
Abstract
Proper repair of double-strand breaks (DSBs) is key to ensure proper chromosome segregation. In this study, we found that the deletion of the SRS2 gene, which encodes a DNA helicase necessary for the control of homologous recombination, induces aberrant chromosome segregation during budding yeast meiosis. This abnormal chromosome segregation in srs2 cells accompanies the formation of a novel DNA damage induced during late meiotic prophase I. The damage may contain long stretches of single-stranded DNAs (ssDNAs), which lead to aggregate formation of a ssDNA binding protein, RPA, and a RecA homolog, Rad51, as well as other recombination proteins inside of the nuclei, but not that of a meiosis-specific Dmc1. The Rad51 aggregate formation in the srs2 mutant depends on the initiation of meiotic recombination and occurs in the absence of chromosome segregation. Importantly, as an early recombination intermediate, we detected a thin bridge of Rad51 between two Rad51 foci in the srs2 mutant, which is rarely seen in wild type. These might be cytological manifestation of the connection of two DSB ends and/or multi-invasion. The DNA damage with Rad51 aggregates in the srs2 mutant is passed through anaphases I and II, suggesting the absence of DNA damage-induced cell cycle arrest after the pachytene stage. We propose that Srs2 helicase resolves early protein-DNA recombination intermediates to suppress the formation of aberrant lethal DNA damage during late prophase I.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In sexually reproducing organisms, meiosis, a specialized form of cell division, produces haploid gametes from diploid germ cells. Following DNA replication, reciprocal recombination takes place to connect the homologous chromosomes and to generate genetic diversity of gametes. With arm cohesion, the connection between the chromosomes, which is cytologically visualized as chiasma, is essential for faithful chromosome segregation during meiosis I by antagonizing the pulling force by spindle microtubules to create tension (Petronczki et al. 2003).
Meiotic recombination is initiated by the generation of DNA double-strand breaks (DSBs) by a meiosis-specific topoisomerase-like protein, Spo11, at recombination hotspots (Keeney et al. 1997). Subsequently, the ends of DSBs are resected to produce 3′-overhanging single-stranded DNAs (ssDNAs). Replication protein A (RPA) binds to the ssDNAs, followed by the loading of Rad51, a homolog of bacterial RecA (Shinohara et al. 1992), with the assistance of auxiliary proteins, such as Rad52, Rad55–Rad57, and Pys3-Csm2-Shu1-Shu2 (aka Shu) (New et al. 1998; Sasanuma et al. 2013b; Shinohara and Ogawa 1998; Sung 1997). Rad51 filaments on ssDNA are active protein machinery for DNA homology search and strand exchange (Ogawa et al. 1993; Sung 1994). Rad51 filament activity is helped by Rad54, which belongs to the SNF2/SWI2 DNA helicase family (Heyer et al. 2006; Shinohara et al. 1997b).
Whereas Rad51 is sufficient for the homology search in recombination during mitosis, meiosis requires a meiosis-specific RecA homolog, Dmc1, for recombination (Bishop et al. 1992). Dmc1 is essential for homology search/strand exchange in inter-homolog recombination during meiosis while Rad51 plays an auxiliary role by assisting Dmc1 assembly (Bishop 1994; Cloud et al. 2012; Shinohara et al. 1997a). Indeed, the Rad51 activity for inter-sister recombination during meiosis is suppressed by the action of a meiosis-specific Rad51 inhibitor, Hed1 (Tsubouchi and Roeder 2004). Like Rad51, Dmc1 forms a nucleo-protein filament on ssDNAs to catalyze the strand invasion of the DNA into its homologous duplex DNA for the formation of an intermediate, D(displacement)-loop (Hong et al. 2001; Sheridan et al. 2008).
In D-loop, DNA synthesis occurs from 3′-end of invading strand as a primer. When the synthesized DNA strand is ejected from the D-loop (Allers and Lichten 2001; Hunter and Kleckner 2001), the ejected synthesized ssDNA is able to anneal with the complementary ssDNA in the other end of the DSB. Annealing induces a second DNA synthesis to complete the recombination by producing noncrossovers. This pathway is called synthesis-dependent strand annealing (SDSA) (Allers and Lichten 2001). On the other hand, when the newly synthesized DNA is stably bound to the D-loop, ongoing DNA synthesis can extend a D-loop with a large displaced ssDNA, which is able to anneal with ssDNA on the opposite DSB ends. Additional processing of the intermediates leads to the formation of double Holliday junction (dHJ) (Schwacha and Kleckner 1994). dHJs are specifically resolved into crossovers. Importantly, meiotic recombination is tightly coupled with chromosome morphogenesis such as the formation of the synaptonemal complex (SC), a meiosis-specific zipper-like chromosome structure, which juxtaposes homologous chromosomes (Cahoon and Hawley 2016).
Srs2 is a 3′-to-5′ SF1 helicase related to bacterial UvrD helicase (Rong et al. 1991). Srs2 protein has some distinct functional domains: DNA helicase domain, Rad51-interaction domain, and also SUMO- and PCNA-binding domains in the C-terminus (Marini and Krejci 2010; Niu and Klein 2017). In mitotic cells, Srs2 plays a critical role in DNA replication, recombination, and DNA repair (Marini and Krejci 2010; Niu and Klein 2017). During S phase, it is involved in post-replicative repair to channel DNA gaps behind the replication fork into error-free gap repair.
Genetic analysis showed positive and negative roles of Srs2 in recombination (Marini and Krejci 2010; Niu and Klein 2017). In mitotic recombination, Srs2 promotes the formation of noncrossovers (thus suppresses crossovers) through the SDSA pathway (Robert et al. 2006; Saponaro et al. 2010). Srs2 antagonizes recombination by removing toxic recombination intermediates. Deletion of SRS2 shows different kinds of genetic interaction with mutants deficient in DNA transactions. The srs2Δ is synthetic lethal with a mutation of the SGS1, encoding a RecQ-type DNA helicase (Gangloff et al. 2000). By forming a complex with Top3 and Rmi1, Sgs1 is known to dissolve the dHJ structure into noncrossovers (Cejka et al. 2010; Wu and Hickson 2003). Moreover, the srs2Δ is synthetic lethal with the deletion of the RAD54 (Klein 2001), suggesting the role of Srs2 in a late stage of the recombination such as the post-invasion step. The lethality is thought to be caused by a fatal defect in the resolution of toxic intermediates in the recombination process. This is supported by the fact that the deletion of RAD51 can suppress the lethality of srs2Δ sgs1Δ and srs2Δ rad54Δ mutants (Gangloff et al. 2000; Schild 1995).
Biochemical studies have demonstrated that purified Srs2 protein can dislodge Rad51 filament on ssDNAs and dramatically inhibits Rad51 joint molecules by direct interaction with Rad51 in vitro (Krejci et al. 2003; Veaute et al. 2003). This biochemical activity of Srs2 supports the idea of Srs2 function as an anti-recombinase. The Rad51-dismantling activity of Srs2 is confirmed by in vivo analysis (Sasanuma et al. 2013a).
Srs2 protein is found only in fungal species including fission yeast (Niu and Klein 2017). On the other hand, in mammals, several proteins with similar Rad51-dismantling activity to Srs2 have been identified. These include RECQL5, FBH1, PARI, and FIGNL1 (Hu et al. 2007; Matsuzaki et al. 2019; Moldovan et al. 2012; Simandlova et al. 2013), which play a role in suppressing crossovers. Like Srs2, PARI, which binds to both RAD51 and PCNA, contains a degenerate UvrD helicase-like domain and dismantles RAD51 filament in a stochastic manner (Moldovan et al. 2012).
Previous genetic studies showed a role of Srs2 in meiosis (Palladino and Klein 1992; Sasanuma et al. 2013b). However, the molecular defects associated with srs2 deletion in meiosis have not been described in detail. Therefore, it remains elusive how Srs2 regulates meiotic recombination. In this study, we analyzed the role of Srs2 helicase in meiotic recombination, particularly looking at dynamics of its interacting partner, Rad51. We found that, in the absence of Srs2, abnormal DNA damage associated with Rad51 aggregation accumulates during late prophase I, after the completion of meiotic recombination. The formation of this DNA damage in the srs2 requires meiotic DSB formation but is independent of chromosome segregation. We also detected thin bridge of Rad51 connecting two adjacent Rad51 foci in early prophase in the absence of Srs2, which is rarely seen in wild-type cells. We propose that Srs2 protects chromosomes in late meiotic prophase I from accumulation of abnormal DNA damage by properly coupling the completion of meiotic recombination with chromosome morphogenesis.
Materials and methods
Yeast strains and medium conditions
All yeast strains used in this article are isogenic derivatives of SK1 and listed in Table S1. pCLB2-SGS1 and RAD54-RFB strains were a gift by Dr. Neil Hunter and Dr. Andreas Hochwagen, respectively. Culture conditions regarding meiosis are described in (Sasanuma et al. 2008).
Antibodies and chemicals
The primary anti-sera were used as following concentrations: anti-Rad51 (guinea pig, 1/500), anti-Dmc1 (rabbit, 1/500), anti-Rad52 (rabbit, 1/300), anti-Rfa2 (rabbit, 1/500), anti-Zip1 (rabbit, 1/500), anti-Red1 (rabbit, 1/500), anti-Hed1 (rabbit, 1/200), and anti-Mei5 (rabbit, 1/500) for cytology. Anti-Hed1 serum from rabbit was prepared for denatured Hed1 protein purified from Escherichia coli. Anti-Nop1 (mouse) is from Encor Biotech (MCA28-F2, 1/500). α-Tubulin is monoclonal antibody of rat that can recognize alpha subunit (AbD Serotec/BioRad, MCA77G). The second antibodies for staining were Alexa-Fluor 488 (goat) and 594 (goat) IgG used at a 1/2000 dilution (Molecular Probes).
Rapamycin (LC-Laboratories, R-5000) and benomyl (methyl 1-[butylcarbamoyl]-2-benzimidazolecarbamate; Sigma-Aldrich, PCode 1002355429) were dissolved in DMSO at a concentration of 1 mM and 30 mg/ml, respectively.
Immunostaining
Chromosome spreads were prepared using the Lipsol method as described previously (Shinohara et al. 2000; Shinohara et al. 2003). Immunostaining was conducted as described (Shinohara et al. 2000). Stained samples were observed using an epifluorescence microscope (BX51; Olympus, Japan) with a × 100 objective (NA1.3). Images were captured by CCD camera (CoolSNAP; Roper, USA) and afterwards processed using IP lab and/or iVision (Sillicon, USA), and Photoshop (Adobe, USA) software tools.
SIM imaging
The structured illumination microscopy was carried out using super-resolution-structured illumination (SR-SIM) microscope (Elyra S.1 [Zeiss], Plan-Apochromat 63x/1.4 NA objective lens, EM-CCD camera [iXon 885; Andor Technology], and ZEN Blue 2010D software [Zeiss]) at Friedrich Miescher Institute for Biomedical Research, Switzerland. Image processing was performed with Zen software (Zeiss, Germany), NIH ImageJ, and Photoshop.
Whole cell staining
Cells were fixed with 1/10 volume of 37% formaldehyde (Wako) and treated with 10 μg/ml Zymolyase 100T (Seikagaku) for 1.5 h. Cells were placed to the poly-L-lysine (Sigma) coated slides and then fixed with cold 100% methanol, cold 100% acetone, and cold 1X PBS. Slides were used for immunostaining.
Western blotting
Western blotting was performed for cell lysates extracted by TCA method. After being harvested and washed twice with 20% TCA, cells were roughly disrupted by Yasui Kikai (Yasui Kikai Co Ltd). Protein precipitation recovered by centrifuge at 1600g for 5 min was suspended in SDS-PAGE sample buffer adjusting to pH8.8 and then boiled at 95 °C for 2 min. The samples were run on a 12.5% SDS polyacrylamide gel and transferred onto a PVDF membrane (Millipore). The proteins were stained with the primary antibody and visualized by the color reaction using the alkaline phosphatase kit (Nacalai).
Southern blotting
Southern blotting analysis was performed with the same procedure as in (Storlazzi et al. 1995). Genomic DNA prepared was digested with both MluI and XhoI (for crossover/noncrossover) and PstI (for meiotic DSB). Probes for Southern blotting were Probe 155 for crossover/noncrossover and Probe 291 for DSB detection as described in (Storlazzi et al. 1995). Image gauge software (Fujifilm Co. Ltd., Japan) was used for quantification for bands of R1, R3, and DSB I.
Statistics
Means ± SD values are shown. Graphs were prepared using Microsoft Excel and GraphPad Prism 7. Datasets were compared using either two-tailed t test or the Mann-Whitney's U test. χ2 test was used for proportion.
Results
SRS2 deletion markedly decreased spore viability
As reported previously (Palladino and Klein 1992; Sasanuma et al. 2013a), the srs2 deletion mutant exhibits reduced spore viability of 36.8%, indicating a critical role of this helicase for meiosis (Fig. 1a). This marked reduction of the spore viability is somehow unexpected given its negative role in recombination.

The srs2 deletion shows defects in meiotic recombination. a Spore viability of wild-type (NKY1303/1543) and the srs2 deletion (HSY310/315) cells was analyzed after 24-h incubation of cells in sporulation medium. After dissecting tetrads, spores were incubated for 3 days for counting. b Representative DAPI-stained image of wild-type (NKY1303/1543) and srs2 (HSY310/315) cells at 12 h. c Meiosis I and II were analyzed by DAPI staining of the wild-type (open circles and open triangle; NKY1303/1543) and srs2 (filled circles and filled triangles; HSY310/315) cells. Circles, meiosis I; triangles, meiosis II. The number of DAPI bodies per nucleus was counted in a minimum of 100 DAPI-positive cells at each time point. d Schematic representation of the HIS4-LEU2 recombination hotspot. e DSB formation (top) and CO/NCO formation (bottom) at the HIS4-LEU2 locus in the wild-type and srs2 strains were verified by Southern blotting. Genomic DNA was digested with PstI for DSB and with XhoI + MluI for CO/NCO. f Kinetic analyses of meiotic DSBs and CO/NCO formation. Parental and DSB bands were quantified and % of DSB (at site I, left) and CO (R1, middle) or NCO (R3, right) was calculated. Graphs show the mean values with variation (n = 2). Wild-type cells (open circles; NKY1303/1543) and srs2 cells (closed circles; HSY310/315). g Immunostaining analysis of a SC protein, Zip1 (red), and an axis protein, Rec8 (green), was carried out in wild-type and mutant cells. Representative images are shown for each strain. Wild type, MSY832/833; srs2, HSY310/315. The bar indicates 2 μm. h Kinetics of SC formation. Zip1 staining in the wild-type and srs2 mutant cells was classified as follows: dot (class I, blue); partial linear (class II, green); full SC (class III, red). Spreads containing Zip1 lines were classified into two classes with less than 5 (class III, pachytene stage) and more than 5 (class II, zygotene stage) Zip1 dots. A minimum of 50 cells were analyzed per time point. i Kinetics of Zip1 polycomplexes in wild-type and srs2 cells. The spreads containing Zip1 polycomplexes were counted at each time point.
We also confirmed the kinetics of meiotic progression in srs2Δ strains by DAPI staining. In the wild-type strain, meiosis I started at 5 h after incubation with sporulation medium (SPM) and was sequentially followed by meiosis II. Finally, ~ 90% of the wild-type cells completed MII at around 8 h (Fig. 1c). In the srs2Δ mutant, the appearance of cells undergoing MI was delayed by ~ 2 h and ~ 75% of cells finished MII at 12 h (Fig. 1c). A similar delay was observed for a srs2 mutant in a different strain background (Palladino and Klein 1992). This indicates a defect during prophase I in the srs2 mutant. In srs2 cells, after sporulation, e.g., 12 h, we often detected fragmented DAPI bodies in a cell/spore (Fig. 1b), indicating a defect in chromosome segregation during the mutant meiosis.
Since Srs2 plays a role in mitotic DNA replication (Niu and Klein 2017), the delay into the MI entry in the srs2 mutant may come from a defect during pre-meiotic S phase. We studied MI progression in the presence of a catalytic-dead spo11 mutation, spo11-Y135F, which abolished meiotic recombination (Bergerat et al. 1997). The spo11 mutant begins to enter MI at 4 h, earlier than wild type (Fig. S1A). Importantly, the spo11-Y135F srs2 double mutant showed similar kinetics of MI progression to the spo11-Y135F single mutant. This indicates that MI delay seen in the srs2 mutant comes from a defect in events in meiotic prophase I, rather than that in pre-meiotic S phase.
The srs2Δ mutant showed a defect in meiotic DSB repair
We analyzed meiotic recombination defects the srs2 deletion mutant in more detail. First, we checked the repair of meiotic DSBs in the mutant by Southern blotting. DSB formation was monitored at the HIS4-LEU2 locus, an artificial meiotic recombination hotspot in chromosome III (Fig. 1d) (Cao et al. 1990). In wild-type cells, DSB frequencies reached its maximum value at 3 h of meiosis (~ 10% of total signals) and then decreased gradually (Fig. 1e, f). The srs2Δ accumulates DSB at higher levels (~ 20%) with more hyper-resection than wild-type and delays the disappearance by ~ 2 h (Fig. 1f), indicating that Srs2 is required for efficient meiotic DSB repair. We also checked the DSB amounts in the rad50S background (Fig. S1B-D), which accumulated DSBs without turnover due to inability to resect DSB ends (Alani et al. 1990). The srs2 rad50S mutant accumulated DSBs at the HIS4-LEU2 to 13.7 ± 2.9% (SD, n = 3) at 8 h, which is similar to the level in the rad50S mutant (15.2 ± 2.1%, P = 0.501, two-tailed t test). These again support the idea that increased steady-state levels of DSBs in the srs2 mutant come from a defect in the processing of DSBs, rather than increased DSB formation per se.
We next checked the formation of two recombinant species, crossover (CO) and noncrossover (NCO) at the same locus. The srs2Δ reduces both CO and NCO to 52% and 64% of the wild-type levels (at 6 h; Fig. 1e, f), respectively. These show that Srs2 is necessary for efficient formation of meiotic recombinants. This is consistent with previous return-to-growth experiment showing delayed recombinant prototroph formation in the srs2Δ mutant (Palladino and Klein 1992). In summary, these results support the idea that Srs2 functions in later steps of meiotic recombination after DSB formation for efficient formation of meiotic recombinants.
During meiotic prophase, homologous recombination is tightly coupled with the formation of the synaptonemal complex (SC), a zipper-like chromosome structure linking two homologous chromosomes. Zip1 is a component of the central region of SC, which serves as a marker for synapsis (Sym et al. 1993). A defect in meiotic recombination results in defective SC formation. We checked the SC formation in the srs2Δ mutant by immunostaining analysis of Zip1 on chromosome spreads as well as a meiosis-specific cohesin component, Rec8 (Fig. 1g). In wild-type cells, ~ 66% of nuclei contained fully elongated Zip1 lines at 4 h and Zip1 signal gradually disappeared from chromosomes. In srs2Δ strains, although Zip1 focus-positive nuclei exceeded 80% at 4 h, the proportion of cells with fully elongated Zip1 was significantly reduced to 13 and 26% at 4 and 5 h, respectively (Fig. 1h). Consistent with this, the proportion of polycomplexes (PCs), which are an aggregate of Zip1 (Sym and Roeder 1995), dramatically increased; ~ 60% of the srs2Δ nuclei contained PCs at 4 h (Fig. 1i). SCs disassembled more slowly in the mutant than wild type (Fig. 1h), consistent with delayed meiotic DSB repair (Fig. 1f).
The srs2Δ mutant accumulated aggregates of Rad51 during late meiotic prophase I
Immunostaining of chromosome spreads can detect recombination proteins such as Rad51 and Dmc1 on the spreads as a foci, which mark sites of ongoing recombination (Bishop 1994). A previous study indicated that the number of Rad51 foci on chromosome spreads in the srs2Δ mutant at 4 h incubation of SPM is slightly reduced compared to those in wild type (Sasanuma et al. 2013b). We performed kinetic analysis of Rad51 and Dmc1 focus formation. In wild-type cells, dotty signals of both Rad51 and Dmc1 peaked at 4 h of meiosis (Figs. 2a and S2A). The appearance of Rad51 foci in cells lacking Srs2 is slightly delayed, and the disappearance of the foci is delayed relative to wild-type cells (Fig. 2b), consistent with delayed DSB repair in the mutant.

The srs2 deletion shows Rad51 aggregates during late prophase I. a Immunostaining analysis of Rad51 (green) and Dmc1 (red) on chromosome spreads in wild-type (NKY1303/1543) and srs2 (HSY310/315) cells. Representative images with or without DAPI (blue) dye at 4, 6, and 8 h for wild-type and the srs2 are shown. The bar indicates 2 μm. b Kinetics of Rad51 focus-positive cells in various yeast strains. A spread with the Rad51 foci is defined as a cell with more than five foci. Spreads containing Rad51 aggregates were also counted. A minimum of 100 cells were analyzed at each time point. Graphs show kinetics of the wild-type cells (top; NKY1303/1543) and srs2 cells (bottom; HSY310/315). Circles and triangles show spreads with Rad51 foci and aggregates, respectively. Graphs show the mean values with standard deviation (SD) as an error bar (n = 3). c Immunostaining analysis of a component of RPA, Rfa2 (green), with Rad51 (red) in wild-type (NKY1303/1543; top) and srs2 (HSY310/315; bottom) cells at 4 h and at 8 h. A dashed square in the merged image is enlarged in the right. The bar indicates 2 μm. d Kinetics of Rfa2 focus-positive cells in wild-type (NKY1303/1543; top, open symbols) and srs2 (HSY310/315; bottom, closed symbols) cells as shown in b. Circles and triangles show spreads with Rfa2 foci and aggregates, respectively. Graphs show the mean values with standard deviation (SD) as an error bar from three independent experiments.
Interestingly, after disappearance of Rad51 foci, we observed reappearance of Rad51 staining with a unique structure after 5 h incubation in the srs2Δ mutant (Figs. 2a and S2A). This staining shows clustering of beads-in-line of Rad51 foci, in which 1–5 bright aggregates of Rad51 are connected to each other through thin threads containing Rad51 as well as a much simple big aggregation of Rad51 (referred to as Rad51 aggregates) (Fig. 2a). The formation of Rad51 aggregates reached a plateau at 6 h, decreases thereafter, but some cells at 10 or 12 h still contained Rad51 aggregates (Fig. 2b), when most of srs2 mutant cells finished MII (Fig. 1c). At 6 and 10 h, 59.3 ± 9.8 and 11.6 ± 6.1 (mean ± standard deviation, SD; n = 3) percent of cells contained aggregates of Rad51, respectively (Fig. 2b, bottom).
This aggregate staining is specific to Rad51, not seen to Dmc1 (Figs. 2a and S2A). Western blots show that Dmc1 and its mediator Mei5 (Hayase et al. 2004) are still present at MI and MII (Fig. S2B). On the other hand, like Rad51 foci, we do see the aggregates of Rad52, a mediator of Rad51 (Shinohara and Ogawa 1998), on chromosomes in the srs2Δ mutant, but not in wild-type cells at late times (Fig. S2C, D). We also found that a Rad51 inhibitor protein, Hed1 (Tsubouchi and Roeder 2006), colocalizes with Rad51 aggregates (Fig. S2E, F). The kinetics of appearance of Rad52 and Hed1 aggregates in the srs2Δ mutant are similar to those of Rad51 (Fig. S2D, F).
In order to know the nature of the late Rad51 foci/aggregates, we also studied the localization of RPA (Rfa2, a middle subunit of RPA) at late prophase I of the srs2Δ mutant. Immunostaining showed that, in addition to early RPA foci (Fig. 2c, d), like Rad51 aggregates, aggregate staining of Rfa2 appeared at late srs2 meiosis, e.g., 6–10 h (Fig. 2d). Closer examination reveals that RPA also exhibits a long-line like staining (Fig. 2c). The kinetics of Rfa2 aggregates in the srs2 mutant is very similar to that of Rad51 (Fig. 2d). Some RPA lines and aggregates co-localized with Rad51 lines and aggregates (Fig. 2c). This suggests the formation of ssDNAs during late prophase I in srs2 cells.
One possibility is that Rad51 aggregates bind to DNA damage in ribosomal DNA (rDNA) region, whose segregation defect is often observed in the recombination-defective mutants (Li et al. 2014). We co-stained Rad51 with anti-Nop1, a marker for an rDNA region (Schimmang et al. 1989). As shown in Fig. S3A, a single Nop1 signal does not co-localize with late Rad51 aggregates as well as early Rad51 foci in the srs2 mutant. This makes it unlikely that late Rad51 aggregates are induced by abnormal recombination in the rDNA repeat.
In order to know the relationship of Rad51 aggregate formation with chromosome segregation, we performed whole cell immunostaining for Rad51 and Dmc1. At early time points, both wild-type and srs2Δ mutant cells showed punctate staining for both Rad51 and Dmc1 with some background diffuse staining in a nucleus (Fig. 3a, b). Rad51-positive nuclei appear at 2 h, peak at 4 h, and then disappear in wild-type cells while the positive nuclei peak at 5 h in the srs2 cells (Fig. 3c). Consistent with results for chromosome spreads (Fig. 2), the srs2Δ cells started to show a big aggregate of Rad51, but not of Dmc1 in nuclei from 5 h and this staining peaked at 8 h (Fig. 3c). Rad51 aggregates in a nucleus often contained thin lines and the number of the aggregate varies up to 2–5 per a nucleus. Importantly, we could also detect Rad51 aggregates in srs2Δ cells with two and four big DAPI bodies in a cell, which correspond with cells finishing MI and MII, respectively (Fig. 3b, d). This suggests that DNA damage associated with Rad51 aggregates does not induce delay or arrest off meiosis. To monitor the DNA damage checkpoint activation at late meiosis of srs2 cells, we analyzed the phosphorylation status of Hop1, which is a substrate of Mec1/ATR and Tel1/ATM (Carballo et al. 2008). We found that the srs2 cells accumulated more phosphorylated Hop1 at 4 h compared to the wild-type cells (Fig. 3e).

Rad51 aggregates in the srs2 deletion pass through Meiosis I and II. a, b Whole cell immunostaining analysis of Rad51 (green) and Dmc1 (red) in wild-type (NKY1303/1543) and srs2 (HSY310/315) cells. Representative images with or without DAPI (blue) at 4 and 8 h in meiosis are shown. In b, cells with Rad51 in prophase I (top), after MI (middle), and after MII (bottom) are shown. The bar indicates 2 μm. c Kinetics of Rad51 focus-positive cells in wild-type and srs2 cells. A focus-positive cell is defined as a cell with more than five foci (closed circles, wild-type, NKY1303/1543; open circles, srs2 cells, HSY310/315). An srs2 mutant cell positive for Rad51 aggregates was measured (open triangles). A minimum of 100 cells were analyzed at each time point. Graphs show the mean values with standard deviation (SD) as an error bar (n = 3). d Kinetics of srs2 cells containing Rad51 aggregates prior to MI (blue; one DAPI body in a cell), after MI (green; two DAPI bodies in a cell), and MII (red; three or more DAPI bodies in a cell) are shown. Mean values with SD from three independent experiments are shown. e Western blotting analysis of Hop1 during meiosis. Cell lysates at different time points in meiosis in wild-type (NKY1303/1543) and srs2 (HSY310/315) cells were probed with anti-Hop 1 (top) and anti-tubulin (bottom) antibodies. Phosphorylated Hop1 (shown as “Hop1-P” on the right) shows slower mobility relative to unphosphorylated Hop1. Relative signal intensity of Hop1 (open and closed circles) and phosphorylated Hop1 (Hop1-P, open and closed triangles) was plotted at each time point. The signal was normalized using values for tubulin as a control. Mean values with SD from three independent experiments are shown.
Rad51 aggregate formation in the srs2 mutant depends on Spo11
To understand the nature of Rad51 aggregates in the srs2 mutant, we looked for the genetic requirement of aggregate formation in the mutant. Rad51 aggregate formation in srs2Δ cells is dependent on DSB formation, since a catalytic-dead spo11 mutation, spo11-Y135F (Bergerat et al. 1997), almost abolishes both early foci and aggregates of Rad51 in the srs2Δ mutant (Fig. S3B). It is likely that early DSB-related events in the srs2Δ cells may trigger Rad51 aggregates during late meiosis.
In mitosis, the sgs1 mutation is synthetic lethal with the srs2 mutation, indicating a redundant role for these two helicases (Gangloff et al. 2000). Sgs1 helicase, together with Top3 and Rmi1, is known to prevent the formation of the untangled chromosomes. The absence of Sgs1 results in abnormal meiosis divisions due to accumulation of resolved recombination products involving multi-chromatids (Jessop and Lichten 2008; Jessop et al. 2006; Oh et al. 2007; Oh et al. 2008; Tang et al. 2015). In mammals, the lack of Sgs1 ortholog, BLM helicase, induces anaphase bridges, which are associated with DNA damage generated during S phase (Biebricher et al. 2013; Chan et al. 2007). We examined the late Rad51 aggregate formation in a meiotic-null allele of sgs1, sgs1-mn (CLB2p-SGS1) (Oh et al. 2007). The sgs1-mn forms early Rad51 foci with delayed disappearance in prophase of MI, but, unlike the srs2, the mutant does not form late Rad51 aggregates (Fig. S3C, D), suggesting that unresolved recombination intermediates formed in the absence of the Sgs1 do not trigger Rad51 aggregate formation.
The effect of Rad54 depletion on the kinetics of Rad51 aggregates in the srs2 mutant
Above results showed that some Rad51 aggregates turned over during meiosis. We deduced that we could expect to stall Rad51 dynamics by blocking late stage of the recombination reaction. We focused on Rad54, which functions after Rad51 assembly (Shinohara et al. 1997b), and tried to examine the effect of RAD54 deletion on Rad51 aggregates. However, it is reported that the rad54 deletion is synthetically lethal with the srs2 deletion (Klein 2001; Palladino and Klein 1992; Schild 1995). To circumvent this, we used Rad54 anchor-away system, which specifically depletes nuclear Rad54 fused with RFB by the addition of the drug rapamycin (Haruki et al. 2008; Subramanian et al. 2016). The srs2 RAD54-RFB cells grow normally in the absence of rapamycin while the srs2 RAD54-RFB cells grow poorly on the plate containing the drug, confirming synthetic lethality of the rad54 and srs2 (Fig. 4a). In order to know the functional relationship between Rad54 and Srs2 during late meiosis, first, we added rapamycin at 4 h to RAD54-RFB and srs2 RAD54-RFB cells and analyzed both spore viability and Rad51 foci. The srs2 RAD54-RFB decreased spore viability to 64% in the absence of the drug. As reported (Shinohara et al. 1997b), RAD54-RFB cells decreased spore viability to 48% in the presence of the drug. Addition of the rapamycin also reduced the spore viability of the srs2 RAD54-RFB to 24%, indicating the additive effect of the srs2 deletion and RAD54 depletion on spore viability (Fig. 4b). RAD54 depletion does not affect delayed MI progression in the srs2 deletion (Fig. 4c). As in wild-type cells, RAD54-RFB cells showed normal assembly and disassembly of Rad51 foci in the absence of the drug (Rapa−; Fig. 4d, e). However, we found that, from 5 h, 1 h after the addition of the drug (Rapa+), a new class of Rad51 staining appeared. This class contains 5–10 brighter foci of Rad51, hereafter called “Rad51 clump,” which is distinct from the typical Rad51 foci and aggregates (Fig. 4d). This Rad51 clump peaks at 6 h and then disappears (Fig. 4e), indicating the role of Rad54 in the post-Rad51 assembly stage. In the absence of rapamycin, the srs2 RAD54-RFB mutant has similar kinetics for both Rad51 focus and aggregate formation as the srs2Δ mutant. By the addition of the rapamycin at 4 h, like RAD54-RFB cells, the srs2 RAD54-RFB mutant formed a Rad51 clump after 5 h and showed the similar kinetics to that in the RAD54-RFB (Rapa+). In addition, Rad51 aggregates appeared at 5 h and accumulated during further incubation. Rad51 aggregate kinetics in the absence of RAD54 (Rapa+) is delayed relative to its presence (Fig. 4e). This result indicates that Rad51 clumps formed without Rad54 are independent of Srs2. And also, Rad51 aggregate kinetics in the srs2 cell is independent of Rad54 function.

Rad54 depletion induces abnormal Rad51 assembly. a Mitotic plate assay to confirm synthetic lethality of srs2 RAD54 anchor on YPAD plates containing 1 μM rapamycin. Tenfold serial dilutions of RAD54-RFB (H7790/7791) and RAD54-RFB srs2 (HYS71/82) cells were spotted. b Spore viability of RAD54-RFB (H7790/7791) and RAD54-RFB srs2 (HYS71/82) cells in the absence (−) and the presence (+) of rapamycin. The number of tetrads dissected is as follows: wild-type, n = 432; RAD54-RFB (−), n = 408; srs2 RAD54-RFB (−), n = 424; RAD54-RFB (+), n = 384; srs2RAD54-RFB (+), n = 388. P values were calculated using the chi-square test. c Kinetics of MI entry in RAD54-RFB (H7790/7791, green circles) and RAD54-RFB srs2 (HYS71/82, red circles) cells in the absence (−, open symbols) and the presence (+, closed symbols) of rapamycin. Rapamycin was added at 4 h at a concentration of 1 μM. Graphs show mean values with SD from three independent experiments. d Immunostaining analysis of Rad51 (green) on chromosome spreads in RAD54-RFB (H7790/7791) and RAD54-RFB srs2 (HYS71/82) cells in the absence and the presence of rapamycin. Rapamycin (1 μM) was added at 4 h in meiosis. Representative images of Rad51 staining with or without DAPI (blue) after the addition of rapamycin are shown. The bar indicates 2 μm. e Kinetics of Rad51 foci-positive cells in RAD54-RFB (top graph; H7790/7791) and RAD54-RFB srs2 (bottom graph; HYS71/82) cells in the absence (−) or the presence (+) of rapamycin. Rad51 foci (circles), Rad51 clumps (square), and Rad51 aggregates (triangles); green or blue open symbols (without rapamycin) and green or red closed symbols (addition of the rapamycin at 4 h). Graphs show mean values with SD (n = 3).
Rad51 aggregate in the srs2 mutant is dependent of pachytene exit but is independent of the onset of meiosis I
Rad51 aggregates in the srs2Δ mutant are formed at late times during prophase I. To know the relationship between the focus formation and the progression of meiosis, we first analyzed the Rad51 aggregate formation in the srs2Δ with the ndt80 mutation, which induces pachytene arrest due to the inability to express genes necessary for exit from mid-pachytene stage (Xu et al. 1995). Staining of chromosome spreads in ndt80 cells reveal accumulation of cells with Rad51 foci (Figs. 5a and S4A), which is induced by persistent DSB formation during pachytene arrest by the ndt80 (Carballo et al. 2013). At later times, the ndt80 mutant showed the reduced number of Rad51 foci compared to early time points (Fig. S4A). However, Rad51 foci seemed to turn over less efficiently in the ndt80 mutant (Fig. S4A, B). Little Rad51 aggregate formation was seen in srs2 ndt80 cells arrested at mid-pachytene both on chromosome spreads and in whole cells (Figs. 5a and S4B). This indicates that the formation of Rad51 aggregates in the srs2 mutant depends on Ndt80, thus after the exit of mid-pachytene stage.

Rad51 bridges are formed between the foci. a Immunostaining analysis of Rad51 on spreads of ndt80 (HSY596/597) and srs2 ndt80 (LPY058/059) cells at 4 and 6 h. The bar indicates 2 μm. b Rad51 bridges in srs2 (HSY310/315) cells at 4 h. Rad51 tail and bridge are shown in arrowheads and arrows, respectively. The bar indicates 1 μm. c A spread with Rad51-tail or Rad51-bridge is classified into three classes; Rad51 focus with tail (left), Rad51 bridge between two foci (middle), and Rad51 bridge among three or more foci (right). On each spread, the number of each class per a spread was counted, and then a count of the spreads in each class is shown. Forty-two spreads of wild-type (NKY1303/1543) and srs2 (HSY310/315) cells at 4 h were analyzed and counted. Graphs show the mean values with SD from three independent experiments. d The length of the Rad51 bridge between two Rad51 foci was measured and plotted. Three horizontal lines from the top indicate the 75, 50 (median), and 25 percentiles, respectively. P = 0.39; Mann-Whitney's U test. e SR-SIM microscopic observation of Rad51 (green) in wild-type (NKY1303/1543) and srs2 (HSY310/315) cells. Representative images of DAPI (blue; left) dye and Rad51 (green, middle) are shown. The bar indicates 2 μm
When the kinetics of Rad51 aggregate formation in the srs2 mutant was compared to kinetics of meiosis I entry, Rad51 aggregate in the srs2 mutant appear 1 h earlier than the entry into meiosis I (Fig. 1c). To confirm this, we blocked the microtubule dynamics by treating cells with a benomyl, a microtubule depolymerization drug. As shown previously (Hochwagen et al. 2005), the addition of benomyl to yeast meiosis at 4 h prior to the formation of the aggregates largely suppressed the entry of meiosis I, thus the onset of anaphase I, in both wild-type and srs2 cells (Fig. 6b). The treatment with benomyl does not affect Rad51 focus kinetics in both wild-type and srs2 mutant (Fig. 6a, c). Moreover, the srs2 cells formed Rad51 aggregates in the presence of benomyl with similar kinetics in its absence (mock treatment with DMSO) (Fig. 6c). This indicates that Rad51 aggregate formation in the srs2 mutant occurs in the absence of microtubule dynamics, thus chromosome segregation, suggesting that Rad51 aggregate formation in srs2 mutants is associated with an event during late prophase I, not with events during the metaphase I or anaphase I.

Rad51 aggregate forms in the absence of chromosome segregation. a Immunostaining analysis of Rad51 (green) in wild-type (NKY1303/1543) and srs2 (HSY310/315) cells in the presence of benomyl. The benomyl was added at 4 h at a concentration of 120 μg/ml. The bar indicates 2 μm. b Kinetics of MI entry in wild-type (green) and srs2 (red) cells in the absence (open symbols) or the presence (closed symbols) of benomyl. Graphs show mean values with SD from three independent experiments. c Kinetics of Rad51 foci and Rad51 aggregates in wild-type (top, green) and srs2 (bottom, red) cells in the absence (open) or the presence (closed) of benomyl. Circles, Rad51 foci; triangles, Rad51 aggregates. Graphs show mean values with SD (n = 3). d Immunostaining analysis of Rad51 and Red1 in CDC20-mn (YFY74/77) and srs2 CDC20-mn (YFY80/83) cells. The chromosome spreads at 5 and 7 h were immunostained against Rad51 (green) as well as chromosome protein Red1 (red). The bar indicates 2 μm. e Kinetics of Rad51 aggregate-positive cells in Red1-positive and Red1-negative spreads. Rad51-focus and Rad51-aggregate positive spreads were classified into Red1-negative (open bars) and Red1-positive (closed bars) at each time point. At each time point, more than 50 spreads were counted. The result is one of representative from two independent experiments.
Rad51 aggregates in the srs2 mutant appear when SC is disassembled
In order to confirm that Rad51 aggregate formation in the srs2 is independent of the onset of anaphase I, we used a meiosis-specific null mutant of the CDC20, which encodes an activator of anaphase-promoting complex/cyclosome (APC/C), the cdc20-mn (CLB2p-CDC20). As reported previously (Lee and Amon 2003), the cdc20-mn shows an arrest at the onset of anaphase I. In the cdc20-mn, Rad51 foci appear and disappear like in wild-type control. As expected from the results with benomyl, the Rad51 aggregate formation occurs after the disappearance of Rad51 foci in the srs2 cdc20-mn double mutant as in the srs2 mutant (Fig. 6d, e). This supports the notion that Rad51 aggregate formation in srs2 mutant is independent of the entry into anaphase I, thus chromosome segregation.
The relationship between the formation of Rad51 aggregates and late meiotic prophase I such as SC disassembly was compared by immunostaining of Rad51 with Zip1 (Fig. S5A). After the pachytene exit, the central region of SCs is dismantled as seen in the loss of Zip1 line signals from chromosomes (Sym et al. 1993). The srs2 cells containing Rad51 aggregates were almost negative for Zip1 lines (Fig. S5A).
We also performed Red1 staining, which is a component of the chromosome axis (Smith and Roeder 1997). Most cells with Rad51 foci at 3–5 h are almost positive for Red1 staining in both cdc20-mn and srs2 cdc20-mn cells (Fig. 6d, e). In contrast, srs2 cdc20-mn cells with Rad51 aggregates were negative for Red1 signal. These results indicate that Rad51 aggregate formation in the srs2 occurs after or during disassembly of Red1 axis. This is confirmed in the background of wild type too (Fig. S5B, C).
We confirmed this by staining of Rec8, a kleisin subunit of cohesin (Klein et al. 1999). At late time points such as 6 h, Rec8 showed dotty staining compared to 5 h (Challa et al. 2019), when most of Rec8 show line staining. Rec8 line positive spreads contained Rad51 foci (Fig. S5D, E). In srs2 cells with or without cdc20-mn, Rad51 aggregates are predominantly seen in cells with Rec8 dots (Fig. S5D, E).
The srs2 mutant accumulated bridge staining of Rad51 between two recombination foci during early prophase I
During our staining analysis, we noticed that the srs2 cells show very unique thin line staining of Rad51 during early prophase (see 4 h in Fig. 5b). The thin Rad51 line in srs2 cells is connected from one Rad51 focus to the other focus/foci, which we refer to as “Rad51 bridge.” At least one clear Rad51 bridge between two Rad51 foci was observed at ~ 40% frequency of srs2 spreads at 4 h (middle graph of Fig. 5c). Few Rad51 bridges were seen in wild type. We also found the Rad51 bridge staining among more than three Rad51 foci in srs2 cells, but not in wild type (right graph of Fig. 5c). Careful examination of Rad51 foci in wild-type spreads often detected a Rad51 focus with “single tail (or whisker)” (left graph of Fig. 5c). Most single Rad51 foci with the tail showed one tail. When measured the length of the bridge between two foci, we found both wild-type and the srs2 cells show similar distribution of the lengths (Fig. 5d). These results indicate that Srs2 suppresses the formation of Rad51 bridges. Indeed, the srs2 cells increased the frequency of the Rad51 bridges and more connections among more than two foci relative to wild-type cells (Fig. 5c).
We then used super-resolution microscopy to analyze Rad51 localization on meiotic chromosomes at high resolution. A structured illumination microscope (SIM) was used to determine Rad51 localization in wild-type and srs2 cells at 4 h (Fig. 5e). As shown above, in the srs2 mutant, we detected both Rad51 bridges and tails more than in wild-type. The wild-type and the srs2 mutant at 4 h show a Rad51 focus with tail/bridge per a Rad51 focus at a frequency of 15.4 ± 4.2% (n = 18) and 54.3 ± 8.5% (n = 20), respectively.
The average length of the bridge is ~ 0.4 μm (Fig. 5d). If the bridge is postulated to consist of a single Rad51 filament on the ssDNA, which is extended twofold relative to the B-form DNA (Ogawa et al. 1993), we can calculate the bridge contains ~ 600 nt (400 nm/2 × 3.3/10.5). This is a reasonable estimate of ssDNA length at a single DSB site with ~ 900 nt (Mimitou et al. 2017; Zakharyevich et al. 2010). The Rad51 bridges described here might be similar to the staining of “ultra-fine bridge” seen in anaphase of damage mammalian cells (Chan and Hickson 2011) (see “Discussion”). The formation of Rad51 thin bridges in early prophase I of the srs2 cells suggests entanglement of recombination intermediates.
The Rad51 bridge was also observed in cells arrested by ndt80 deletion (Fig. S4C). As in wild-type background, srs2 cells in ndt80 background accumulate more bridges than ndt80 cells, indicating that Rad51 bridges appear prior to mid-pachytene stage.
Discussion
In mitotic recombination, Srs2 promotes NCO formation through the SDSA pathway and indirectly decreases CO formation (Niu and Klein 2017). On the other hand, in this study, the srs2Δ mutant shows decreased levels of CO and NCO during meiosis relative to the wild type, indicating a positive role of Srs2 in meiotic recombination (pro-recombination role). This weak defect in the recombination is consistent with delayed DSB repair (delayed disassembly of Rad51 foci) as well as defective SC formation in the mutant. Given that Rad51-Dmc1 balance determines a pathway choice to inter-homolog or inter-sister recombination (Hong et al. 2013; Lao et al. 2013; Schwacha and Kleckner 1997), we speculate that the srs2 deletion might stabilize Rad51 assembly, but not Dmc1 assembly (Crickard et al. 2018; Sasanuma et al. 2013a). This imbalance of Rad51-Dmc1 function in the mutant may lead to the reduction of inter-homolog recombination. On the other hand, studies in an accompanying paper (Hunt et al. accompanying paper) showed normal CO and NCO formation at a different hotspot in the srs2 mutant, suggesting a locus-specific role of Srs2 in meiotic recombination. Further analysis is needed to elucidate the role of Srs2 helicase in CO/NCO or inter-homolog/inter-sister pathway choice during meiosis.
The “weak” defect in the meiotic recombination cannot explain reduced spore viability of the mutant, since the mutants with 50% reduction of CO show high spore viability, e.g., spo11, xrs2, msh4/5 hypomorphic mutants (Martini et al. 2006; Nishant et al. 2010; Shima et al. 2005). Consistent with low spore viability of the mutant, we and others detected abnormal chromosome segregation in srs2 meiosis, suggesting the presence of DNA abnormality in the mutant (this study and Hunt et al. accompanying paper). In this study, we described “unusual” DNA damage formed in the absence of Srs2 helicase during meiosis. This damage is marked with the association of the recombination protein, Rad51, in a large quantity, which we refer to as “Rad51 aggregate.” The Rad51 aggregates are not protein aggregates since they contain another recombination protein, Rad52, as well as RPA, but not meiosis-specific recombination proteins such as Dmc1. The presence of RPA strongly suggests the presence of ssDNAs. Indeed, thin line-like staining of Rad51 and RPA emanating from the aggregate are often observed.
The formation of Rad51 aggregates in srs2Δ mutant requires Spo11 catalytic activity, thus DSB formation. On the other hand, kinetic analysis revealed that Rad51 aggregates in srs2Δ mutant appear in late prophase I after the disappearance of Spo11-dependent Rad51 foci associated with meiotic recombination. Rad51 aggregates appear just after the disappearance of “normal” Rad51 foci. This suggests that the formation of Rad51 aggregates occur after the completion of DSB repair such as Rad51-mediated strand invasion. Consistent with this, the ndt80 mutation, which induces an arrest at mid-pachytene stage, blocks the aggregate formation in the srs2Δ mutant. The ndt80 mutant accumulates dHJ as a product of completion of Rad51-dependent strand invasion (Allers and Lichten 2001) and also shows persistent formation of Spo11-dependent meiotic DSBs (Carballo et al. 2013). Therefore, persistent DSBs and dHJs are unlikely to be directly linked with Rad51 aggregate formation.
Mutant analysis shows the formation of Rad51 aggregates in the srs2Δ requires pachytene exit but occurs prior to the transition of metaphase I to anaphase I, chromosome segregation. Moreover, Rad51 aggregate formation occurs even when chromosome segregation was inhibited by treatment with a microtubule polymerization inhibitor or the CDC20 depletion, which delays and blocks the onset of anaphase I, respectively. These indicate that the aggregate formation is induced around the disassembly of meiotic chromosome structure, e.g., diplotene or diakinesis.
One possibility to explain Rad51 aggregate formation in the srs2 mutant is that, after the exit of Ndt80 execution point, there might be unrepaired damage, which could be repaired by Rad51-dependent pathway (but not Dmc1-dependent pathway) during late prophase I. The srs2 mutant might be specifically defective in this DSB repair after the pachytene exit. In this pathway, Srs2 may be essential for Rad51 removal, which may prevent the accumulation of unrepaired ssDNAs. However, this is unlikely since even DSB ends formed during pachytene are bound and could be repaired by Dmc1 as well as Rad51. However, the Rad51 aggregates in the srs2 mutant do not contain Dmc1 even when Dmc1 protein is present in a cell.
Alternatively, Rad51 aggregates and/or its associated DNA damage are formed in two-step process. First, DSB repair in the absence of Srs2 may result in the formation of aberrant recombination products/intermediates such as entangled duplexes DNAs (see Fig. 7b). Second, this aberrant product/intermediate might be converted into DNA damage with Rad51 aggregates in late prophase I. Consistent with this two-step model, we found a novel structure called Rad51 bridge (or tail/whisker), thin lines of Rad51 which connect Rad51 foci. This bridge is seen at early prophase I of the srs2 mutant more frequently than in wild type.

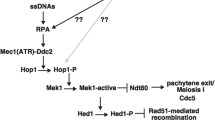
A schematic model of Rad51-bridge and Rad51-aggregate formation in wild-type and srs2 cells. a A model of Rad51 focus comprised of the Rad51 filament. Rad51 filament is accommodated into a three-dimensional structure. b A recombination pathway with Rad51 focus and filaments is described. In left, the second-end capture by a displaced strand from D-loop, which is mediated by the Rad51 filament may trigger the disassembly of Rad51 filament (focus) on the second end, which is promoted by Srs2. Srs2 also prevents the formation of a multi-invasion intermediate. These abnormal recombination intermediates are processed into a final product, which may contain a pathological damage that may lead to the formation of a long ssDNAs with Rad51 aggregates during late prophase I
The presence of Rad51 bridge and Rad51 tail/whisker from Rad51 focus suggests that Rad51 focus is not a simple Rad51 filament, rather may contain a three-dimensional configuration of a Rad51 filament (Fig. 7a). Rad51 bridge line staining is reminiscent of anaphase bridge or ultra-fine bridge of chromosomes in mammalian cells (Chan et al. 2007). The formation of anaphase bridges in mammalian cells is a two-step process. Although these bridges are formed during M phase with onset of anaphase, the initiation event leading to the bridge formation occurs during S phase. These bridges are induced by the treatment of the cell with DNA replication inhibitor(s) or in the absence of DNA repair protein such as BLM helicase. The bridges are decorated with repair proteins such as BLM and RPA, but not Rad51.
Rad51 aggregate formation in srs2 meiosis is clearly different from the anaphase bridge in the following two aspects. First, the formation does not require chromosome segregation. Second, the initiation event should be Spo11-dependent DSB formation in early prophase I (meiotic G2 phase). If the two-step model as described above is true, the conversion of the aberrant product/intermediate into the aggregate in srs2Δ mutant should occur after mid-pachytene exit. Given drastic chromosome morphogenesis such as chromosome compaction and disassembly of the meiosis-specific chromosome structure, SC, occur during late meiotic prophase I (Challa et al. 2019), these events might induce the aberrant DNA damage with Rad51 aggregates.
Given that Rad51 bridge in the srs2 mutant is formed between Rad51 foci, Srs2 might play a role to process this kind of Rad51-associated DNA entanglement between the two DSB sites (Fig. 7b). One likely intermediate is multiple invasions (Piazza et al. 2017), which are formed by Rad51-mediated strand invasion into multiple loci. Therefore, Srs2 might play a role in resolution of multiple invasion by controlling Rad51 filament dynamics using its Rad51-dismantling activity.
Bishop and his colleagues show that a pair of Rad51 foci during early meiotic pro-phase I are formed in the two ends of a single DSB site (Brown et al. 2015). Thus, it is likely that the Rad51 bridge we observed is formed between a pair of Rad51 foci on the two DSB ends. If so, one likely possibility is that the bridge is a ssDNA between two DSB ends. One way to connect two DSB ends is to bridge by the annealing of ejected ssDNA from the D-loop after the DNA synthesis (Fig. 7b). Since the bridge is mainly seen in the absence of Srs2, we propose that Rad51-dismantling activity of Srs2 promotes the removal of Rad51 from the rejected ssDNA. Moreover, it is likely that Srs2 also remove Rad51 in the other end of the DSB during the second-end capture. This idea could explain the formation of the bridge between two Rad51 foci in the srs2 but rare in wild type. In wild-type cells, Srs2 seems to remove Rad51 assembly from the intermediates for the second-end capture. Importantly, genetic analysis of mitotic recombination in the srs2 mutant suggests the role of Srs2 to facilitate the annealing of the newly synthesized strand to second resected ends by removing Rad51 from the second end (Ira et al. 2003; Liu et al. 2017; Mitchel et al. 2013).
We still cannot figure out the molecular nature of recombination products/intermediates formed in the absence of Srs2, which trigger the formation of the Rad51 aggregate. 1D gel analysis has shown that there is a slightly reduced level of abnormal recombination intermediate such as multiple chromatid joint molecules in an srs2 mutant relative to wild-type cells (Hunt et al., accompanying paper). Thus, multiple chromatid joint molecules are unlikely. Consistent with this, the sgs1 mutant, which accumulates the intermediates, does not form Rad51 aggregate. Rather, there might be a topological entanglement of DNA strands after the completion of the meiotic recombination (Fig. 7b). This intermediate seems to be related to a lethal recombination intermediate formed in the srs2 mutant during mitosis (Elango et al. 2017).
In either scenario, our analysis reveals a novel pathway that protects meiotic cells in late prophase I from the formation of aberrant DNA damage induced by Spo11. This repair pathway heavily depends on Srs2 function, which is almost essential for meiosis. As described above, we would like to point out that Rad51 aggregates in the srs2 mutant are related to lethal recombination intermediates in srs2 mitotic cells, which is postulated to form through two-step model (Elango et al. 2017). Rad51 aggregate-associated DNA damage seems unrepaired during meiosis. During late prophase I, there should be sister chromatid or other recombination partners to repair the damage, suggesting the presence of the inhibition on recombination-mediated repair of the damage. This inhibition might be due to the presence of Rad51 inhibitor Hed1, which clearly suppresses Rad51-mediated DNA repair (Tsubouchi and Roeder 2006).
In the absence of Srs2, DNA damage with Rad51 aggregates is formed and passed into MI and MII with activation of DNA damage checkpoint, which leads more catastrophic events such as chromosome fragmentation with DSB formation in spores. This may explain the reduction of spore viability of the srs2 diploid despite reasonable levels of meiotic recombination. The absence of DNA damage-induced delay in late meiosis I in the srs2 cells is quite different from cell cycle delay induced by the recombination (pachytene) checkpoint during early prophase I (MacQueen and Hochwagen 2011; Tsubouchi et al. 2018). In the recombination checkpoint, DSBs and associated ssDNA activate sensor kinases Tel1(ATM) and Mec1(ATR), respectively (Clerici et al. 2004; Usui et al. 2001). Activated Mec1 and Tel1 induce the activation of a meiosis-specific kinase, Mek1, the Rad53 homolog, by phosphorylating its partner protein Hop1 (Carballo et al. 2008; Ontoso et al. 2013). High Mek1 activity downregulates Ndt80 activity, thus blocking the exit of mid-pachytene stage (Hollingsworth and Gaglione 2019). During meiosis, the activation of mitotic DNA damage downstream kinases, Rad53 and Chk1, is blocked through an unknown mechanism (Clerici et al. 2004). At late times in the srs2 mutant, we did not see prolonged phosphorylation of Hop1, thus little activation of Mec1 (and Tel1). This strongly suggests that DNA damage with Rad51 aggregates in the srs2 is masked for the checkpoint activation or there is no such surveillance mechanism in late G2 phase of meiotic cells. Alternatively, although not exclusive with the above, Srs2 may function in the activation of the checkpoint during this phase.
Upon Rad54 depletion after the assembly of Rad51 on the ssDNA during meiosis, we found a novel staining of Rad51 called Rad51 clump, which is different from typical Rad51 foci in wild type and aggregates in the srs2 cells. The presence of Rad51 clumps supports the idea of a role of Rad54 after the assembly of Rad51 filaments. Moreover, Rad54 may function in the Rad51-mediated repair of DSBs in late prophase I for inter-sister recombination as suggested (Niu et al. 2009). On the other hand, previous cytological analysis of the rad54 deletion does not show the formation of Rad51 clump in late meiosis I (Shinohara et al. 2000; Shinohara et al. 1997b). In current study, we used the anchor-away for Rad54 depletion, which may leave residual Rad54 protein in a nucleus. This residual Rad54 may affect the dynamics of Rad51 in a late stage in the meiotic recombination. A more detailed analysis to understand the role of Rad54 in meiotic inter-sister recombination.
Note: in the accompanying paper, Goldman and his colleagues described the formation of Rad51 aggregates during srs2 meiosis.
References
Alani E, Padmore R, Kleckner N (1990) Analysis of wild-type and rad50 mutants of yeast suggests an intimate relationship between meiotic chromosome synapsis and recombination. Cell 61:419–436
Allers T, Lichten M (2001) Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 106:47–57
Bergerat A, de Massy B, Gadelle D, Varoutas PC, Nicolas A, Forterre P (1997) An atypical topoisomerase II from archaea with implications for meiotic recombination. Nature 386:414–417. https://doi.org/10.1038/386414a0
Biebricher A, Hirano S, Enzlin JH, Wiechens N, Streicher WW, Huttner D, Wang LHC, Nigg EA, Owen-Hughes T, Liu Y, Peterman E, Wuite GJL, Hickson ID (2013) PICH: a DNA translocase specially adapted for processing anaphase bridge DNA. Mol Cell 51:691–701. https://doi.org/10.1016/j.molcel.2013.07.016
Bishop DK (1994) RecA homologs Dmc1 and Rad51 interact to form multiple nuclear complexes prior to meiotic chromosome synapsis. Cell 79:1081–1092
Bishop DK, Park D, Xu L, Kleckner N (1992) DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 69:439–456
Brown MS, Grubb J, Zhang A, Rust MJ, Bishop DK (2015) Small Rad51 and Dmc1 complexes often co-occupy both ends of a meiotic DNA double strand break. PLoS Genet 11:e1005653. https://doi.org/10.1371/journal.pgen.1005653
Cahoon CK, Hawley RS (2016) Regulating the construction and demolition of the synaptonemal complex. Nat Struct Mol Biol 23:369–377. https://doi.org/10.1038/nsmb.3208
Cao L, Alani E, Kleckner N (1990) A pathway for generation and processing of double-strand breaks during meiotic recombination in S. cerevisiae. Cell 61:1089–1101
Carballo JA, Johnson AL, Sedgwick SG, Cha RS (2008) Phosphorylation of the axial element protein Hop1 by Mec1/Tel1 ensures meiotic interhomolog recombination. Cell 132:758–770. https://doi.org/10.1016/j.cell.2008.01.035
Carballo JA, Panizza S, Serrentino ME, Johnson AL, Geymonat M, Borde V, Klein F, Cha RS (2013) Budding yeast ATM/ATR control meiotic double-strand break (DSB) levels by down-regulating Rec114, an essential component of the DSB-machinery. PLoS Genet 9:e1003545. https://doi.org/10.1371/journal.pgen.1003545
Cejka P, Plank JL, Bachrati CZ, Hickson ID, Kowalczykowski SC (2010) Rmi1 stimulates decatenation of double Holliday junctions during dissolution by Sgs1-Top3. Nat Struct Mol Biol 17:1377–1382. https://doi.org/10.1038/nsmb.1919
Challa K, Fajish VG, Shinohara M, Klein F, Gasser SM, Shinohara A (2019) Meiosis-specific prophase-like pathway controls cleavage-independent release of cohesin by Wapl phosphorylation. PLoS Genet 15:e1007851. https://doi.org/10.1371/journal.pgen.1007851
Chan KL, Hickson ID (2011) New insights into the formation and resolution of ultra-fine anaphase bridges. Semin Cell Dev Biol 22:906–912. https://doi.org/10.1016/j.semcdb.2011.07.001
Chan KL, North PS, Hickson ID (2007) BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J 26:3397–3409. https://doi.org/10.1038/sj.emboj.7601777
Clerici M, Baldo V, Mantiero D, Lottersberger F, Lucchini G, Longhese MP (2004) A Tel1/MRX-dependent checkpoint inhibits the metaphase-to-anaphase transition after UV irradiation in the absence of Mec1. Mol Cell Biol 24:10126–10144. https://doi.org/10.1128/mcb.24.23.10126-10144.2004
Cloud V, Chan YL, Grubb J, Budke B, Bishop DK (2012) Rad51 is an accessory factor for Dmc1-mediated joint molecule formation during meiosis. Science 337:1222–1225. https://doi.org/10.1126/science.1219379
Crickard JB, Kaniecki K, Kwon Y, Sung P, Greene EC (2018) Meiosis-specific recombinase Dmc1 is a potent inhibitor of the Srs2 antirecombinase. Proc Natl Acad Sci U S A 115:E10041–e10048. https://doi.org/10.1073/pnas.1810457115
Elango R, Sheng Z, Jackson J, DeCata J, Ibrahim Y, Pham NT, Liang DH, Sakofsky CJ, Vindigni A, Lobachev KS, Ira G, Malkova A (2017) Break-induced replication promotes formation of lethal joint molecules dissolved by Srs2. Nat Commun 8:1790. https://doi.org/10.1038/s41467-017-01987-2
Gangloff S, Soustelle C, Fabre F (2000) Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat Genet 25:192–194
Haruki H, Nishikawa J, Laemmli UK (2008) The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell 31:925–932. https://doi.org/10.1016/j.molcel.2008.07.020
Hayase A, Takagi M, Miyazaki T, Oshiumi H, Shinohara M, Shinohara A (2004) A protein complex containing Mei5 and Sae3 promotes the assembly of the meiosis-specific RecA homolog Dmc1. Cell 119:927–940
Heyer WD, Li X, Rolfsmeier M, Zhang XP (2006) Rad54: the Swiss Army knife of homologous recombination? Nucleic Acids Res 34:4115–4125. https://doi.org/10.1093/nar/gkl481
Hochwagen A, Wrobel G, Cartron M, Demougin P, Niederhauser-Wiederkehr C, Boselli MG, Primig M, Amon A (2005) Novel response to microtubule perturbation in meiosis. Mol Cell Biol 25:4767–4781. https://doi.org/10.1128/mcb.25.11.4767-4781.2005
Hollingsworth NM, Gaglione R (2019) The meiotic-specific Mek1 kinase in budding yeast regulates interhomolog recombination and coordinates meiotic progression with double-strand break repair. Curr Genet 65:631–641. https://doi.org/10.1007/s00294-019-00937-3
Hong EL, Shinohara A, Bishop DK (2001) Saccharomyces cerevisiae Dmc1 protein promotes renaturation of single-strand DNA (ssDNA) and assimilation of ssDNA into homologous super-coiled duplex DNA. J Biol Chem 276:41906–41912
Hong S, Sung Y, Yu M, Lee M, Kleckner N, Kim KP (2013) The logic and mechanism of homologous recombination partner choice. Mol Cell 51:440–453. https://doi.org/10.1016/j.molcel.2013.08.008
Hu Y, Raynard S, Sehorn MG, Lu X, Bussen W, Zheng L, Stark JM, Barnes EL, Chi P, Janscak P, Jasin M, Vogel H, Sung P, Luo G (2007) RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev 21:3073–3084. https://doi.org/10.1101/gad.1609107
Hunter N, Kleckner N (2001) The single-end invasion: an asymmetric intermediate at the double-strand break to double-Holliday junction transition of meiotic recombination. Cell 106:59–70
Ira G, Malkova A, Liberi G, Foiani M, Haber JE (2003) Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115:401–411
Jessop L, Lichten M (2008) Mus81/Mms4 endonuclease and Sgs1 helicase collaborate to ensure proper recombination intermediate metabolism during meiosis. Mol Cell 31:313–323. https://doi.org/10.1016/j.molcel.2008.05.021
Jessop L, Rockmill B, Roeder GS, Lichten M (2006) Meiotic chromosome synapsis-promoting proteins antagonize the anti-crossover activity of Sgs1. PLoS Genet 2:e155. https://doi.org/10.1371/journal.pgen.0020155
Keeney S, Giroux CN, Kleckner N (1997) Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88:375–384
Klein HL (2001) Mutations in recombinational repair and in checkpoint control genes suppress the lethal combination of srs2Delta with other DNA repair genes in Saccharomyces cerevisiae. Genetics 157:557–565
Klein F, Mahr P, Galova M, Buonomo SB, Michaelis C, Nairz K, Nasmyth K (1999) A central role for cohesins in sister chromatid cohesion, formation of axial elements, and recombination during yeast meiosis. Cell 98:91–103. https://doi.org/10.1016/S0092-8674(00)80609-1
Krejci L, van Komen S, Li Y, Villemain J, Reddy MS, Klein H, Ellenberger T, Sung P (2003) DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423:305–309
Lao JP, Cloud V, Huang CC, Grubb J, Thacker D, Lee CY, Dresser ME, Hunter N, Bishop DK (2013) Meiotic crossover control by concerted action of Rad51-Dmc1 in homolog template bias and robust homeostatic regulation. PLoS Genet 9:e1003978. https://doi.org/10.1371/journal.pgen.1003978
Lee BH, Amon A (2003) Role of Polo-like kinase CDC5 in programming meiosis I chromosome segregation. Science 300:482–486. https://doi.org/10.1126/science.1081846
Li P, Jin H, Yu HG (2014) Condensin suppresses recombination and regulates double-strand break processing at the repetitive ribosomal DNA array to ensure proper chromosome segregation during meiosis in budding yeast. Mol Biol Cell 25:2934–2947. https://doi.org/10.1091/mbc.E14-05-0957
Liu J et al. (2017) Srs2 promotes synthesis-dependent strand annealing by disrupting DNA polymerase delta-extending D-loops eLife 6 doi:https://doi.org/10.7554/eLife.22195
MacQueen AJ, Hochwagen A (2011) Checkpoint mechanisms: the puppet masters of meiotic prophase. Trends Cell Biol 21:393–400. https://doi.org/10.1016/j.tcb.2011.03.004
Marini V, Krejci L (2010) Srs2: the “odd-job man” in DNA repair. DNA Repair (Amst) 9:268–275
Martini E, Diaz RL, Hunter N, Keeney S (2006) Crossover homeostasis in yeast meiosis. Cell 126:285–295. https://doi.org/10.1016/j.cell.2006.05.044
Matsuzaki K, Kondo S, Ishikawa T, Shinohara A (2019) Human RAD51 paralogue SWSAP1 fosters RAD51 filament by regulating the anti-recombinase FIGNL1 AAA+ ATPase. Nat Commun 10:1407. https://doi.org/10.1038/s41467-019-09190-1
Mimitou EP, Yamada S, Keeney S (2017) A global view of meiotic double-strand break end resection. Science 355:40–45. https://doi.org/10.1126/science.aak9704
Mitchel K, Lehner K, Jinks-Robertson S (2013) Heteroduplex DNA position defines the roles of the Sgs1, Srs2, and Mph1 helicases in promoting distinct recombination outcomes. PLoS Genet 9:e1003340. https://doi.org/10.1371/journal.pgen.1003340
Moldovan GL, Dejsuphong D, Petalcorin MI, Hofmann K, Takeda S, Boulton SJ, D'Andrea AD (2012) Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol Cell 45:75–86
New JH, Sugiyama T, Zaitseva E, Kowalczykowski SC (1998) Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature 391:407–410. https://doi.org/10.1038/34950
Nishant KT, Chen C, Shinohara M, Shinohara A, Alani E (2010) Genetic analysis of baker’s yeast Msh4-Msh5 reveals a threshold crossover level for meiotic viability. PLoS Genet 6:e1001083. https://doi.org/10.1371/journal.pgen.1001083
Niu H, Klein HL (2017) Multifunctional roles of Saccharomyces cerevisiae Srs2 protein in replication, recombination and repair FEMS yeast research 17 doi:https://doi.org/10.1093/femsyr/fow111
Niu H, Wan L, Busygina V, Kwon YH, Allen JA, Li X, Kunz RC, Kubota K, Wang B, Sung P, Shokat KM, Gygi SP, Hollingsworth NM (2009) Regulation of meiotic recombination via Mek1-mediated Rad54 phosphorylation. Mol Cell 36:393–404. https://doi.org/10.1016/j.molcel.2009.09.029
Ogawa T, Yu X, Shinohara A, Egelman EH (1993) Similarity of the yeast RAD51 filament to the bacterial RecA filament. Science 259:1896–1899
Oh SD, Lao JP, Hwang PY, Taylor AF, Smith GR, Hunter N (2007) BLM ortholog, Sgs1, prevents aberrant crossing-over by suppressing formation of multichromatid joint molecules. Cell 130:259–272. https://doi.org/10.1016/j.cell.2007.05.035
Oh SD, Lao JP, Taylor AF, Smith GR, Hunter N (2008) RecQ helicase, Sgs1, and XPF family endonuclease, Mus81-Mms4, resolve aberrant joint molecules during meiotic recombination. Mol Cell 31:324–336. https://doi.org/10.1016/j.molcel.2008.07.006
Ontoso D, Acosta I, van Leeuwen F, Freire R, San-Segundo PA (2013) Dot1-dependent histone H3K79 methylation promotes activation of the Mek1 meiotic checkpoint effector kinase by regulating the Hop1 adaptor. PLoS Genet 9:e1003262. https://doi.org/10.1371/journal.pgen.1003262
Palladino F, Klein HL (1992) Analysis of mitotic and meiotic defects in Saccharomyces cerevisiae SRS2 DNA helicase mutants. Genetics 132:23–37
Petronczki M, Siomos MF, Nasmyth K (2003) Un menage a quatre: the molecular biology of chromosome segregation in meiosis. Cell 112:423–440
Piazza A, Wright WD, Heyer WD (2017) Multi-invasions are recombination byproducts that induce chromosomal rearrangements. Cell 170:760–773.e715. https://doi.org/10.1016/j.cell.2017.06.052
Robert T, Dervins D, Fabre F, Gangloff S (2006) Mrc1 and Srs2 are major actors in the regulation of spontaneous crossover. EMBO J 25:2837–2846. https://doi.org/10.1038/sj.emboj.7601158
Rong L, Palladino F, Aguilera A, Klein HL (1991) The hyper-gene conversion hpr5-1 mutation of Saccharomyces cerevisiae is an allele of the SRS2/RADH gene. Genetics 127:75–85
Saponaro M, Callahan D, Zheng X, Krejci L, Haber JE, Klein HL, Liberi G (2010) Cdk1 targets Srs2 to complete synthesis-dependent strand annealing and to promote recombinational repair. PLoS Genet 6:e1000858. https://doi.org/10.1371/journal.pgen.1000858
Sasanuma H, Hirota K, Fukuda T, Kakusho N, Kugou K, Kawasaki Y, Shibata T, Masai H, Ohta K (2008) Cdc7-dependent phosphorylation of Mer2 facilitates initiation of yeast meiotic recombination. Genes Dev 22:398–410. https://doi.org/10.1101/gad.1626608
Sasanuma H, Furihata Y, Shinohara M, Shinohara A (2013a) Remodeling of the Rad51 DNA strand-exchange protein by the Srs2 helicase. Genetics 194:859–872. https://doi.org/10.1534/genetics.113.150615
Sasanuma H, Tawaramoto MS, Lao JP, Hosaka H, Sanda E, Suzuki M, Yamashita E, Hunter N, Shinohara M, Nakagawa A, Shinohara A (2013b) A new protein complex promoting the assembly of Rad51 filaments. Nat Commun 4:1676. https://doi.org/10.1038/ncomms2678
Schild D (1995) Suppression of a new allele of the yeast RAD52 gene by overexpression of RAD51, mutations in srs2 and ccr4, or mating-type heterozygosity. Genetics 140:115–127
Schimmang T, Tollervey D, Kern H, Frank R, Hurt EC (1989) A yeast nucleolar protein related to mammalian fibrillarin is associated with small nucleolar RNA and is essential for viability. EMBO J 8:4015–4024
Schwacha A, Kleckner N (1994) Identification of joint molecules that form frequently between homologs but rarely between sister chromatids during yeast meiosis. Cell 76:51–63
Schwacha A, Kleckner N (1997) Interhomolog bias during meiotic recombination: meiotic functions promote a highly differentiated interhomolog-only pathway. Cell 90:1123–1135
Sheridan SD, Yu X, Roth R, Heuser JE, Sehorn MG, Sung P, Egelman EH, Bishop DK (2008) A comparative analysis of Dmc1 and Rad51 nucleoprotein filaments. Nucleic Acids Res 36:4057–4066
Shima H, Suzuki M, Shinohara M (2005) Isolation and characterization of novel xrs2 mutations in Saccharomyces cerevisiae. Genetics 170:71–85. https://doi.org/10.1534/genetics.104.037580
Shinohara A, Ogawa T (1998) Stimulation by Rad52 of yeast Rad51-mediated recombination. Nature 391:404–407
Shinohara A, Ogawa H, Ogawa T (1992) Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell 69:457–470
Shinohara A, Gasior S, Ogawa T, Kleckner N, Bishop DK (1997a) Saccharomyces cerevisiae recA homologues RAD51 and DMC1 have both distinct and overlapping roles in meiotic recombination. Genes Cells 2:615–629
Shinohara M, Shita-Yamaguchi E, Buerstedde JM, Shinagawa H, Ogawa H, Shinohara A (1997b) Characterization of the roles of the Saccharomyces cerevisiae RAD54 gene and a homologue of RAD54, RDH54/TID1, in mitosis and meiosis. Genetics 147:1545–1556
Shinohara M, Gasior SL, Bishop DK, Shinohara A (2000) Tid1/Rdh54 promotes colocalization of Rad51 and Dmc1 during meiotic recombination. Proc Natl Acad Sci U S A 97:10814–10819
Shinohara M, Sakai K, Ogawa T, Shinohara A (2003) The mitotic DNA damage checkpoint proteins Rad17 and Rad24 are required for repair of double-strand breaks during meiosis in yeast. Genetics 164:855–865
Simandlova J, Zagelbaum J, Payne MJ, Chu WK, Shevelev I, Hanada K, Chatterjee S, Reid DA, Liu Y, Janscak P, Rothenberg E, Hickson ID (2013) FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. J Biol Chem 288:34168–34180. https://doi.org/10.1074/jbc.M113.484493
Smith AV, Roeder GS (1997) The yeast Red1 protein localizes to the cores of meiotic chromosomes. J Cell Biol 136:957–967
Storlazzi A, Xu L, Cao L, Kleckner N (1995) Crossover and noncrossover recombination during meiosis: timing and pathway relationships. Proc Natl Acad Sci U S A 92:8512–8516
Subramanian VV, MacQueen AJ, Vader G, Shinohara M, Sanchez A, Borde V, Shinohara A, Hochwagen A (2016) Chromosome synapsis alleviates Mek1-dependent suppression of meiotic DNA repair. PLoS Biol 14:e1002369. https://doi.org/10.1371/journal.pbio.1002369
Sung P (1994) Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science 265:1241–1243
Sung P (1997) Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J Biol Chem 272:28194–28197
Sym M, Roeder GS (1995) Zip1-induced changes in synaptonemal complex structure and polycomplex assembly. J Cell Biol 128:455–466
Sym M, Engebrecht JA, Roeder GS (1993) ZIP1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell 72:365–378
Tang S, Wu MKY, Zhang R, Hunter N (2015) Pervasive and essential roles of the Top3-Rmi1 decatenase orchestrate recombination and facilitate chromosome segregation in meiosis. Mol Cell 57:607–621. https://doi.org/10.1016/j.molcel.2015.01.021
Tsubouchi H, Roeder GS (2004) The budding yeast Mei5 and Sae3 proteins act together with dmc1 during meiotic recombination. Genetics 168:1219–1230. https://doi.org/10.1534/genetics.103.025700
Tsubouchi H, Roeder GS (2006) Budding yeast Hed1 down-regulates the mitotic recombination machinery when meiotic recombination is impaired. Genes Dev 20:1766–1775. https://doi.org/10.1101/gad.1422506
Tsubouchi H, Argunhan B, Tsubouchi T (2018) Exiting prophase I: no clear boundary. Curr Genet 64:423–427. https://doi.org/10.1007/s00294-017-0771-y
Usui T, Ogawa H, Petrini JH (2001) A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell 7:1255–1266
Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F (2003) The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423:309–312
Wu L, Hickson ID (2003) The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 426:870–874. https://doi.org/10.1038/nature02253
Xu L, Ajimura M, Padmore R, Klein C, Kleckner N (1995) NDT80, a meiosis-specific gene required for exit from pachytene in Saccharomyces cerevisiae. Mol Cell Biol 15:6572–6581
Zakharyevich K, Ma Y, Tang S, Hwang PY, Boiteux S, Hunter N (2010) Temporally and biochemically distinct activities of Exo1 during meiosis: double-strand break resection and resolution of double Holliday junctions. Mol Cell 40:1001–1015. https://doi.org/10.1016/j.molcel.2010.11.032
Acknowledgements
We are grateful for Drs. Alastair Goldman and Michael Lichten for sharing unpublished results prior to publication. We thank Dr. Neil Hunter (UC, Davis) for pCLB2-SGS1 yeast and Dr. Andreas Hochwagen (New York University) for the anchor-away yeast strains. We thank Ms. H. Matsumoto, S. Hashimoto, C. Watanabe, and H. Wakabayashi for excellent technical assistance.
Funding
This work was supported by the Japanese Society for Promotion of Science (JSPS) KAKENHI Grant Numbers 22125001, 22125002, 15H05973, and 16H04742 to A.S.; 21770005 to H.S.; 15H05973, M.S. H.S.M.S. was supported by Institute for Protein Research.
Author information
Authors and Affiliations
Contributions
H.S., M.S., and A.S. designed the experiments. H.S., H.S.M.S., Y.F., K.C., L.P., and A.S. performed experiments. M.S. provided reagents. H.S., H.S.M.S., and A.S. analyzed the data. A.S. prepared manuscripts with help by H.S., H.S.M.S., S.G., and M.S.
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of a Special Issue on Recent advances in meiosis from DNA replication to chromosome segregation edited by Valérie Borde and Francesca Cole, co-edited by Paula Cohen and Scott Keeney
Electronic supplementary material
ESM 1
(PDF 706 kb)
Rights and permissions
About this article

Cite this article
Sasanuma, H., Sakurai, H.S.M., Furihata, Y. et al. Srs2 helicase prevents the formation of toxic DNA damage during late prophase I of yeast meiosis. Chromosoma 128, 453–471 (2019). https://doi.org/10.1007/s00412-019-00709-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-019-00709-5