters are hypermethylated, but the methylation pattern is much more complex. Normal cell or tissue hTERT promoter is mostly methyl-free or only hypomethylated [13]. However, in the stem cells, germ cells, and some normal somatic cells such as human oral fibroblasts, the hTERT promoter is methylated [14]. Previously, studies on methylation of hTERT gene focused on the mechanism of tumor occurrence and development, diagnosis, and treatment. There are few reports on the connection between methylation of hTERT gene and age. In this work, the hTERT gene was used to investigate the relationship of methylation and age.
Materials and methods
Sample collection and DNA extraction
The study was approved in a Chinese Han population. Blood samples were collected by the members of the Department of Clinical Laboratory, Shanghai Xuhui District Dahua Hospital. All volunteers signed the written informed consent statements after explaining the goals of the study. Blood samples from 90 individuals (female 44, male 46) aged between 1 and 79 years were developed in our DNA methylation assay. The collected blood samples were placed in a collection tube containing EDTA frozen at − 20 °C until DNA extraction.
Genomic DNA was extracted from 200 μl peripheral blood using a DNeasy Blood & Tissue Kit (QIAGEN) according to the manufacturer’s protocol in a final volume of 100 μl. Finally, the integrity was separated by 1% agarose gel electrophoresis.
Bisulfite conversion
Genomic DNA (200 ng) in a volume of 20 μl was converted using the EZ DNA Methylation-Gold Kit (Cat. No. D5005) as per the manufacturer’s instructions, where unmethylated cytosine was converted to uracil and methylated cytosine did not change. There was complete conversion of GC-rich DNA in 3 hours. The two heat denaturation reaction steps simplify the conversion of unmethylated cytosine to uracil. The total reaction volume was 10 μl. The DNA can be used for immediate analysis store − 20 °C for later use in a short-time period or − 70 °C for long-term future use.
Methylation-specific polymerase chain reaction
The methylated cytosine in the DNA sequence after vulcanization remains unchanged, while the unmethylated cytosine is converted to uracil, and uracil can be replaced by deoxythymidine during the PCR reaction. MSP technology is designed for two pairs of different primers to detect the methylation status in the DNA sequence, which has the advantages of having minimal amount of DNA required, no need for specific restriction sites, and high sensitivity. The reaction conditions include denaturation at 98 °C for 10 s, annealing at 60 °C for 30 s, and extension at 72 °C for 1 min for a total of 40 cycles. The specific primers designed by Meth-Primer and their sequences are shown in Table 1. The amplified product was electrophoresed on a 2% agarose gel containing ethidium bromide and observed under a UV detector. The presence of M-primer amplification products indicates the presence of DNA methylation. The presence of amplification products in U-primer is the absence of DNA methylation. If amplification products are present at the same time, partial DNA methylation is considered. Finally, a prediction model for age was established by comparing the optical density values of the M and U bands.
Real-time PCR
Real-time PCR was used to perform relative quantification and absolute quantification of the hTERT gene. The PCR reaction system contained SYBR@Premix Ex TaqTM (2×) of about 12.5 μl, 0.5 μl of the upstream and downstream primers, and 200 ng of template DNA. The volume of reaction was supplemented with double distilled water to 25 μl and subjected to two-step PCR amplification. Cycle parameters are 95 °C, 30 s; 95 °C, 5 s; 60 °C, 30 s; 40 cycles; 95 °C, 15 s; 60 °C,1 min; 95 °C 15 s. Two replicate holes are provided for each specimen. According to Methprimer software, few methylation was found in a certain region of the promoter of the housekeeping gene β-actin. Therefore, β-actin was chosen as an internal reference gene. The degree of methylation of hTRET gene was obtained by comparing the optical density of hTERT gene with β-actin. Their sequences are shown in Table 1.
The hTERT gene was absolutely quantified by methylation standard, and the mixture was diluted 10 times with double distilled water as the solvent, and the concentration was 100, 80, 60, 40, 200 ng/μl, and stored at 4 °C. Methylation-positive control DNA with different concentrations as the reaction substrate was optimized with a concentration gradient. A standard curve was prepared by taking the Ct value as the abscissa and the amount of the standard DNA as the ordinate. Two models can be established by relative quantification and absolute quantification to detect the degree of methylation of the hTERT gene.
Validation of the developed models in blood and statistics
Thirty blood samples as the validation set was allowed to examine the age prediction model. The average error from the chronological age was calculated to evaluate the accuracy of our prediction assay with linear regression model. Finally, by comparing the average error of the three age prediction models, the final age prediction model was determined.
Results
Methylation-specific polymerase chain reaction
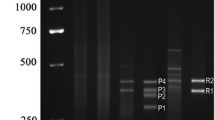
The DNA sequence of the hTERT gene promoter region contains a large number of CpG islands, and the DNA sequence detected in this experiment spans the transcription initiation site of the hTERT gene. The length of the methylated and unmethylated bands was 151 bp and 153 bp, respectively. We used 2% agarose gel electrophoresis and ethidium bromide staining to show the methylated and unmethylated DNA obtained by MSP (Fig.1). From the randomly selected 24 specimens, we can see that the unmethylated band gradually deepens with age. It can be preliminarily judged that the methylation status of the hTERT gene promoter region gradually decreases with age. Based on the ratio of the optical densities of the methylated bands to the unmethylated bands of a total of 60 blood samples, a predictive model of the relationship between the degree of methylation of the hTERT gene and age was obtained. We found that the methylation of the hTERT gene is negatively correlated with age shown in Fig.2a (Pearson r = −0.9923). As the age increased, the degree of methylation of the hTERT gene was gradually reduced, which is consistent with the electropherogram results of the obtained MSP. Figure 2 b presents a plot with chronological age vs. predicted age. The developed model explained 91% of variation in age (corrected R2 = 0.9106) with an average error of 6.60 years.

Electropherogram of MSP product in partial sample of hTERT gene. M, product of methylation primer amplification (151 bp); U, product of unmethylated primer amplification (153 bp). The top number represents age

Development of the statistical model for age prediction using MSP. a The relationship of age and methylation (linear correlation R2 = 0.9846). b Predicted vs. chronological age (years) for all 60 individuals used in this study (linear correlation R2 = 0.9106, average error = 6.60 years)
Real-time PCR
Real-time PCR was based on the addition of MSP using SYBR Green I dye to determine the amount of initial template by detecting an increase in fluorescence signal. The relative quantification of real-time was by comparing the optical density values of the methylated band with the internal reference gene β-actin. Absolute quantitative real-time PCR was based on methylation-positive control to establish a standard curve to predict the methylation level of hTERT gene in different age groups. Two real-time PCR results were shown in Figs. 3a and 4a. The results are consistent with those obtained by MSP, but result is that methylation is negatively correlated with age (Pearson r = − 0.9656 and Pearson r = − 0.9842). As shown in Figs. 3b and 4b, the predictive model established by relative quantitative real-time PCR with age information and DNA methylation pattern of the 60 training set samples explained 93.83% of the total variance (R2 = 0.9383) with an average error of 5.31 years and one established by absolute quantitative real-time PCR explained 95.64% of the total variance in the 60 individuals (R2 = 0.9564) with an average error of 4.36 years.

Development of the statistical model for age prediction using real-time relative quantification PCR. a The relationship of age and methylation (linear correlation R2 = 0.9324). b Predicted vs. chronological age (years) for all 60 individuals used in this study (linear correlation R2 = 0.9383, average error = 5.31 years)

Development of the statistical model for age prediction using real-time absolute quantification PCR. a The relationship of age and methylation (linear correlation R2 = 0.9687). b Predicted vs. chronological age (years) for all 60 individuals used in this study (linear correlation R2 = 0.9564, average error = 4.36 years)
Validation of the developed models in blood
To validate the model’s prediction performance, 30 blood samples were allowed to examine the age prediction model. As shown in Fig. 5, the accuracy of the model established by real-time absolute quantitative PCR is higher than that of MSP and real-time relative quantitative PCR. This age prediction model from MSP had only slightly worse parameters, explaining 91.5% of age variance with an average error of 6.60 years. The model from real-time relative quantification PCR had an R value of 0.974 and the developed model explained 94.88% of variation in age (corrected R2 = 0.9488) with an average error of 5.19 years. Figure 5c presents a plot with chronological age vs. predicted age (R2 = 0.9634) with the average error of 4.29 years. From Fig. 5, we can also see that R2 is constantly improving, which also shows the model from the real-time absolute quantitative PCR can improve the accuracy of predicted age.

Validation of the developed models. a MSP: predicted vs. chronological age (years) for all 30 individuals used in this study (linear correlation R2 = 0.9153, average error = 6.60 years). b The relative quantification of real-time: predicted vs. chronological age (years) for all 30 individuals used in this study (linear correlation R2 = 0.9488, average error = 5.19 years). c The relative quantification of real-time: predicted vs. chronological age (years) for all 60 individuals used in this study (linear correlation R2 = 0.9634, average error = 4.29 years)
Discussion
DNA methylation is a paramount component of epigenetics which plays an indispensable role in maintaining normal cell function, genetic imprinting, embryonic development, and human tumorigenesis. Some cells undergo age-related changes during aging. For example, de novo methylation of a CpG island shuts down a gene, causing loss of physiological functions associated with this gene; likewise, loss of methylation also activates genes that are normally silenced, resulting in inappropriate ectopic expression. DNA methylation is characterized by age, with major causes including changes in DNA methylation that accumulate during the body’s growth and changes in methylation status at specific sites in the genome due to gene expression requirements [15]. With the increasing of age, due to the progressive loss of DNA methyltransferase DNMT1a enzyme activity, the overall level of DNA methylation presented a downward trend, while increased at some special sites, revealing certain correlation [16].
Human telomerase reverse transcriptase is the center of human telomerase activity and is one of the most imperative and common tumor-specific biochemical markers. It is a significant protease involved in tumorigenesis and development [17]. In recent years, the hTERT promoter has been used to regulate anti-tumor genes such as oncolytic virus, apoptosis gene, tumor suppressor gene, and suicide gene all of which have obvious anti-tumor effects in experiments. So far, studies on methylation of hTERT gene focused on the mechanisms of tumor development, diagnosis, and treatment. The latest report on the methylation status of an hTERT gene promoter in peripheral blood leukocytes can be used as a molecular marker for the progression of head and neck cancer [18]. Since previous reports on the hTERT gene have concentrated on cancer and are rarely related to age [18], it is used as a gene of interest to better correlate age and disease. As demonstrated by results, the degree of methylation of the hTERT gene decreases with age. The degree of methylation is illustrated negatively correlated with age. Hence, the hTERT gene is a promising gene that also corresponds with age. Compared with the studies of Spólnicka et al. [11], hTERT is different from ELOVL2 and C1orf132, which is both related to disease and age. How to coordinate the correlation between disease and age of hTERT methylation remains to be further studied in the future.
Currently, methylation detection methods include the following types: (1) polymorphism detection by methylation-sensitive restriction endonuclease combined with polyacrylamide gel electrophoresis (MSAP), (2) utilization of sulfurous acid hydrogen salt modification method, and (3) chip technology. The most commonly used is the bisulfite modification method. In order to verify the conversion efficiency of bisulfite, we referred to the methods mentioned in the paper from Ogino et al. [19] to ensure the same conversion efficiency for each sample. We also compare the three methods based on the principle of bisulfite modification. After modification, the unmethylated cytosine is converted to uracil, and the methylated cytosine remains unchanged. In contrast, bisulfite conversion retains intact DNA sequences and requires a small amount of DNA template, making it a usual method for forensic DNA methylation analysis for qualitative and quantitative analysis of methylation. However, the transformation conditions are harsh otherwise it is inclined to trigger DNA degradation. At the same time, there may be incomplete conversions that result in overestimation of the proportion of DNA methylation. In the subsequent PCR process, the converted base U is regarded as T by the DNA polymerase, and thus may also cause non-specific amplification. For both cases, MSP designed different primers to amplify methylated and unmethylated DNA and determined methylation at specific sites based on electrophoresis results. Real-time PCR is based on MSP, using SYBR Green I dye to increase the amount of initial template by detecting an increase in fluorescence signal. Although MSP is more commonly used, the common MSP has low sensitivity and can only be used for qualitative analysis, while electrophoresis always generates environmental pollution. Real-time PCR can perform real-time monitoring and quantitative analysis of DNA methylation. From our results, compared with the models established by methylation-specific PCR and real-time relative quantitative PCR, real-time absolute quantitative PCR model predicts the smallest age error (4.29 years). Therefore, real-time absolute quantitative PCR builds up a more accurate quantification of the degree of methylation.
Horvath et al. [15] used 353 CpG sites to predict chronological age with a mean absolute deviance of 3.6 years, while many of the CpG sites individually show only a weak correlation with age. Although the predicted age error of our study is slightly higher, we only use one gene as a marker, which is more convenient. For another, Freire-Aradas et al. [20] have analyzed a total of 209 individuals from 2 to 18 years old using EpiTYPER® DNA methylation analysis. From a total of ten selected genes, six were used to develop a preliminary age prediction model [20], which is more applicable to some children who commit crimes and cannot be well applied to all age groups. Jung et al. [21] have developed age prediction models built separately for each sample type using the DNA methylation values at the 5 CpG sites showed high prediction accuracy with a mean absolute deviation from the chronological age (MAD) of 3.478 years in blood, 3.552 years in saliva and 4.293 years in buccal swab samples, while the studies are based on targeted methylation detection via complex procedures including pyrosequencing, melting curve analysis, and the EpiTYPER system. In our study, the method is easy, convenient, economical, and without age restrictions. Therefore, it has potential application in age prediction.
In summary, as an important marker for forensic genetics, the detection of DNA methylation, especially the specific gene methylation is very useful. It is expected that the real-time absolute quantitative PCR of hTERT gene methylation will be applied as a tool in the age prediction of victims in the field of forensic science in the future.
References
Marroquin T, Karkhanis S, Kvaal SI et al (2017) Age estimation in adults by dental imaging assessment systematic review. Forensic Sci Int 275:203–211
Bürkle A, Moreno-Villanueva M, Bernhard J, Blasco M, Zondag G, Hoeijmakers JHJ, Toussaint O, Grubeck-Loebenstein B, Mocchegiani E, Collino S, Gonos ES, Sikora E, Gradinaru D, Dollé M, Salmon M, Kristensen P, Griffiths HR, Libert C, Grune T, Breusing N, Simm A, Franceschi C, Capri M, Talbot D, Caiafa P, Friguet B, Slagboom PE, Hervonen A, Hurme M, Aspinall R (2015) MARK-AGE biomarkers of ageing. Mech Ageing Dev 151:2–12
Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, Warren ST (2012) Age-associated DNA methylation in pediatric populations. Genome Res 22(4):623–632
Ehrlich M, Gamasosa MA, Huang LH et al (1982) Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 10(8):2709–2721
Chouliaras L, Mastroeni D, Delvaux E, Grover A, Kenis G, Hof PR, Steinbusch HWM, Coleman PD, Rutten BPF, van den Hove DLA (2013) Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol Aging 34(9):2091–2099
Day K, Waite LL, Thalacker-Mercer A, West A, Bamman MM, Brooks JD, Myers RM, Absher D (2013) Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol 14(9):R102
Vidaki A, Daniel B, Court DS (2013) Forensic DNA methylation profiling-potential opportunities and challenges. Forensic Sci Int Genet 7(5):499–507
Vidaki A, Ballard D, Aliferi A, Miller TH, Barron LP, Syndercombe Court D (2017) DNA methylation-based forensic age prediction using artificial neural networks and next generation sequencing. Forensic Sci Int Genet. 28:225–236
Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda SV, Klotzle B, Bibikova M, Fan JB, Gao Y, Deconde R, Chen M, Rajapakse I, Friend S, Ideker T, Zhang K (2013) Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 49(2):359–367
Zbieć-Piekarska R, Spólnicka M, Kupiec T, Parys-Proszek A, Makowska Ż, Pałeczka A, Kucharczyk K, Płoski R, Branicki W (2015) Development of a forensically useful age prediction method based on DNA methylation analysis. Forensic Sci Int Genet. 17:173–179
Spólnicka M, Pośpiech E, Pepłońska B, Zbieć-Piekarska R, Makowska Ż, Pięta A, Karłowska-Pik J, Ziemkiewicz B, Wężyk M, Gasperowicz P, Bednarczuk T, Barcikowska M, Żekanowski C, Płoski R, Branicki W (2017) DNA methylation in ELOVL2 and C1orf132 correctly predicted chronological age of individuals from three disease groups. Int J Legal Med 132(1):1–11
Guilleret I, Benhattar J (2003) Demethylation of the human telomerase catalytic subunit (hTERT) gene promoter reduced hTERT expression and telomerase activity and shortened telomeres. Exp Cell Res 289(2):326–334
Wang Z, Xu J, Geng X, Zhang W (2010) Analysis of DNA methylation status of the promoter of human telomerase reverse transcriptase in gastric carcinogenesis. Arch Med Res 41(1):1–6
Santourlidis S, Wernet P, Ghanjati F, Graffmann N, Springer J, Kriegs C, Zhao X, Brands J, Araúzo-Bravo MJ, Neves R, Koegler G, Uhrberg M (2011) Unrestricted somatic stem cells (USSC) from human umbilical cord blood display uncommitted epigenetic signatures of the major stem cell pluripotency genes. Stem Cell Res 6(1):60–69
Horvath S (2013) DNA methylation age of human tissues and cell types. Genome Biol 14(10):R115
Ciccarone F, Tagliatesta S, Caiafa P, Zampieri M (2018) DNA methylation dynamics in aging: how far are we from understanding the mechanisms? Mech Ageing Dev 174:3–17
Leão R, Apolónio JD, Lee D, Figueiredo A, Tabori U, Castelo-Branco P (2018) Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: clinical impacts in cancer. J Biomed Sci 25(1):22
Sobecka A, Blaszczak W, Barczak W, Golusinski P, Rubis B, Masternak MM, Suchorska WM, Golusinski W (2018) hTERT promoter methylation status in peripheral blood leukocytes as a molecular marker of head and neck cancer progression. J Appl Genet 59(4):453–461
Ogino S, Kawasaki T, Brahmandam M, Cantor M, Kirkner GJ, Spiegelman D, Makrigiorgos GM, Weisenberger DJ, Laird PW, Loda M, Fuchs CS (2006) Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J Mol Diagn 8(2):209–217
Freire-Aradas A, Phillips C, Girón-Santamaría L, Mosquera-Miguel A, Gómez-Tato A, Casares de Cal MÁ, Álvarez-Dios J, Lareu MV (2018) Tracking age-correlated DNA methylation markers in the young. Forensic Sci Int Genet. 36:50–59
Jung SE, Lim SM, Hong SR, Lee EH, Shin KJ, Lee HY (2019) DNA methylation of the ELOVL2, FHL2, KLF14, C1orf132/MIR29B2C, and TRIM59 genes for age prediction from blood, saliva, and buccal swab samples. Forensic Sci Int Genet. 3:1–8
Funding
This work was sponsored by grants from the National Natural Science Foundation of China (31671309), the Opening Project of Shanghai Key Laboratory of Crime Scene Evidence (2018XCWZK12), and the Development Project of Qinghai Provincial Key Laboratory (2017-ZJ-Y10).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethical approval
“All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.”
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xin, Y., Dong, K., Cao, F. et al. Studies of hTERT DNA methylation assays on the human age prediction. Int J Legal Med 133, 1333–1339 (2019). https://doi.org/10.1007/s00414-019-02076-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-019-02076-3