
Abstract
Short tandem repeats (STRs) are DNA regions with a variable number of repeated units (2–6 bp) and attractive to genetic application like forensic, population genetics, and anthropological studies. The Huaxia Platinum System allows the multiplex amplification of 23 autosomal STR loci and two gender determination loci. In the present study, genetic polymorphisms of these 23 autosomal STR loci were first obtained from 309 Sichuan Han individuals, southwest China. No evidence of deviation from the Hardy-Weinberg equilibrium (HWE) was identified. The combined match probability (CMP), combined power of discrimination (CPD), and combined power of exclusion (CPE) are 1.087 × 10−27, 0.999999999999999999999999999, and 0.9999999996 in the Sichuan Han Chinese population, respectively. The inter-population differentiation, PCA, MDS, and phylogenetic analysis revealed that Sichuan Han had a closer genetic relationship with neighboring Han Chinese populations. Our findings indicated that the 23 autosomal STR loci are polymorphic and informative in studied southwestern Chinese Han population and can be utilized as a powerful tool for forensic applications. In addition, population comparisons demonstrated that the genetic similarities widely existed among Han Chinese populations distributed in different administrative divisions, and genetic differences had existed between minorities (most prominently for Uyghur and Kazakh) and Han groups.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
At present, short tandem repeats (STRs) consisted of different DNA motifs, as a polymorphic genetic marker widely scattered through the human genome, played an important role in the personal identification, parentage testing, and evolutionary research [1, 2]. Autosomal STR typing has been becoming an indispensable and routine part of criminological practice after 30 years of technical development in the DNA genotyping and detection [2, 3]. To improve the discriminating power and facilitate the international DNA database establishment, the European Standard Set (ESS) [4], the Combined DNA Index System (CODIS) [5], and the Chinese National Database had successively updated the included STR loci. Recently, a great number of commercial PCR amplification kits combined with different numbers of STR loci had developed, such as the PowerPlex® 21 System [6], AGCU 21+1 [7], and Goldeneye™ DNA ID system 20A kit [8]. However, the international coordination of the aforesaid amplification system has not been achieved yet. The Huaxia Platinum System (Thermo Fisher Scientific) was specifically developed to facilitate international data sharing [9]. The system is a 25-loci, six-dye, multiplex that simultaneously amplifies 23 autosomal STR loci (D3S1358, vWA, D16S539, CSF1PO, TPOX, D8S1179, D21S11, D18S51, Penta E, D2S441, D19S433, TH01, FGA, D22S1045, D5S818, D13S317, D7S820, D6S1043, D10S1248, D1S1656, D12S391, D2S1338, and Penta D) as well as gender identification loci of amelogenin and Y-InDel (rs2032678) [9]. However, the forensic characteristics of these sets of STR genetic markers in southwest Chinese populations remain unclear [10].
China, with a population of approximately over 1.3 billion based on the 2010 national population census, has repeatedly been a research hotspot to elucidate demographic processes in forensic science, population genetics, and molecular anthropology studies. The Chinese population substructure is further complicated. Han Chinese population, with a population over 1.22 billion, was widely distributed in 34 administrative regions. In the present study, a cohort of 309 unrelated healthy Han individuals residing in Sichuan province were recruited and genotyped using the Huaxia Platinum System. Allele frequencies and statistical parameters of forensic interest were evaluated. Additionally, genetic affinity based on 19 overlapped STR loci (D8S1179, D21S11, D7S820, CSF1P0, D3S1358, THOl, D13S317, D16S539, D2S1338, D19S433, vWA, TPOX, D18S51, D5S818, FGA, D16S1043, Penta D, Penta E, and D21S391) between the investigated population and 26 previous investigated Chinese populations which had been typed with the PowerPlex® 21 System or Goldeneye™ DNA ID system 20A kit was investigated in the subsequent comprehensive genetic analysis.
A total of 309 unrelated healthy Han Chinese individuals (172 males and 137 females) residing in Chengdu city of Sichuan province (southwest China) were collected (Figure S1). One milliliter of peripheral blood was obtained in tubes with EDTA. The ancestors of all subjects must live in the present region at least three generations. The humane and ethical research principles recommended by Sichuan University were followed in this study. All participants signed the written informed consents before sample collection. Our study design was approved by the medical ethics committee of Sichuan University. The QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) was used to extract the genomic DNA based on the manufacturer’s recommendations. A 7500 real-time PCR system (Thermo Fisher Scientific) was employed to determine the DNA concentration using Quantifiler Human DNA Quantification Kit. The DNA was diluted to 1 ng/μL and stored at − 20 °C until amplification.
Twenty-three autosomal STR loci and two gender determination loci of amelogenin and Y-InDel (rs2032678) included in the Huaxia Platinum System were co-amplified in one multiplex PCR reaction on a ProFlex PCR System (Thermo Fisher Scientific) according to the manufacturer’s instructions. Separation and detection of PCR-amplified products were conducted on a 3500 genetic analyzers (Applied Biosystems, Foster City, CA, USA). Allele designation was conducted using the software of GeneMapper ID-X v.1.4 by comparison with the allele ladder provided by the corresponding kit. Our laboratory is accredited with ISO 17025 and China National Accreditation Service for Conformity Assessment (CNAS). The guidelines published by the International Journal of Legal Medicine [11] were followed in the overall experimental procedure. The positive control of DNA 007 and negative control of ddH2O in each batch of genotyping were conducted.
Allele frequencies and forensic statistical parameters of 23 autosomal STR loci were calculated in the modified PowerStates spreadsheet. The aforementioned parameters contained the power of discrimination (PD), power of exclusion (PE), and match probability (MP). Subsequently, the observed heterozygosity (Ho), expected heterozygosity (He), probability values (p) of the Hardy-Weinberg equilibrium, and linkage disequilibrium testing were assessed in the Arlequin V3.5 software [12]. Inter-population differentiation between our investigated population and other 26 Chinese reference populations was performed in the Arlequin V3.5 using locus-by-locus comparisons based on 19 overlapped STR loci. Principal component analysis (PCA) and multidimensional scaling analysis (MDS) were conducted in SPSS software (IBM SPSS, version 19.0, Chicago). Nei’s standard genetic distance (Rst) was calculated using Phylip3.695, and a phylogenetic tree was delineated in the Molecular Evolutionary Genetics Analysis 7.0 (MEGA 7.0) software [13].
The genotype data of 23 autosomal STR loci included in the Huaxia Platinum System in the Han Chinese are listed in Table S1. The p values of pairwise linkage of Sichuan Han population are presented in Table S2. Linkage analysis results showed that no evidence of linkage inheritance was observed in the pairs of loci. In addition, no deviation from the Hardy-Weinberg disequilibrium was detected in the studied population at all loci (Table S3). Allele frequencies of our investigated Han population are listed in Table S4. A total of 255 alleles were identified with corresponding allele frequencies spanned from 0.0016 to 0.5291. As shown in Table S3, the MP, PD, PE, TPI, Ho, and He varied from 0.0158 to 2044, 0.7956 to 0.9842, 0.3256 to 0.8080, 1.3435 to 5.3276, 0.6278 to 0.9062, and 0.6181 to 0.9158, respectively. The combined match probability (CMP), combined power of discrimination (CPD), and combined power of exclusion (CPE) are 1.0872 × 10−27, 0.999999999999999999999999999, and 0.9999999996, respectively.
To explore the population genetic similarities and differences among diverse nationalities in different administrative divisions, analysis of molecular variance analysis (AMOVA) was performed. The Fst and corresponding p values between the investigated population and six minority groups (two Manchus, one Hui, one Uyghur, one Kazakh, and one Bai) as well as 20 Han Chinese populations based on allele frequency distributions were calculated. The detailed information of geographic position of the aforementioned populations is presented in Figure S1. In our locus-by-locus comparisons, significant genetic differences were observed between the Sichuan Han population and previously investigated Chinese Uyghur at four loci, Xinjiang Kazakh at one locus, and Jiangxi Han at one locus after the Bonferroni correction (p < 0.0017). No significant difference was identified between the Sichuan Han and other groups (Table S5).
To further investigate the genetic background of the studied population, principal component analysis was conducted based on the overall genetic variation of 19 STR loci. As listed in Figure S2, the first principal component defined 98.158% of the total variance. The second principal component accounted for 0.697%. The Xinjiang groups (Uyghur and Kazakh), which had similar culture, history, and language group, were located together in the upper side. Twenty-one Han Chinese populations residing in different administrative divisions, Bai, Manchu, and Hui populations, were located on the upper side. These distribution patterns indicated a far genetic relationship between the minority groups (Xinjiang Uyghur) and other reference populations, and the close genetic relationship among Han Chinese populations came from different geographic regions.
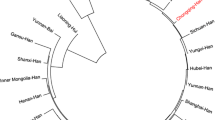
Nei’s standard genetic distances among the 27 Chinese populations are presented in Table S6. The largest genetic distance with our studied population was identified between the Sichuan Han and Xinjiang Kazakh (Rst = 0.0534), while the least genetic distance was observed between Sichuan Han and Hubei Han (Rst = 0.0038). Based on the genetic distance matrix, multidimensional scaling plot (MDS) was depicted in the SPSS and submitted in Figure S3. The population structure pattern was in line with the findings in the PCA. Phylogenetic tree was constructed using the unweighted pair group method with arithmetic means method (UPGMA) and presented in the Figure S4. Two main branches were clustered in the dendrogram. The under branch was consisted of Uyghur and Kazakh populations which belong to Turkic language family groups. The upper branch was made up of representatives of one Hui, two Manchu, one Bai, and 21 Han Chinese populations. Our investigated Sichuan Han was first clustered with Shanghai Han. The genetic relationship revealed by phylogenetic analysis was in agreement with the results of PCA and MDS. Distinctive genetic background and population origins were discovered in this study. Some minority groups, including most prominently Kazakh and Uyghur, demonstrated significant genetic distinction from the Han and other groups, while genetic differentiations between Chinese Han populations distributed in different geographic regions were less pronounced. To better understand the genetic background of Sichuan Han population, it will be necessary to investigate the population relationships between the targeted population distributed in different regions based on a relatively larger sample size and more relative reference populations in the future.
In summary, genetic polymorphisms of 23 autosomal STR loci included in the Huaxia Platinum System were first obtained in the Sichuan Han. Our findings demonstrated that the 23 autosomal STRs were highly polymorphic and informative in the investigated population and can be used as a useful tool for forensic individual identification and parentage testing, and even a powerful tool in population genetic study. The inter-population differentiation, PCA, and MDS as well as phylogenetic analysis revealed that the Sichuan Han clustered together with ethno-origin populations (Han Chinese populations from different administrative regions).
References
Ellegren H (2004) Microsatellites: simple sequences with complex evolution. Nat Rev Genet 5:435–445. https://doi.org/10.1038/nrg1348
Kayser M, de Knijff P (2011) Improving human forensics through advances in genetics, genomics and molecular biology. Nat Rev Genet 12:179–192. https://doi.org/10.1038/nrg2952
Jobling MA, Gill P (2004) Encoded evidence: DNA in forensic analysis. Nat Rev Genet 5:739–751. https://doi.org/10.1038/nrg1455
Gill P, Fereday L, Morling N, Schneider PM (2006) The evolution of DNA databases—recommendations for new European STR loci. Forensic Sci Int 156:242–244. https://doi.org/10.1016/j.forsciint.2005.05.036
Hares DR (2012) Expanding the CODIS core loci in the United States. Forensic Sci Int Genet 6:e52–e54. https://doi.org/10.1016/j.fsigen.2011.04.012
He G-L, Chen P-Y, Gao B et al (2017) Phylogenetic analysis among 27 Chinese populations and genetic polymorphisms of 20 autosomal STR loci in a Chinese Uyghur ethnic minority group. Aust J Forensic Sci:1–10. https://doi.org/10.1080/00450618.2016.1275793
Zhu BF, Zhang YD, Shen CM et al (2015) Developmental validation of the AGCU 21+1 STR kit: a novel multiplex assay for forensic application. Electrophoresis 36:271–276. https://doi.org/10.1002/elps.201400333
Zou X, Li Y, Li P et al (2017) Genetic polymorphisms for 19 autosomal STR loci of Chongqing Han ethnicity and phylogenetic structure exploration among 28 Chinese populations. Int J Legal Med. https://doi.org/10.1007/s00414-017-1577-7
Wang Z, Zhou D, Jia Z et al (2016) Developmental validation of the Huaxia Platinum System and application in 3 main ethnic groups of China. Sci Rep 6:31075. https://doi.org/10.1038/srep31075
Yang L, Zhang X, Zhao L et al (2017) Population data of 23 autosomal STR loci in the Chinese Han population from Guangdong Province in southern China. Int J Legal Med. https://doi.org/10.1007/s00414-017-1588-4
Poetsch M, Bajanowski T, Pfeiffer H (2012) The publication of population genetic data in the International Journal of Legal Medicine: guidelines. Int J Legal Med 126:489–490. https://doi.org/10.1007/s00414-012-0700-z
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Acknowledgements
We would like to thank the volunteers who contributed samples for this study.
Funding
This study was supported by grants from the National Key R&D Program of China (2016YFC0800703) and National Natural Science Foundation of China (81501635, 81330073).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The humane and ethical research principles recommended by Sichuan University were followed in this study. All participants signed the written informed consents before sample collection. Our study design was approved by the medical ethics committee of Sichuan University.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Figure S1
The geographic position of the investigated population and reference populations. (JPEG 611 kb).
Figure S2
Principal component analysis based on 19 overlapped STR loci of our studied population and 26 reference populations. (JPEG 153 kb).
Figure S3
Multidimensional Scaling plots showed the genetic relationships between the investigated population (bold font and red) and 26 reference populations. All Han Chinese populations distributed in different regions just marked their region name in the MDS. (JPEG 129 kb)
Figure S4
The phylogenetic relationship among the studied population and 26 reference populations, which was constructed by the unweighted pair-group method with arithmetic averages (UPGMA) based on 19 overlapped STR loci in the Mega7.0 software. (JPEG 305 kb).
Table S1
(XLSX 74 kb).
Table S2
(XLSX 12.5 kb).
Table S3
(XLSX 11.4 kb).
Table S4
(XLSX 15.4 kb).
Table S5
(XLSX 20.1 kb).
Table S6
(XLSX 16.3 kb).
Rights and permissions
About this article

Cite this article
He, G., Wang, M., Liu, J. et al. Forensic features and phylogenetic analyses of Sichuan Han population via 23 autosomal STR loci included in the Huaxia Platinum System. Int J Legal Med 132, 1079–1082 (2018). https://doi.org/10.1007/s00414-017-1679-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-017-1679-2