
Abstract
Despite the natural abundance of pyrite and marcasite and their intergrowth, and a wealth of information they can provide on the physical–chemical conditions of mineral deposits, a complete mechanistic and kinetic study on the phase transformation from the thermodynamically metastable polymorph marcasite to the stable polymorph pyrite is yet to be made. This limits the application of marcasite as an indicator mineral for low-temperature geological environments. Here, we report results from in situ synchrotron powder X-ray diffraction and ex situ anneal/quench experiments at 400–540 °C, demonstrating that the mechanism and kinetics of this transformation depend not only on temperature, but also on particle size, the presence of water vapor, and the presence of pyrite inclusions in marcasite. Under dry conditions, the transformation is limited by surface nucleation and occurs via epitaxial nucleation of pyrite on marcasite, with {100}pyrite//{101}marcasite and {001}pyrite//{010}marcasite. In contrast, in the presence of water vapor, there is little crystallographic orientation relationship between the two phases; the transformation is still limited by surface nucleation, but modification of the surface properties by water vapor results in a different nucleation mechanism, and consequently different kinetics. Kinetic analysis estimates a half-life of 1.5 Ma at 300 °C for the transformation under dry conditions with small and pyrite-free marcasite grains, but this estimation should be used with extreme caution due to the complexity of the transformation. From synchrotron X-ray fluorescence elemental mapping, trace elements (As and Pb) play an insignificant role in the transformation. However, the presence of a fluid phase changes the behavior of Pb. Under dry conditions randomly oriented particles of galena formed in pyrite, while under water vapor conditions arrays of nano- to microparticles of galena precipitated in pores. This study highlights that although the natural occurrence of marcasite can indicate low-temperature environments, precise estimation of temperature should not be made without considering the influences from various reaction parameters.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Pyrite and marcasite, the two polymorphs of FeS2, are among the most abundant metal sulfides in the Earth’s upper crust, and play key roles in the global cycles of Fe and S. Commonly, these two minerals form intergrowths or alternating concentric layers (Kullerud and Yoder 1959), and are found in many types of ore deposits, including porphyry, epithermal, orogenic gold, volcanic-hosted massive sulfide (VHMS), and sedimentary rock-hosted and supergene environments (e.g., Large et al. 2014; Franchini et al. 2015; Murray et al. 1989; Rickard and Luther 2007). While only pyrite is found in modern marine sediments, marcasite is formed in ancient sediments as a result of circulating acidic groundwaters and marine waters (Rickard and Luther 2007). Both pyrite and marcasite are important hosts for precious metals, particularly gold and hazardous elements such as mercury and arsenic, up to weight percent levels (e.g., Xing et al. 2019; Wu et al. 2019; Kusebauch et al. 2019; Cook and Chryssoulis 1990; Large et al. 2007; Rickard and Luther 2007; Deditius et al. 2014). Because the oxidation of these minerals generates sulfuric acid, they are key contributors to supergene metal enrichment process in ore deposits, promoting the dissolution of other base metal sulfides and their re-deposition in the supergene zone. The weathering of pyrite and marcasite from sulfide-bearing rocks also negatively impacts the environment, as the process lowers the pH of groundwater and releases hazardous elements into aquifers (Burton et al. 2008). In recent years, polymorphs of FeS2 have been extensively studied in materials science, due to their natural abundance and their favorable electronic and optical properties. For example, marcasite has been recognized as potential anode materials in Li-ion batteries (Fan et al. 2017; Li et al. 2015), pyrite as photovoltaic absorber materials in solar cells (Khalid et al. 2018), and both FeS2 phases as thermoelectric materials (Gudelli et al. 2013). Hence, understanding the reactivity of pyrite and marcasite and the mechanisms of their transformation is important not only to Earth sciences, but also has great implications for economic geology, environmental management, and materials science.
The formation of pyrite and marcasite under hydrothermal conditions has been studied extensively (Murowchick 1992; Schoonen and Barnes 1991a, b, c; Qian et al. 2011), and it is generally agreed that marcasite forms under acidic conditions (pH < 5), or in S(-II)-deficient solutions (saturation index << 1000 with respect to either pyrite or marcasite), while pyrite is formed at relatively higher pH or S(-II)-rich solutions with saturation index > 1000 (with respect to either pyrite or marcasite) (Murowchick and Barnes 1986; Qian et al. 2011). Thermodynamically, marcasite is the metastable phase relative to pyrite in a wide temperature range from 5 to 700 K (Gronvold and Westrum 1976). The transformation of marcasite to pyrite was observed in natural specimen (Murowchick 1992) and in experiments (Fleet 1970; Rising 1973; Lennie and Vaughan 1992). Early combustion experiments showed that compared with pyrite, marcasite has a higher (by 5.6 kcal mol−1) heat of formation at 25 °C (Lipin et al. 1942). The thermodynamic stability of pyrite relative to marcasite at ambient conditions is also supported by recent computational experiments using density functional theory (DFT) (Spagnoli et al. 2010). Consequently, the formation and preservation of marcasite under certain hydrothermal conditions has been attributed to kinetic factors.
The transformation from marcasite to pyrite is kinetically hindered at ambient conditions, but has been predicted to occur at high pressures (e.g., 3.7 GPa) by first principle calculations (Gudelli et al. 2013), and has been observed at elevated temperature by several experimental studies (Anderson and Chesley 1933; Fleet 1970; Rising 1973; Kjekshus and Rakke 1975; Murowchick 1992; Lennie and Vaughan 1992). However, some controversy remains about the rate of the transformation. Anderson and Chesley (1933) heated marcasite at 405 °C, 415 °C, and 425 °C for 5 h in sealed glass tubes under H2S atmosphere (PH2S = 1.0) and characterized the samples using pinhole diffraction. The authors observed that the rate of transformation increased with temperature and reported that complete transformation from marcasite to pyrite occurred at 425 °C, partial transformation (~ 50%) at 415 °C, but no transformation at 405 °C. Fleet (1970) heated marcasite fragments in sealed glass capillaries and characterized the samples by single crystal diffraction. He observed a slower transformation rate than Anderson and Chesley (1933). After heating marcasite at 425 °C for 12 h, only partial transformation was achieved while the process was completed after heating at 475 °C for about 4 h (Fleet 1970). Rising (1973) studied this phase transformation at lower temperature (157–340 °C) in a 4 M NH4Cl aqueous solution and found that the transformation was fast: complete transformation took only less than 9 days at 315 °C and 14 days at 280 °C. These rates are in strong contrast with those obtained at similar temperatures but under dry conditions (evacuated silicate tubes) by Kjekshus and Rakke (1975): no transformation was observed after heating marcasite for up to 14 months at or below 300 °C, and complete transformation was achieved after heating for 4 months at 400 °C. Murowchick (1992) reported very different transformation rates for marcasite from two different localities, and for two different grains from the same locality. After heating marcasite in sealed gold capsules at 500 °C for 48 h, the polycrystalline marcasite (Elmo mine, Shullsburg, Wisconsin) was completely transformed to pyrite; in the case of two euhedral crystals of marcasite from Picher, Oklahoma, one was completely transformed to pyrite, but only a small fraction of the other grain was transformed. So far, the only quantitative kinetic study on this polymorphic phase transformation was carried out by Lennie and Vaughan (1992). These authors heated marcasite in evacuated silica tubes at four temperatures, and used infrared spectroscopy to quantify marcasite and pyrite. They observed that complete transformation took about 7.5 h at 462 °C, 12 h at 450 °C, 26 h at 437 °C, and 42 h at 425 °C. The results are comparable to the reported data by Fleet (1970), but very different to the other early studies.
The discrepancy between the reaction rates reported in early studies suggests that the polymorphic transformation from marcasite to pyrite may not be a simple process, and various factors may contribute to the mechanism and kinetics of the transformation. However, so far there has been no systematic study investigating the influence of physical–chemical parameters on the mechanism and kinetics of this reaction. To this end, we carried out a detailed study investigating the effects of pyrite inclusion and trace elements (using two specimens from different localities), temperature, particle size, and the presence of water vapor on the mechanism and kinetics of this transformation. We used in situ synchrotron powder X-ray diffraction (PXRD) to monitor the time dependence of the phase transformation to provide reliable kinetic parameters. We also carried out ex situ experiments for detailed microstructure and chemical studies of partially transformed grains, which provided complementary insights into our understanding of the mechanism of the transformation from marcasite to pyrite.
Materials and methods
The starting marcasite samples
The marcasite samples are from two different localities, one is from Oumjrane mines, Alnif, Er Rachidia, Meknès-Tafilalet, Morocco (hereafter denoted as marcasite-M), and another is from Czech Republic (South Australian Museum Registration No. G12711) (hereafter denoted as marcasite-CR). The samples were characterized by PXRD, electron probe microanalysis (EPMA), synchrotron X-ray fluorescence microscopy (SXFM), and reflected-light optical microscopy. The results are summarized in Table 1 and Fig. 1.

Results of sample characterization of (a–f) marcasite-M and (g–l) marcasite-CR. a, g Rietveld refinement analyses of synchrotron-based powder X-ray diffraction patterns, b, h synchrotron X-ray fluorescence microscopy (SXFM) elemental maps, c, i cross-polarized reflective optical micrographs showing the same regions of SXFM, and (d–f, j–l) regions of interest showing marcasite twinning in both marcasite-M and marcasite-CR as well as pyrite inclusions in marcasite-CR. Note that the dark areas surrounding the flat FeS2 surface in the optical micrographs are shadows of the grains below the surface of transparent epoxy resin. Mrc marcasite, Py pyrite. (For color figure, the reader is referred to the web version of this paper.)
For Marcasite-M, Rietveld refinement and quantitative phase analysis of the PXRD pattern showed that it contains 99.6 wt.% marcasite with unit cell parameters of a = 4.445 Å, b = 5.425 Å, and c = 3.387 Å, and a small quantity of pyrite of 0.4 wt% (Fig. 1a). Based on electron probe microanalysis (EPMA) (n = 219), the chemical composition of marcasite-M is FeS2.010±0.002, with traces of As (~ 1300 ppm), Pb (~ 1100 ppm), Hg (~ 600 ppm), Cu (~ 300 ppm), Ni (~ 300 ppm), Te (~ 300 ppm), and Cd (~ 300 ppm) (Table 1). The distributions of the trace elements (As and Pb) are heterogeneous (Fig. 1b). Twinning was observed in some grains (Fig. 1d–f).
Marcasite-CR contains 95.2 wt% marcasite with unit cell parameters a = 4.445 Å, b = 5.426 Å, and c = 3.387 Å, and 4.8 wt% pyrite (Fig. 1g). From EPMA analyses (n = 43), the composition of marcasite-CR is FeS2.020±0.002. It contains much less trace elements, including Si (~ 660 ppm), Zn (~ 210 ppm), and Pb (100 ppm) (Table 1) than Marcasite-M. Like Marcasite-M, the distributions of the trace elements (As and Pb) are heterogeneous (Fig. 1h), and twinning was observed in some grains (Fig. 1j–l). In some grains, As is concentrated in zones (Fig. 1h), which do not reflect morphology or twinning (Fig. 1i). Pyrite inclusions were observed in some of the grains (Fig. 1j–l).
The marcasite samples were crushed and sieved; the size fractions < 38 μm, 38–53 μm, and 106–150 μm were used in this study. The crushed marcasite was cleaned in absolute ethanol (> 99.7%, Merck) and dried under ambient condition before the experiments.
In situ synchrotron PXRD experiments
In situ PXRD experiments are able to monitor the reactions in real time under reaction conditions, thus eliminating the quenching step necessary for ex situ studies, providing more reliable kinetic data. The advantage of in situ PXRD for kinetic studies has been demonstrated in several solid-state and solid-solution reactions (Song et al. 2015; Xia et al. 2014; Webster et al. 2013; Peterson et al. 2016).
The in situ synchrotron PXRD experiments were carried out at the powder diffraction beamline at the Australian Synchrotron in Melbourne, Australia, using an X-ray energy of either 18 keV (λ = 0.6886 Å) or 21 keV (λ = 0.5909 Å); the beam energy was calibrated using an LaB6 standard (NIST SRM 660b). The schematic setup of the experiments is shown in Fig. 2. About 5 mg marcasite-M (< 38 μm) was loaded into the sealed end of a quartz glass capillary (1.0 mm in outer diameter, 0.1 mm in wall thickness, and 35 mm in length), and then a pile of silica glass wool plug was inserted next to the powder to prevent the powder moving out of the X-ray beam during data collection. A PTFE ferrule was glued onto the open end of the capillary and then the capillary was flushed with nitrogen gas and the open end was sealed using a Swagelok fitting (Fig. 2b). The fitting holding the capillary was fixed onto the goniometer head in a way that the capillary is positioned horizontally with the sample section just at the beam spot. The capillary was then aligned. The heating was provided by a hot air blower and the temperature was monitored by a K-type thermocouple. Precise temperature control was achieved within 0.1 °C. Before all in situ experiments, the thermocouple temperature was calibrated based on the polymorphic phase transition from orthorhombic to trigonal KNO3 at 128 °C, and from α-quartz to β-quartz at 573 °C. For studying the marcasite transformation, we first conducted an in situ slow heating experiment (13 °C min−1 to 600 °C) to detect the onset temperature for the marcasite transformation. Based on the results of the slow heating experiment, the subsequent isothermal in situ experiments were conducted at 520 °C and 540 °C. These temperatures were chosen so that the transformation was not too fast to capture various stages of the transformations and within the pyrite stability region below 743 °C (Kullerud and Yoder 1959). In each of the isothermal experiments, the hot air blower was pre-heated to the target temperature away from the capillary, and then was quickly moved underneath the capillary with the help of a motorized X–Y stage. As a result, the sample was heated very rapidly (within few seconds) from room temperature to the experiments’ target temperatures. Such procedure is required for reliable isothermal kinetic analyses. A room temperature dataset was also collected just before this rapid heating. The in situ diffraction patterns were collected using a high-resolution position-sensitive Mythen detector over the 2θ range of 1°–81°, under the Debye–Scherrer geometry, with the capillary spinning at a speed of 60 rpm. Diffraction patterns were collected for 48 s and in two positions and merged to remove the gaps between modules in the detector. To study the effect of water vapor on the phase transformation, the sample preparation was slightly different (Fig. 2c). After loading 5 mg powdered sample into the capillary, about 1 mg of Milli-Q water was added into the powder before inserting the silica glass wool. A small section of Milli-Q water was then carefully injected into the capillary in the non-heated section but close to the silica wool. This is to minimize condensation loss at the non-heated section of the capillary. After heating, the water droplet in the sample section became water vapor and was kept within about 1.5 cm between the sealed end of the capillary and the cool water section.

Schematics showing a the setup for the in situ powder X-ray diffraction experiments, and sample loading in the closed quartz glass capillary for b dry and c water vapor experiments. (For color figure, the reader is referred to the web version of this paper.)
Ex situ experiments
Because in situ PXRD is only applicable for small particle size (< 38 μm), complementary ex situ experiments were carried out using larger particle sizes (38–53 μm and 106–150 μm) under both dry and water vapor conditions to enable detailed microscopic examinations of the newly formed pyrite and the determination of crystallographic orientation relationships between marcasite and pyrite. Both marcasite-M and marcasite-CR were studied to elucidate the effects of impurities (pre-existing pyrite inclusions and trace elements) on the transformation processes. Ex situ experiments were conducted using the standard silica tube method (Xia et al. 2008; Etschmann et al. 2004). For experiments under dry conditions, marcasite particles (0.1 g) were sealed into silica tubes (14.2 mm in outer diameter, 2.2 mm in wall thickness, and 120 mm in length) under vacuum using a high-temperature flame torch. Each of the charges was then placed in a pre-heated muffle furnace at either 400 °C, 462 °C, or 520 °C, and heated for 10 days, 3 h, or 20 min, respectively. After heating, the charges were quenched in a large amount of cold water. After quick cooling, the tube was cut open and the reacted particles were recovered. For experiments under water vapor conditions, sample preparation was slightly different. After adding 0.1 g marcasite, 0.02 g water was added into the sample. Then a pile of silica wool was inserted into the tube near the minerals. Then, the bottom section of the tube was immersed in a dry ice bath to freeze the water, and finally while keeping the tube in the bath vertically, the sample-free section at the top was sealed under vacuum. In this way, water loss due to vacuum was at minimum. The heating schemes and quenching were the same as the dry experiments. The recovered samples were characterized by synchrotron PXRD (λ = 0.5909 Å; see Sect. 2.2 for instrumental parameters), scanning electron microscopy (SEM), electron backscatter diffraction (EBSD), optical microscopy, and synchrotron X-ray fluorescence microscopy (SXFM).
Rietveld-based quantitative phase analyses
Rietveld-based quantitative phase analyses were carried out for all in situ and ex situ PXRD data, using Topas Academic v6. The instrumental zero shift and peak profiles were obtained from refining the LaB6 standard (NIST SRM 660b) dataset, using empirical peak functions considering both Gaussians and Lorentzian contributions. For the refinement of in situ and ex situ PXRD patterns, the background was modeled using a fifth-order polynomial function. For each mineral phase, the unit cell parameters, scale factors, and peak broadening due to crystallite size effect were refined. The starting crystal structures were from the ICSD database, #55699 for pyrite (Wu et al. 2004) and #109374 for marcasite (Rieder et al. 2007). For in situ PXRD datasets, the refinements were run in batches where the output file from one refinement became the input file of the next dataset in the sequence. This approach minimizes parameter adjustment in the following dataset refinement, hence minimizing errors arising from correlated parameters. Once good fitting was achieved, the percentages of the involved phases were calculated by the equation,
where Wp is the relative weight percentage of phase p, S the scale factor, Z the number of formula units per unit cell, M the molecular weight of the formula unit, and V the volume of the unit cell. i represents each phase in the mixture (Pecharsky and Zavalij 2003; Hill and Howard 1987).
Scanning electron microscopy (SEM)
The cross sections of the reacted samples were examined using a Verios XHR SEM, located at the Center for Microscopy, Characterisation and Analysis (CMCA), at the University of Western Australia (UWA). Mineral grains were embedded in epoxy resin (Epofix from Struers), then ground using 1200 grit silicon carbide abrasive paper to expose the cross sections, and finally polished using 3 μm and 1 μm diamond pastes. The polished surface was cleaned and coated with a thin film of carbon. Backscattered electron (BSE) micrographs were collected using an acceleration voltage of 10 kV.
Electron backscatter diffraction (EBSD)
To determine the crystallographic orientation relationships between pyrite and marcasite, electron backscatter diffraction (EBSD) patterns were acquired with a Zeiss UltraPlus FEG SEM equipped with a Bruker eFlash EBSD detector, at CSIRO, Kensington. Operating conditions were an accelerating voltage of 20 kV, a sample tilt angle of 70°, and a working distance of 23 mm. Pole figures of pyrite and marcasite were obtained. Sample preparation was similar to SEM, but the polishing included a final polishing step using colloidal silica (0.04 μm) suspension to remove the layer damaged by the mechanical polishing. EBSD data were acquired using the Bruker Esprit 2.2 software and exported as a.ctf file for post processing. Missing data were interpolated using EBSDInterp (Pearce 2015) which uses the EBSD pattern quality (band contrast) maps to prevent artifacts (Prior et al. 2009). Pole figures show the orientation of specific crystallographic directions and are plotted using the raw data for single and bi-crystal. Where many crystals are present within a grain aggregate, data are contoured using a Gaussian with a 15° half-width at half-maximum to simplify interpretation to the most volumetrically significant orientations. Crystallographic misorientations between marcasite and pyrite, which have different space groups, are analyzed using the method described by Krakow et al. (2017). This method shows patterns in large misorientation datasets to identify systematic relationships between any two crystals regardless of their space group. Data are reduced to one point per boundary (Storey and Prior 2005) to eliminate the effect of grain size and boundary length on the analysis. For all datasets plotted using this methodology, the standard (first) settings are used as set out in Krakow et al. (2017).
Electron probe microanalysis (EPMA)
Electron probe microanalysis (EPMA) of the marcasite samples were carried out using a field-emission JEOL 8530F hyperprobe at CMCA, UWA. The analyses were undertaken using a take-off angle of 40°, an accelerating voltage of 20 kV, and a beam current of 40 nA. X-ray lines, analyzing crystals, counting time, and standards used for each element were: S Kα (PETJ, 20 s, pyrite), As Kα (TAP, 40 s, Asp200), Fe Kα (LiF, 20 s, pyrite), Au Kα (LiF, 40 s, Au metal), Pb Kα (PETH, 40 s, galena), Cu Kα (LiF, 40 s, Cu metal), Se Kα (TAP, 40 s, Bi2Se3), Ni Kα (LiF, 40 s, Ni metal), Mn Kα (LiF, 40 s, Mn metal), Ag Kα (PETJ, 40 s, Ag metal), Hg Lα (LiF, 40 s, coloradoite), Si Kα (TAP, 20 s, wollastonite), Sb Lα (PETH, 40 s, Sb metal), Te Lα (PETH, 40 s, Te metal), Cd Lα (PETJ, 40 s, Cd metal), and Zn Kα (LiF, 40 s, Zn metal). Mean atomic number (MAN) background corrections were employed throughout (Donovan and Tingle 1996). Unknown and standard intensities were corrected for dead time and the ZAF algorithm was used for matrix absorption (Armstrong 1988). Detection limits ranged from 0.007 wt% for Sb to 0.046 wt% for Au.
Synchrotron X-ray fluorescence microscopy (SXFM)
Synchrotron X-ray fluorescence elemental mapping was collected at the X-ray fluorescence microscopy (XFM) beamline at the Australian Synchrotron in Melbourne, Australia (Paterson et al. 2011; Li et al. 2016). The beam energy was set at 18,500(5) eV, and the beam was focused to a spot size of ~ 2 × 2 μm using Kirkpatrick–Baez mirrors. Fluorescence data were collected using the massively parallel 384-element Maia detector system (Kirkham et al. 2010; Ryan et al. 2010a, b, 2014). The detector was placed in front of the sample at a distance of 1 mm, which enabled the collection of nearly 50% of a full hemisphere of the X-rays emitted by the sample. The incident X-rays travelled through a central hole in the detector. Standard foils (Pt, Mn, Fe) were used to (1) constrain detector efficiency and geometry (e.g., distance from the sample), (2) constrain any filters that are used in front of the detector, and (3) convert from ion chamber counts to flux (photons/s; Ryan et al. 2010a). The area of each sample was mapped using a scan speed of 1 mm/s, corresponding to a dwell time of 1.00 ms/pixel.
The SXFM data were analyzed with GeoPIXE II (Ryan et al. 2005) using the dynamic analysis (DA) method to project quantitative elemental images from the full fluorescence spectra (Ryan 2000; Ryan et al. 2009). The DA technique is a matrix transform algorithm that unfolds overlaps and subtracts background, escape peaks and other detector artifacts. The advantage of this method is that by fitting multiple lines per element, it benefits from better counting statistics and it is possible to distinguish between elements that have overlapping X-ray lines, which are traditionally difficult to separate using the region of interest approach.
Kinetic analysis
The isothermal kinetics of a wide range of phase transformations follow the Avrami–Erofeev equation (Avrami 1939; Khanna and Taylor 1988; Xia et al. 2014; Song et al. 2015):
where α is the reaction extent, k (s−1) is the rate constant, t (s) is the reaction time, t0 (s) is the induction time, and n is the Avrami exponent indicative of reaction mechanisms, which are summarized in Table 2 (Hulbert 1969). Induction time t0 is the time between the start of heating and the onset of pyrite formation evidenced from PXRD, and reaction extent α is calculated as:
where wt, w0 and we are the weight fractions of marcasite at an arbitrary reaction time t, initially (t = 0) and at equilibrium (t = ∞, in this case we = 0), respectively (Wang et al. 2005; Xia et al. 2007, 2009, 2012). These weight fractions were determined from Rietveld quantitative phase analysis of the PXRD datasets (Eq. 1). The Avrami–Erofeev equation can be rewritten as the Sharp–Hancock equation (Hancock and Sharp 1972; Lasaga 1998):
By plotting ln[−ln(1− α)] as a function of ln(t–t0) and carrying out a linear fit through the data points, k and n were obtained from the intercept and slope of the linear fit.
The rate of the transformation is temperature dependent, and it is assumed to follow the Arrhenius equation:
where A(s−1) is the pre-exponential factor, T (K) is the absolute temperature, R (8.314 J mol−1 K−1) is the ideal gas constant, and Ea (kJ mol−1) is the activation energy of the reaction.
Rewriting Eq. 5, we have:
By plotting ln(k) as a function of 1/T and carrying out a linear fit through the data points, Ea was obtained from the slope of the linear straight line.
Results
In situ PXRD results
The results of the four in situ PXRD experiments are presented in Fig. 3. In the slow heating experiment with a heating rate of 13 °C min−1, the transformation from marcasite to pyrite did not occur until the temperature reached 550 °C (Fig. 3a). During temperature ramping, the marcasite peaks continuously shift toward lower diffraction angles (Fig. 3a), documenting the expansion of the unit cell with increasing temperature. From linear fits of the unit cell parameters against temperature (Fig. 4), the linear thermal expansion coefficients are 1.41 × 10–5 K−1 along a direction, 1.09 × 10–5 K−1 along b direction, and 7.38 × 10–6 K−1 along c direction, and the volumetric thermal expansion coefficient is 3.26 × 10–5 K−1. It is also seen from Fig. 3a that unidentified diffraction peaks appeared during heating, but none of them could be assigned to phases in the Fe–S–O system. These peaks could belong to phases rich in As or Pb because the starting marcasite contains measurable amounts of these elements (see EPMA results in Table 1), but unfortunately, none of the phases in the latest Crystallography Open Database (COD) inorganic database (update date 10.09.2019) could match these peaks (Gražulis et al. 2009). Pyrrhotite appeared at 580 °C, most likely due to the decomposition of the newly formed pyrite, FeS2 → FeSx + (2−x)S(g) (1 ≤ x ≤ 1.143), as has been reported previously (Lambert et al. 1998).

In situ PXRD patterns (viewed down the intensity axis) for the transformation of marcasite-M to pyrite under a slow heating dry condition from room temperature to 600 °C (13 °C min−1), b isothermal dry conditions at 540 °C, c isothermal dry condition at 520 °C, and d isothermal water vapor condition at 520 °C. (For color figure, the reader is referred to the web version of this paper.)

The changes of marcasite unit cell parameters (a, b, c, V) with increasing temperature during the slow-heating experiment. The slopes of the linear fittings give the thermal expansion coefficients. Error bars of the data points are within the size of the symbols
The in situ PXRD data of isothermal experiments at 520 °C and 540 °C are presented in Fig. 3b–d, and the reaction extents obtained from quantitative analysis of the diffraction datasets are plotted against time and shown in Fig. 5a. The PXRD patterns showed that the progressive transformation from marcasite to pyrite occurred at both temperatures and under both dry (Fig. 3b, c) and water vapor (Fig. 3d) conditions. The sudden shifts of the marcasite peaks to lower angles after the first room temperature dataset are clearly seen in the zoomed-in plots (the 14.4°–14.8° plots in Fig. 3b–d), indicating almost instant heating of the sample from room temperature to the target temperatures when the capillaries were moved to the top of the pre-heated hot air blower. This ensures accurate kinetic analyses using the isothermal kinetic models.

Kinetic results of the isothermal in situ PXRD experiments using marcasite-M as the starting materials. a Reaction extent as a function of time; b Avrami plot; and c Arrhenius plot giving the activation energy of the transformation under dry conditions. Error bars of the data points are within the size of the symbols
Comparing the results of the dry experiments at 520 and 540 °C (Figs. 3b, c, 5a), it is clear that the transformation was faster at higher temperatures. Pyrite appeared as soon as the temperature reached 540 °C and marcasite disappeared almost completely after 18 min (Figs. 3b, 5a). However, for the dry experiment at 520 °C, pyrite appeared after heating for nearly 7 min (induction time) and marcasite was still detected after heating for nearly 90 min (Figs. 3c, 5a).
From the isothermal experiments at 520 °C under dry and water vapor conditions (Fig. 3c, d), we see that water vapor promoted the transformation from marcasite to pyrite. Pyrite peaks appeared as soon as the temperature reached 520 °C, compared with the dry experiment with a 7-min induction time (Figs. 3c, 5a).
The transformation rates are directly compared in Fig. 5a. The rate of transformation was fastest at 540 °C under dry condition, slower at 520 °C under water vapor condition, and slowest at 520 °C under dry condition.
The rate constant (k) and Avrami exponent (n) obtained based on kinetic analyses using the Avrami–Erofeev method are plotted in Fig. 5b. The values are k = 21.72 ± 8.6 × 10–4 s−1 and n = 1.52 ± 0.06 for the experiment at 540 °C under dry condition, k = 5.25 ± 0.90 × 10–4 s−1 and n = 1.72 ± 0.03 for the experiment at 520 °C under dry condition, and k = 2.64 ± 0.86 × 10–4 s−1 and n = 1.12 ± 0.04 for the experiment at 520 °C under water vapor condition. Because the value of Avrami exponent indicates the reaction mechanism (Table 2) (Hulbert 1969), similar values of n for the two experiments under dry condition suggest the same reaction mechanism prevailed. However, under water vapor condition, n is significantly lower than for the dry experiments, suggesting a different mechanism. Based on the Arrhenius plot (Fig. 5c), the activation energy of the transformation under dry condition is 380 ± 15 kJ mol−1, and the pre-exponential factor A is 8.39 × 1020 s−1.
Ex situ experimental results
For studying the effects of temperature, water vapor, particle size, and impurities (pyrite inclusions and trace elements in marcasite grains) on the mechanism and kinetics of the transformation, 24 ex situ experiments were carried out and the results are presented in Figs. 6, 7, 8, 9, 10, 11, 12and 13.

Reaction extent of the ex situ experiments as a function of temperature, water vapor, particle size, and marcasite locality. Standard deviations of reaction extent are shown in the brackets. (For color figure, the reader is referred to the web version of this paper.)
Transformation rate
The reaction extents were summarized in Fig. 6. The effects of the investigated experimental parameters on the rate of transformation are summarized below:
- 1.
The effect of temperature. Under dry and water vapor conditions, the transformation of both marcasite-M and marcasite-CR was facilitated by temperature. This is illustrated in Fig. 6, where the columns represent snapshots with increasing temperature (400 °C, 462 °C, and 520 °C) at rapidly decreasing times (14,400, 180, and 20 min). For marcasite-M, the effect of temperature is clear when comparing the following data groups: dry and small particles (38–53 µm) (Fig. 6a, c, e); water vapor and small particle (Fig. 6b, d, f); dry and large particles (106–150 µm) (Fig. 6g, i, k), and water vapor and large particles (Fig. 6h, j, l). For marcasite-CR, although the reaction extent was generally slightly higher for the 400 °C experiments than for the 462 °C experiments, the reaction time at 400 °C is 10 days, 80 times more than the time (3 h) at 462 °C. Hence, the positive effect of temperature on the transformation was found for marcasite-CR as well.
- 2.
The effect of water vapor was found to depend on particle size. For the large particles (106–150 μm), the experiments under water vapor condition were generally faster than the dry counterparts, which is clearly seen when we compare the following pairs in Fig. 6: (i and j), (k and l), (s and t), (u and v), and (w and x). The only exception is the experiments at 400 °C using marcasite-M (Fig. 6g, h), but considering the very low reaction extents and the associate errors (0.017 ± 0.003 and 0.014 ± 0.003) they can be considered as identical. Interestingly, for the small particles (38–53 μm), the transformations with water vapor were generally slower than their dry counterparts, when comparing the following pairs in Fig. 6: (a and b), (c and d), (m and n), (o and p), and (q and r). The only exceptions are the experiments at 520 °C using marcasite-M (Fig. 6e, f), but the values 0.128 ± 0.002 and 0.134 ± 0.002 are nearly identical and likely inconclusive.
- 3.
The effect of particle size depends on whether water vapor is present. Under dry condition, the transformation was faster with small particles (38–53 μm) for both marcasite-M and marcasite-CR and the three temperatures studied. This is clear when comparing the following pairs in Fig. 6: (a and g), (c and i), (e and k), (m and s), (o and u), and (q and w). However, in the presence of water vapor, the transformation was generally faster with large particle size, which is clearly seen when comparing the following pairs in Fig. 6: (d and j), (f and l), (n and t), (p, and v), and (r and x). The only exception is the experiments at 400 °C using marcasite-M (Fig. 6b, h). However, due to the small reaction extents and the associate error (0.022 ± 0.003 and 0.014 ± 0.003), this comparison may be considered invalid.
- 4.
The effect of impurities (pre-existing pyrite inclusions and trace elements) in marcasite grains: from PXRD and microscopy analyses (Fig. 1), it is known that marcasite-CR contains 4.8 wt% of pyrite inclusions, much higher than marcasite-M which contains only 0.4 wt% pyrite. Additionally, from EPMA analyses (Table 1), marcasite-M contains much more trace elements (especially, As and Pb) than marcasite-CR. In almost all experimental conditions, the transformation of the marcasite-CR was faster than the marcasite-M, evident from pairs in Fig. 6: (a and m), (b and n), (c and o), (e and q), (f and r), (g and s), (h and t), (i and u), (j and v), (k and w), and (l and x). The only exception is small particle at 462 °C under water vapor condition (Fig. 6d, p); however, due to similar reaction extents and the associate errors (0.036 ± 0.003 and 0.035 ± 0.003), the results are considered identical.
SEM results
Four samples were examined by SEM, to study the effect of water vapor on the transformation using either marcasite-M or marcasite-CR. The results are presented in Fig. 7 (marcasite-M) and Fig. 8 (marcasite-CR).

Backscatter electron images of marcasite-M grains (106–150 μm) reacted at 520 °C for 20 min, under (a, b) dry, and (c, d) water vapor conditions, corresponding to k and l in Fig. 6, respectively. Gn = galena

Backscatter electron images of marcasite-CR grains (106–150 μm) reacted at 520 °C for 20 min, under (a, b) dry, and (c, d) water vapor conditions, corresponding to w and x in Fig. 6
In experiments using marcasite-M, few large pyrite grains were formed within the marcasite grain with sharp phase boundaries between pyrite and marcasite (Fig. 7). Porosity formed in pyrite phase (Fig. 7), because the molar volume of pyrite (23.94 cm3 mol−1) is smaller than that of marcasite (24.58 cm3 mol−1). The reduction in molar volume for this transformation is − 2.6%. However, the pore texture is very different in samples obtained under dry and water vapor conditions. In the case of dry conditions, the pyrite phase contains heterogeneously distributed pores of a wide size range from less than 50 nm to about 400 nm (Fig. 7a, b). In the case of water vapor condition, much narrow pore size distribution was observed with a diameter of about 300 nm and the pores are homogeneously distributed in a very ordered way: they are aligned as straight lines with even gaps (Fig. 7c, d). In both cases, nano- and micro-inclusions of galena were observed as a by-product. Under dry condition, galena was present in the non-porous region of pyrite (Fig. 7b), while under water vapor condition, galena both fills the pores and presents in the non-porous region. It appears that particles of galena are distributed in ordered fashion following the trace of pores (Fig. 7d). This presence of galena particles might be related to the concentration of Pb in the starting material (Fig. 10j). The formation of galena was confirmed by synchrotron PXRD (Fig. 9), and is likely due to the relatively high concentration of Pb (Table 1) in the starting marcasite-M.

Synchrotron-based PXRD patterns of the samples after heating marcasite-M (106–150 μm) at 520 °C for 20 min under dry and water vapor conditions. Note that the intensity has been taken square root to make the small galena peak visible. See Fig. 6k and l for reaction extent of these two experiments. (For interpretation of the references to color in this figure, the reader is referred to the web version of this article.)
In experiments using marcasite-CR, numerous small pores in pyrite were observed in both samples (dry and water vapor conditions) (Fig. 8). Compared with the experiments using marcasite-M under water vapor condition (Fig. 7d), the experiments using marcasite-CR (Fig. 8) show that: (i) the pores have a polymodal size distribution with a size range of about 100–1000 nm; (ii) no by-product phases were observed, consistent with smaller amounts of trace elements in marcasite-CR (Table 1); and (iii) the phase boundary between pyrite and marcasite is highly irregular. Under dry conditions, the area of pores in newly formed pyrite was larger under wet conditions than pyrite formed in dry system; this is ~ 12.5% and 8%, respectively (Fig. 8b, d).
SXFM results
Synchrotron X-ray florescence mapping was performed to compare the spatial distributions of As and Pb in iron disulfides and assess the effect of trace elements on the transformation from marcasite to pyrite. Reflected-light optical images, presented together with elemental maps, were used to identify pyrite and marcasite. The results are shown in Fig. 10 (marcasite-M) and Fig. 11 (marcasite-CR). It is worth nothing that the gained shapes from the optical microscopy and SXFM maps are slightly different because they represent different sections of the grains (Figs. 10, 11); optical images represent the top surface since pyrite and marcasite are opaque to visible light, while X-ray in SXFM penetrate relatively deep with the sample; 86% of the incident X-ray reach a depth of 15 μm, and about 50% of the As Kα X-rays can escape through 15 μm of pyrite; hence, the SXFM maps represent an average composition over a significant depth of the pyrite/marcasite grains. However, this does not significantly influence the comparison due to the relatively large size of the pyrite and marcasite crystals.

Cross-polarized reflective optical images and SXRF maps (RGB and As, Fe, Pb) showing marcasite-M grains (106–150 μm) reacted at 520 °C for 20 min, under (a–e) dry, and (f–j) water vapor conditions, corresponding to k and l in Fig. 6. Note that the dark areas surrounding the flat FeS2 surface in the optical micrographs are shadows of the grains below the surface of transparent epoxy resin. (For color figure, the reader is referred to the web version of this paper.)

Cross-polarized reflective optical images and SXRF maps (RGB and As, Fe) showing marcasite-CR grains (106–150 μm) reacted at 520 °C for 20 min, under a–d dry, and e–h water vapor conditions, corresponding to w and x in Fig. 6. Note that the dark areas surrounding the flat FeS2 surface in the optical micrographs are shadows of the grains below the surface of transparent epoxy resin. (For color figure, the reader is referred to the web version of this paper.)
In samples using marcasite-M as the starting material, heterogeneous distributions of As and Pb were observed in both samples from dry and water vapor experiments (Fig. 10c, e, h, j). The concentrations and distributions of As and Pb are positively correlated (Fig. 10c, e; 10 h, j). Importantly, comparing optical images with SXFM maps, there is no correlation between the distribution of iron disulfide polymorphs and the amount of either As or Pb, under both dry (Fig. 10a, c, e) and water vapor conditions (Fig. 10f, h, j), and both As and Pb have no clear correlation with Fe (Fig. 10b–e, g–j). For example, under dry condition (Fig. 10a–e), quite homogeneous elemental distributions were observed in Grain 1 (Fig. 10c, e), but in this grain, pyrite and marcasite form alternating zoning (Fig. 10a); both Grains 2 and 3 show no sign of transformation, yet Grain 2 has high concentrations of As and Pb, and in Grain 3 half contains little As and Pb and the other half contains relatively high As and Pb. Similarly, under water vapor conditions (Fig. 10f–j), the untransformed grains include those with little As and Pb (e.g., Grains 4, 11, 13) and with high concentrations of As and Pb (e.g., Grain 8); and the transformed or partially transformed grains include those with low concentrations of As and Pb (e.g., Grain 5), high concentrations of As and Pb (e.g., Grain 7), low As and moderate to high Pb (e.g., Grain 9, 12), moderate As and high Pb (e.g., Grain 10), and uneven distributions of As and Pb (e.g., Grain 6).
In samples using marcasite-CR as the starting material, the grains are mostly Pb free (Pb maps not shown), and the As maps also show heterogeneous distribution among the grains and within the grains (Fig. 11c, g) and no clear correlation was found between the As and Fe maps (cf. Figure 11c, d; cf. Figure 11g, h). It seems that there is no spatial relationship between As concentration and iron disulfide polymorphs. For example, under dry conditions (Fig. 11a–d), untransformed grains appear to be intact, and it contains grains with no As (Grains 7 and 8), moderate amounts of As (Grain 6), oscillation zones of high and low As contents (Grain 3), areas with high As rim near grain surface (Grain 2), and high As patches near grain surface (Grains 1 and 9). On the other hand, the transformed grains included some with low As (Grain 4) and high As concentrations (Grain 5). Similarly, under water vapor conditions (Fig. 11e–h), untransformed intact grains contain low As (Grain 10), uneven moderate As (Grain 11), and unevenly high As (Grain 13); and transformed or mostly near completely transformed grains include some with no As (Grains 12, 14, 16–18) and high As concentrations (Grain 15).
EBSD results
Electron backscatter diffraction (EBSD) analyses were carried out for partially transformed marcasite-M gains from dry and water vapor experiments at 520 °C using large particle size (106–150 μm) (correspond to Fig. 6k, l). The results are presented in Fig. 12 (dry condition) and Fig. 13 (water vapor condition). The aim of this analysis was to investigate the effect of water vapor on the transformation mechanism.

EBSD analysis of the cross section of the grains after heating marcasite-M (106–150 μm) at 520 °C for 20 min under dry condition (see Fig. 6k). a EBSD map showing the distribution of marcasite (blue) and pyrite (red), and b contoured pole figures generated from 606 (pyrite) and 3104 (marcasite) diffraction patterns for particle 1, 151 (pyrite) and 1139 (marcasite) diffraction patterns for particle 2, and 548 (pyrite) and 1431 (marcasite) diffraction patterns for particle 3. c 3D misorientation space showing misorientations between neighboring pyrite and marcasite crystals for all grains in the map using one point per boundary. Py pyrite; Mrc marcasite. (For color figure, the reader is referred to the web version of this paper.)

EBSD analysis of the cross-section of the grains after heating marcasite-M (106–150 μm) at 520 °C for 20 min under water vapor condition (see Fig. 6l). a EBSD map showing the distribution of marcasite (blue) and pyrite (red), and b contoured pole figures generated from 11,614 (pyrite) and 646 (marcasite) diffraction patterns for particle 1, 2805 (pyrite) and 6625 (marcasite) diffraction patterns for particle 2, and 4517 (pyrite) and 4809 (marcasite) diffraction patterns for particle 3. c 3D misorientation space showing misorientations between neighboring pyrite and marcasite crystals for all grains in the map using one point per boundary. Py pyrite; Mrc marcasite. (For color figure, the reader is referred to the web version of this paper.)
For the sample from the dry experiment, the EBSD map showed that no transformation occurred in more than half of the total marcasite grains, but in grains where partially transformation occurred, significant amount of marcasite was transformed to pyrite with sharp phase boundaries (Fig. 12a). Pyrite formed bands (zones) in the marcasite grains (e.g., Grains 1, 2, 3 in the EBSD map in Fig. 12a), or occupied one side of the initial grain. Pole figures of pyrite and marcasite were generated for the three selected grains (Fig. 12b). In all three grains, a clear orientation relationship between pyrite and marcasite is confirmed, i.e., {100}pyrite//{101}marcasite and {010}pyrite//{010}marcasite (Fig. 12b). The linear traces of the phase boundaries in most of the grains are consistent with the interfaces being planar, suggesting a control of the parent grain not only on orientation of the product, but also on the reaction interface geometry. Both the {100}pyrite and {101}marcasite comprise multiple symmetric equivalents. The specific variant that is shared between the pyrite and marcasite is consistent with that particular plane also being the interface between the two minerals. Also, in Grain 3, the two spatially separated pyrite crystals (zones) have identical crystallographic orientations showing that the crystallographic control of marcasite on nucleating pyrite is repeatable for any given marcasite grain. All reacted domains in the dataset shown in Fig. 12a are analyzed in Fig. 12c. Each point represents a misorientation between a marcasite crystal and a pyrite crystal. The data cluster tightly at ± 52.7° along the b-axis of the plot. This axis corresponds to both {010}marcasite and {010}pyrite and is the misorientation axis expected for the relationship outlined above, i.e., the angle between {100}marcasite and {101}marcasite. The tight clustering shows that most of the pyrite–marcasite boundaries analyzed have the same misorientation.
For the sample exposed to water vapor, the EBSD map revealed that the transformation from marcasite to pyrite had occurred in majority of the grains. However, the reaction extent varied significantly from grain to grain, from complete transformation to a few percent fraction (Fig. 13a). In most cases, more than two newly formed pyrite grains can be observed in a single marcasite grain. Pole figures of pyrite and marcasite were generated for the three selected grains (Fig. 13b). In all three grains, particularly Grains 1 and 3, we see that the pyrite crystals formed in a single marcasite grain have different crystallographic orientations (pairs in pyrite pole figures). Also, in all three grains, no clear crystallographic orientation relationship between pyrite and marcasite can be found in the pole figures (Fig. 13b). While specific examples are shown in the pole figures, all the data in Fig. 13a are analyzed using the misorientation analysis (Fig. 13c). There is a small cluster at 52.7° along the b-axis of the plot showing that some of the misorientations are consistent with those observed in the dry samples. However, the wide scatter of points within the misorientation figure shows that there is not generally a strict crystallographic correspondence between the starting marcasite and the reacted pyrite for the wet sample.
Discussion
At first sight, the polymorphic transformation from marcasite to pyrite appears to be a simple reaction to transform a thermodynamically less stable polymorph to the stable form. However, our experimental results suggest that the reaction is influenced by various factors and can proceed via different mechanisms. Distinctly different textures were observed in reactions under dry and water vapor conditions, suggesting that different mechanisms control the phase transformation in the two cases. In the following, we will discuss these mechanisms based on the observed reaction rates and sample textures.
The transformation under dry condition
The transformation rate from marcasite to pyrite under dry conditions was reported to increase with temperature (Anderson and Chesley 1933; Fleet 1970; Kjekshus and Rakke 1975; Lennie and Vaughan 1992). This was also observed in the present study (Figs. 5, 6). However, the reported transformation rates are not consistent. For example, the transformation rate of a big euhedral crystal (5–7 × 5 × 1–2 mm) in Murowchick (1992) was slower than what was reported by Fleet (1970) and Lennie and Vaughan (1992) who used smaller particles. Anderson and Chesley (1933) reported the fastest transformation amongst these five studies, but they used the smallest particles of less than 74 μm. In the present study, the transformation is slower than most of the reported results (Anderson and Chesley 1933; Fleet 1970; Lennie and Vaughan 1992). For example, after heating the 106–150 μm particles at 462 °C for 3 h, only 3.2(3)% and 6.7(2)% was transformed into pyrite using marcasite-M and marcasite-CR, respectively (Fig. 6i, u); this compares with ~ 50% transformation reported by Lennie and Vaughan (1992) at the same temperature and time, and complete transformation by Anderson and Chesley (1933) at a lower temperature of 425 °C for similar time (5 h). However, these inconsistencies are expected because the results of this study revealed that in addition to temperature and time, this mineral transformation is controlled by particle size and inclusions of pre-existing pyrite in marcasite (Fig. 6). Unfortunately, there is no data about the chemical composition of marcasite in the early studies, and the particle size was described as ‘coarsely ground’ (Lennie and Vaughan 1992), or can only be inferred from the diameter of the silica glass tube container (0.3 mm) (Fleet 1970), or no information about particle size is available (Kjekshus and Rakke 1975).
Here, it is suggested that under dry conditions, the transformation rate is inversely proportional to particle size (Fig. 6), which can be explained by means of the rate-limiting step between nucleation and crystal growth. For marcasite-M with negligible amount of pre-existing pyrite inclusions, the EBSD maps (Fig. 12a) revealed that only few marcasite grains are transformed to pyrite (many grains show no pyrite), suggesting that a very limited number of pyrite nuclei were formed initially. Thus, the transformation is limited by nucleation rather than crystal growth. Considering the nucleation sites, grain surface is likely the preferred location rather than the interior of the grains because surface atoms are not fully bonded. This agrees with the large activation energy (380 ± 15 kJ mol−1) of the transformation, suggesting that the transformation involves breaking of chemical bonds (Lasaga 1998; Xia et al. 2012). The notion of surface nucleation is also supported by microscopic observations showing that at least a part of the pyrite crystals occupy the grain surface (Figs. 7a, 12a). This suggests that nucleation occurred on the grain surface and subsequently the transformation progressed into the marcasite grains. If nucleation occurred within the grains, then some pyrite inclusions would be entirely embedded in marcasite, but this was not observed. The reason for the faster rate of transformation for smaller grains of marcasite is supported by their higher specific surface area, and hence the higher numbers of nucleation sites. For marcasite-CR, the inverse relationship between particle size and transformation rate was also observed, but the transformation was faster than marcasite-M (Fig. 6). This agrees with the nucleation control mechanism. Apart from surface nucleation due to the size effect, the higher amount of pre-existing pyrite inclusions in marcasite-CR must have acted as seeds, providing additional growing sites. It is important to note that the difference in transformation rate in marcasite-M and marcasite-CR is mostly likely due to the pre-existing pyrite, because the SXFM results strongly suggest that trace elements (As and Pb) play insignificant roles (Figs. 10, 11).
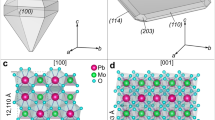
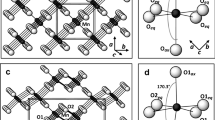
The crystallographic orientation relationship between marcasite and pyrite, {100}pyrite//{101}marcasite and {001}pyrite//{010}marcasite (Fig. 12b), suggests that the transformation is facilitated by epitaxial growth of {100}pyrite on {101}marcasite. Similar orientation relationship between pyrite and marcasite was determined experimentally under dry conditions (Fleet 1970) and in natural samples (Gait and Dumka 1986). The epitaxial growth of pyrite on marcasite is possible due to similar crystal structures. Marcasite has an orthorhombic symmetry (space group Pnnm) with lattice parameters of a = 4.445 Å, b = 5.425 Å, and c = 3.387 Å (Rieder et al. 2007; Hyde and O’Keeffe 1996) (Fig. 14a), while cubic pyrite (space group Pa \(\stackrel{-}{3}\)) has the lattice parameters of a = b = c = 5.417 Å (Bayliss 1977; Hyde and O’Keeffe 1996) (Fig. 14b). In both structures, the Fe atoms are coordinated with six nearest-neighbored sulfur dumbbells (S22−) forming distorted octahedra. The length of the edge of pyrite unit cell is very similar to that of marcasite (in b direction) with a small mismatch of 0.15%. In addition, the Fe–Fe atomic distance (5.588 Å) in the {101} lattice plane of marcasite is also similar to that of pyrite: 3.16% longer than pyrite in b direction (Fig. 14c, d). Hence, lattice mismatch between {100}pyrite and {101}marcasite is small.

Crystal structures of marcasite and pyrite, showing a marcasite unit cell, b pyrite unit cell, c marcasite {101} plane, and d pyrite {100} plane. (For color figure, the reader is referred to the web version of this paper.)
The crystallographically controlled growth of pyrite on marcasite on the {101} lattice plane suggests that pyrite growth may be restricted in other directions, leading to two-dimensional growth. This oriented growth is also suggested by the EBSD maps showing that some pyrite grains are present as bands/zones in the marcasite grains (Fig. 12a). This is further inferred from the value of Avrami exponent (n), which are 1.52 ± 0.06 and 1.72 ± 0.03 for the transformations under dry conditions (Fig. 5), in agreement with the phase boundary-controlled mechanism with zero nucleation rate (few nuclei formed initially) and between one-dimensional (n = 1) and two-dimensional (n = 2) growth models (Table 2). Because the lattice mismatch is larger on one edge (3.16% mismatch) than the other (0.15% mismatch) on the marcasite {101} plane, as illustrated in Fig. 14c, d, the growth rate of pyrite along the direction with smaller lattice mismatch is likely to be higher than along the direction with larger lattice mismatch on the marcasite {101} plane. Consequently, pyrite forms bands as seen in some of the grains (Fig. 12a). Such growth may be regarded as a growth model between one- and two-dimensional growth.
The transformation under water vapor condition
Under water vapor conditions, ex situ experiments suggest that the transformation rate is proportional to the particle size (Fig. 6), which suggests different mechanism(s) than under dry condition. Similar to the dry condition, the EBSD map (Fig. 13a) documents few pyrite grains in each of the reacted marcasite grains and suggest that the reaction is likely limited by nucleation. However, different from the dry reactions, there is little clear crystallographic orientation relationship between marcasite and pyrite (Fig. 13b, c). This means that epitaxial nucleation played an insignificant role and that the transformation involved breaking of chemical bonds. From the pyrite pole figures (Fig. 13a), several grain orientations (pairs of poles) can be identified. However, within each of the pyrite domains, the orientation of the individual grains is identical (Fig. 13b). This means that once a pyrite nucleus forms, further growth of pyrite is due to epitaxial growth on the pyrite nucleus, rather than on the parent marcasite grain.
It is likely that water vapor facilitates the breaking of chemical bonds and provides a diffusion medium, accelerating pyrite nucleation. The tube contained 0.1 g water, which can cover around 40 m2 assuming single molecular coverage, much greater than the total surface area of marcasite in the tube. Assuming spherical shape, and using geometric mean sizes, 44.88 μm for the 38–53 μm fraction and 126.09 μm for the 106–150 μm fraction, the total surface area of 0.1 g marcasite in the tube is ~ 0.0274 m2 for small fraction (38–53 μm) or ~ 0.0098 m2 for large fraction (106–150 μm). Hence, in either case, water vapor is expected to cover the entire surface of the grains and form multiple layers. This means that the specific surface area may play an insignificant role in the presence of water vapor. A possible explanation is that, the nucleation is proportional to the surface area of individual grains. Larger grains have larger surface area per grain and hence are more likely to form nucleus compared with small grains, and once nucleation starts, the full grain is replaced quickly, enhancing the overall transformation rate.
Under water vapor conditions, pyrite growth is not controlled by the marcasite structure, and it can be random and two dimensional on the grain surface and/or along the fractures, or three dimensional progressing into the marcasite grains. The value of the Avrami exponent (n) from the in situ PXRD experiment under water vapor condition is 1.12 ± 0.04, which is between two-dimensional (n = 1.0) and three-dimensional (n = 1.5) growth models under diffusion-controlled and zero nucleation rate categories (Table 2).
Ordered arrays of narrowly dispersed pores were observed only under water vapor conditions (Fig. 7d). The ordered pores also shed light on the role of water vapor on the transformation process. It is plausible that during the progress of the transformation, the water vapor in pores may prevent their disappearance because the shrinking of pore size means increasing vapor pressure within the pores, and after reaching a certain pressure, the pyrite growth toward the center of the pores may stop or slow down significantly. Such a process could explain the nearly monodispersed size distribution of the pores, as the threshold vapor pressure is likely to be pore size dependent. On the other hand, under dry vacuum condition, there is no resistance from the vapor phase that would prevent the pore from annealing out and/or coarsening, a process leading to surface energy minimization (Putnis et al. 2004). Hence, under dry conditions, some areas of pyrite grains are pore free (Fig. 7b), and others host relatively large pores with non-uniform size (Fig. 7b). In the dry experiments, no pyrrhotite was detected by PXRD, which suggests minimum amount of sulfur vapor (if present). Sulfur vapor would also prevent the pores from annealing out.
The effect of small amount of water on mineral reactions is complicated and still poorly understood. Our results suggest that compared with dry conditions, the presence of water vapor can either promote or inhibit the transformation from marcasite to pyrite depending on the particle size of marcasite grains (Figs. 5, 6). As discussed above, this is due to the different controlling mechanisms for the transformation under dry and water vapor conditions, and the opposite dependence of the mechanisms on particle size under dry and water vapor conditions. Recent experimental studies have demonstrated that very small amount of water can significantly accelerate mineral reactions (Zhao et al. 2017; Milke et al. 2013). However, the mechanisms are different. For example, in a silicate rock, the small amount of water (20 ppm) keeps the reaction-induced pores open under confining pressure (1.9 GPa) and the open pores act as effective diffusion pathways for the reaction between olivine and quartz, forming orthopyroxene (Milke et al. 2013). In sulfide reactions, a bornite–digenite solid solution (bdss), produced from hydrothermal mineral replacement of chalcopyrite, contains a small amount of fluid residue within the bdss phase, and the fluid significantly enhances the unmixing (exsolution) reaction in bdss upon cooling, producing bornite and digenite with lamella microstructure (Zhao et al. 2017). The coarsening of bornite and digenite phases during the unmixing reaction is about 1000 times faster than the same reaction under fluid-free condition, and has been interpreted as enhanced healing of the open porous microstructure in the presence of a fluid phase (Zhao et al. 2017).
The kinetics of the transformation
The only quantitative kinetic study on the transformation of marcasite to pyrite in the literature was carried out by Lennie and Vaughan (1992) under dry conditions. The authors reported an activation energy of 253 ± 8 kJ mol−1. Their extrapolations suggested that the half-life for this transformation is 3 × 106 years at 200 °C and 1.3 × 109 years at 160 °C. In the present study, we found a higher activation energy of 380 ± 15 kJ mol−1, and based on the same extrapolation, the half-life for the transformation is much longer, 3.2 × 1013 years at 200 °C and 2.4 × 1017 years at 160 °C. Even at a higher temperature of 300 °C, the half-life is long, 1.5 × 106 years. The difference in the estimated reaction rates from the two studies demonstrates that the extrapolation of the results obtained at higher temperatures using kinetic parameters for the estimations of reaction rates at lower temperatures may not be correct. This is particularly true for the transformation from marcasite to pyrite, where the reaction mechanism depends on several factors such as particle size, the presence of water vapor, and pyrite inclusions in the primary marcasite. There might be additional factors influencing the mechanism and kinetics of the transformation. For example, marcasite from mineral replacement reactions of pyrrhotite is often porous (Qian et al. 2011), but the role of these pores, which may act as traps for fluids, on the transformation from marcasite to pyrite is yet to be understood.
Trace elements mobilization during the transformation
Marcasite and pyrite incorporate from few parts per million (ppm) up to wt% levels of trace and/or minor elements (e.g., Deditius et al. 2014; Franchini et al. 2015). Subsequently, these elements can be liberated from the FeS2 structure due to recrystallization and/or replacement reactions (e.g., Sung et al. 2009; Deditius et al. 2011). This study revealed that the presence of water vapor and the amount of Pb in the starting marcasite affect the behavior of the metal during transformation to pyrite. No galena particles were found during the transformation of marcasite-CR containing 100–300 ppm Pb, irrespective of the water content (Fig. 8). When the content of Pb reached 1000 s of ppm in marcasite-M (mean value ~ 1100 ppm), nano- and microparticles of PbS were detected in the newly formed pyrite (Fig. 7). Under dry conditions the particles were randomly distributed in pyrite (Fig. 7b), while in the presence of water vapor galena particles precipitated in ordered arrays in the porous pyrite (Fig. 7d). The random distribution of PbS particles in pyrite formed under dry conditions at 520 °C suggests that they grew according to the Ostwald-type ripening process similar to the growth of Au nanoparticles in pyrite, which started at comparable temperature of 450 °C (Reich et al. 2006). Lack of association between galena particles and pores suggests that porosity does not induce the formation of PbS particles in the newly formed pyrite (Fig. 7b). These observations suggest that some of the metallic inclusions reported in pyrite formed under higher-temperature conditions could have formed during relatively dry metamorphism of marcasite/pyrite and during the transformation into more stable polymorph. It appears that porosity does play a role in the formation of galena and possibly other metallic particles during the transformation of trace element-rich marcasite to pyrite if hydrothermal fluid (vapor) is present (Fig. 7d). Such conditions are likely to occur in epithermal-porphyry systems during the injection of hotter hydrothermal fluids overprinting lower-temperature mineral assemblages (e.g., Williams-Jones and Heinrich 2005). The arrangement of galena particles along or within pores suggests that large Pb2+ ion (crystal radius 0.133 nm in octahedral coordination; Shannon 1976) as incompatible element was removed from the structure of pyrite and precipitated as galena in pores. These results demonstrate that the presence of a fluid phase can change the mechanism of metal mobilization in ore minerals.
Geological implications
Although marcasite is reported to form in sedimentary settings (Schieber 2007, 2011; Ruppert et al. 2005), epithermal deposits (Franchini et al. 2015), Carlin-type gold deposits (Fleet and Mumin 1997; Cline 2001), supergene deposits (Peterson 1965; Kelly and Turneaure 1970; Nickel et al. 1974; Einaudi 1977), VHMS deposits (Maslennikov et al. 2013; Tessalina et al. 2017), and deep sea hydrothermal vent mineralization (Goldfarb 1983; Koski et al. 1984; Hannington and Scott 1985; Schoonen and Barnes 1991a, b, c), its abundance in the Earth’s crust is much less than pyrite. Previous studies highlighted that pyrite and marcasite form under different hydrothermal conditions, i.e., low temperature, low pH, and S(-II)-deficient solutions (saturation index << 1000 with respect to either marcasite or pyrite) promote the formation of marcasite, while the opposite conditions (for example: pH > 5; saturation index > 1000 with respect to either marcasite or pyrite) favor the formation of pyrite (Qian et al. 2011; Schoonen and Barnes 1991a). The conditions for the formation of marcasite are not uncommon in natural environments, yet only minor amounts of marcasite were reported in the literature (Arehart 1996; Arehart et al. 1993; Su et al. 2009). This might reflect the fact that some studies underestimate the abundance of marcasite, as it is undistinguishable from pyrite when investigated by electron microscopy and chemical composition alone. The present study shows that the rapid transformation from marcasite to pyrite at higher temperatures (> 300 °C) may be the reason for limiting the occurrence of marcasite in geological records.
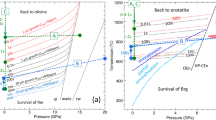
The occurrence of marcasite (Buerger 1934; Grønvold and Westrum 1976) is a useful indicator of low-temperature environments. Transition from porphyry to epithermal style of mineralization provides one of the best examples. Pyrite is the only FeS2 polymorph crystallizing during the early potassic and phyllic stages of alteration (350–550 °C). Marcasite becomes more abundant during later epithermal stages of mineralization in hydrothermal breccias, as exemplified by the Agua Rica deposit in Argentina (Franchini et al. 2015) (Fig. 15a). This means that a significant portion of marcasite survived after the cease of hydrothermal activity ~ 5 Ma ago in Agua Rica (Landtwing et al. 2002), due to the slow transformation from marcasite to pyrite at low temperatures (< 300 °C). The slow kinetics of the transformation from marcasite to pyrite at low temperatures explains the occurrence of marcasite in supergene deposits (Peterson 1965; Kelly and Turneaure 1970; Nickel et al. 1974; Einaudi 1977) and in marine sediments (Schieber 2007) (Fig. 15b), and also explains the complex intergrowth of marcasite and pyrite (Fig. 15c) in the 212–215 Ma Daqiao orogenic gold deposits (China) (Wu et al. 2018, and references therein) at temperatures below 310 °C (Liu et al. 2011, based on fluid inclusions) and particularly after the subsequent Au deposition at 100–240 °C (based on the geothermal gradient) (Xu et al. 2015; Wu et al. 2018, 2019). In 363–363 Ma old VHMS deposit at Yaman-Kasy (Russia) (Tessalina et al. 2017), the appearance of marcasite and coexistence with colloform pyrite indicate lower-temperature conditions (80–200 °C) of the zoned black smokers where hydrothermal fluid mixed with seawater. Lack of subsequent higher-grade metamorphism and thermal overprint at Yaman-Kasy allowed for the preservation of marcasite in this deposit. On the other hand, the pseudomorphosis of pyrite after marcasite may suggest the higher- temperature environment followed initial low-temperature condition (Gait and Dumka 1986) and explains the dominance of pyrite in some metamorphic rocks (Vokes 1993).

Reflective light microphotographs showing marcasite-pyrite intergrowths in natural samples from a porphyry-high sulfidation epithermal deposit at Catamarca, Argentina (modified after Franchini et al. 2015), b marine lag deposit at Eastern US (the white arrows point to corrosion front; modified after Schieber 2007), and c orogenic gold deposit at West Qinling Orogen, China (modified after Wu et al. 2018). Mrc marcasite, Py pyrite, Qz quartz, Cv covellite. (For color figure, the reader is referred to the web version of this paper.)
The present study also suggests that the solid-state phase transformation from marcasite to pyrite can produce textures that resemble those of fluid-mediated mineral replacement reactions. The presence of porosity in the product phase and the sharp phase boundary between parental and product phases (Figs. 7, 8) are strong indicators of fluid-mediated mineral replacement reactions (Putnis 2009; Altree-Williams et al. 2015), but such sample texture is clearly seen in the solid-state transformation from marcasite to pyrite. Hence, interpretation of the reaction mechanism based solely on the textures may lead to incorrect conclusions.
Conclusions
This study combines in situ and ex situ investigations to provide new insights into the solid-state transformation from marcasite to pyrite. The kinetic analysis using the Avrami–Arrhenius method gives an activation energy of 380 ± 15 kJ mol−1 for the transformation under dry conditions, and the extrapolation to lower temperatures (300 °C, 200 °C, and 160 °C) suggests that marcasite may be considered as a kinetically stable phase below 300 °C at the geological time scale. However, it is important to acknowledge that, in addition to temperature, the mechanism and the consequent kinetics of this phase transformation are also dependent on particle size, the presence of water vapor, and pyrite inclusions in marcasite grains. Under dry conditions, the transformation is likely to be initiated by epitaxial pyrite nucleation on the marcasite surface and thus the growth of pyrite is likely to be controlled by phase boundary reactions, and hence the rate of transformation is inversely proportional to particle size. In the presence of water vapor, the transformation is also initiated by limited nucleation, but is not facilitated by epitaxial growth. Instead, diffusion in water vapor may play a more important role in determining the transformation kinetics. The presence of water vapor facilitates the precipitation of metal-bearing nano- to microparticles in pores in sulfides under high-temperature conditions and thus facilitates the mobilization of trace metals in ore deposits. This study highlights the complexity of the transformation from marcasite to pyrite and suggests that, although the natural occurrence of marcasite can indicate low-temperature environments, precise estimation of temperature is difficult.
References
Altree-Williams A, Pring A, Ngothai Y, Brugger J (2015) Textural and compositional complexities resulting from coupled dissolution–reprecipitation reactions in geomaterials. Earth Sci Rev 150:628–651. https://doi.org/10.1016/j.earscirev.2015.08.013
Anderson HV, Chesley KG (1933) X-ray study of the transformation of marcasite into pyrite. Am J Sci 25:315–324. https://doi.org/10.2475/ajs.s5-25.148.315
Arehart GB (1996) Characteristics and origin of sediment-hosted disseminated gold deposits: a review. Ore Geol Rev 11(6):383–403. https://doi.org/10.1016/S0169-1368(96)00010-8
Arehart GB, Eldridge CS, Chryssoulis SL, Kesler SE (1993) Ion microprobe determination of sulfur isotope variations in iron sulfides from the Post/Betze sediment-hosted disseminated gold deposit, Nevada, USA. Geochim Cosmochim Acta 57(7):1505–1519. https://doi.org/10.1016/0016-7037(93)90010-T
Armstrong JT (1988) Quantitative analysis of silicates and oxide minerals: comparison of Monte-Carlo, ZAF and Phi-Rho-Z procedures. In: Newbury DE (ed) Microbeam analysis: San Francisco. San Francisco Press, California, pp 239–246
Avrami M (1939) Kinetics of phase change. I General theory. J Chem Phys 7:1103–1112. https://doi.org/10.1063/1.1750380
Bayliss P (1977) Crystal structure refinement of a weakly anisotropic pyrite. Am Mineral 62:1168–1172
Buerger MJ (1934) The pyrite-marcasite relation. Am Mineral 19:37–61
Burton ED, Bush RT, Sullivan LA, Johnston SG, Hocking RK (2008) Mobility of arsenic and selected metals during re-flooding of iron-and organic-rich acid-sulfate soil. Chem Geol 253(1–2):64–73. https://doi.org/10.1016/j.chemgeo.2008.04.006
Cline JS (2001) Timing of gold and arsenic sulfide mineral deposition at the Getchell Carlin-type gold deposit, north-central Nevada. Econ Geol 96:75–89. https://doi.org/10.2113/gsecongeo.96.1.75
Cook NJ, Chryssoulis SL (1990) Concentrations of invisible gold in the common sulfides. Can Mineral 28:1–16
Deditius AP, Utsunomiya S, Reich M, Kesler SE, Ewing RC, Hough R, Walshe J (2011) Trace metal nanoparticles in pyrite. Ore Geol Rev 42:32–46. https://doi.org/10.1016/j.oregeorev.2011.03.003
Deditius AP, Reich M, Kesler SE, Utsunomiya S, Chryssoulis SL, Walshe J, Ewing RC (2014) The coupled geochemistry of Au and As in pyrite from hydrothermal ore deposits. Geochim Cosmochim Acta 140:644–670. https://doi.org/10.1016/j.gca.2014.05.045
Donovan JJ, Tingle TN (1996) An improved mean atomic number correction for quantitative microanalysis. J Micros 2(1):1–7. https://doi.org/10.1017/S1431927696210013
Einaudi MT (1977) Environment of ore deposition at Cerro de Pasco. Peru Econ Geol 72(6):893–924. https://doi.org/10.2113/gsecongeo.72.6.893
Etschmann BE, Pring A, Putnis A, Grguric BA, Studer A (2004) A kinetic study of the exsolution of pentlandite (Ni, Fe)9S8 from the monosulfide solid solution (Fe, Ni)S. Am Mineral 89(1):39–50. https://doi.org/10.2138/am-2004-0106
Fan HH, Li HH, Huang KC, Fan CY, Zhang XY, Wu XL, Zhang JP (2017) Metastable marcasite–FeS2 as a new anode material for lithium ion batteries: CNFs-improved lithiation/delithiation reversibility and Li-storage properties. ACS Appl Mater Inter 9(12):10708–10716. https://doi.org/10.1021/acsami.7b00578
Fleet ME (1970) Structural aspects of the marcasite–pyrite transformation. Can Mineral 10:225–231
Fleet ME, Mumin AH (1997) Gold-bearing arsenian pyrite and marcasite and arsenopyrite from Carlin Trend gold deposits and laboratory synthesis. Am Mineral 82:182–193. https://doi.org/10.2138/am-1997-1-220
Franchini M, McFarlane C, Maydagán L, Reich M, Lentz DR, Meinert L, Bouhier V (2015) Trace metals in pyrite and marcasite from the Agua Rica porphyry-high sulfidation epithermal deposit, Catamarca, Argentina: Textural features and metal zoning at the porphyry to epithermal transition. Ore Geol Rev 66:366–387. https://doi.org/10.1016/j.oregeorev.2014.10.022
Gait RI, Dumka D (1986) Morphology of pyrite from the Nanisivik mine, Baffin Island, Northwest territories. Can Mineral 24:685–688
Goldfarb MS (1983) The genesis of hot spring deposits on the East Pacific Rise 21ºN. The Kuroko and Related Volcanogenetic Massive Deposits. Econ Geol Monograph 5:184–197
Gražulis S, Chateigner D, Downs RT, Yokochi AFT, Quirós M, Lutterotti L, Manakova E, Butkus J, Moeck P, Le Bail A (2009) Crystallography open database—an open-access collection of crystal structures. J Appl Crystallogr 42:726–729. https://doi.org/10.1107/S0021889809016690
Grønvold F, Westrum EF (1976) Heat capacities of iron disulfides Thermodynamics of marcasite from 5 to 700 K, pyrite from 300 to 780 K, and the transformation of marcasite to pyrite. J Chem Thermodyn 8:1039–1048. https://doi.org/10.1016/0021-9614(76)90135-X
Gudelli VK, Kanchana V, Appalakondaiah S, Vaitheeswaran G, Valsakumar MC (2013) Phase stability and thermoelectric properties of the mineral FeS2: an Ab initio study. J Phys Chem C 117(41):21120–21131. https://doi.org/10.1021/jp406928v
Hancock JD, Sharp JH (1972) Method of comparing solid-state kinetic data and its application to the decomposition of kaolinite, brucite, and BaCO3. J Am Ceram Soc 55:74–77. https://doi.org/10.1111/j.1151-2916.1972.tb11213.x
Hannington MD, Scott SD (1985) A caldera-hosted silicasulfate-sulfide deposit, Axial seamount, central Juan de Fuca Ridge, N. E. Pacific Ocean. Geol Assoc Canada 10:24 (abstr.)
Hill RJ, Howard CJ (1987) Quantitative phase analysis from neutron powder diffraction data using the Rietveld method. J Appl Crystallogr 20:467–474. https://doi.org/10.1107/S0021889887086199
Hulbert SF (1969) Models for solid-state reactions in powdered compacts—a review. J Brit Ceram Soc 6:11–20
Hyde B, O'Keeffe M (1996) Marcasite and pyrite (FeS2). Aust J Chem 49:867–872. https://doi.org/10.1071/CH9960867
Kelly WC, Turneaure FS (1970) Mineralogy, paragenesis and geothermometry of the tin and tungsten deposits of the eastern Andes. Bolivia Econ Geol 65(6):609–680. https://doi.org/10.2113/gsecongeo.65.6.609
Khalid S, Ahmed E, Khan Y, Riaz KN, Malik MA (2018) Nanocrystalline pyrite for photovoltaic applications. Chem Select 3(23):6488–6524. https://doi.org/10.1002/slct.201800405
Khanna YP, Taylor TJ (1988) Comments and recommendations on the use of the Avrami equation for physico-chemical kinetics. Polym Eng Sci 28:1042–1045. https://doi.org/10.1002/pen.760281605
Kirkham R, Dunn P, Kuczewski A, Siddons D, Dodanwela R, Moor-head G, Ryan C, De Geronimo G, Beuttenmuller R, Pinelli D (2010) The Maia spectroscopy detector system: engineering for integrated pulse capture, low-latency scanning and real-time processing. AIP Conf Ser 1234:240–243. https://doi.org/10.1063/1.3463181
Kjekshus A, Rakke T (1975) Compounds with the marcasite type crystal structure. XI. High temperature studies of chalcogenides. Acta Chem Scand 29(a):443–452
Koski RA, Clague DA, Oudin E (1984) Mineralogy and chemistry of massive sulfide deposits from the Juan de Fuca Ridge. Geol Soc Am Bull 95(8):930–945. https://doi.org/10.1130/0016-7606(1984)95%3c930:MACOMS%3e2.0.CO;2
Krakow R, Bennett RJ, Johnstone DN, Vukmanovic Z, Solano-Alvarez W, Lainé SJ, Einsle JF, Midgley PA, Rae CMF, Hielscher R (2017) On three-dimensional misorientation spaces. Proc R Soc A 473(2206):20170274. https://doi.org/10.1098/rspa.2017.0274
Kullerud G, Yoder HS (1959) Pyrite stability relations in the Fe–S system. Econ Geol 54(4):533–572. https://doi.org/10.2113/gsecongeo.54.4.533
Kusebauch C, Gleeson SA, Oelze M (2019) Coupled partitioning of Au and As into pyrite controls formation of giant Au deposits. Sci Adv 5(5):5891. https://doi.org/10.1126/sciadv.aav5891
Lambert JM, Simkovich G, Walker PL (1998) The kinetics and mechanism of the pyrite-to-pyrrhotite transformation. Metall Mater Trans B 29(2):385–396. https://doi.org/10.1007/s11663-998-0115-x
Landtwing M, Dillenbeck E, Leake M, Heinrich C (2002) Evolution of the breccia-hosted porphyry-Cu–Mo–Au deposit at Agua Rica, Argentina: progressive unroofing of a magmatic–hydrothermal system. Econ Geol 97:1273–1292. https://doi.org/10.2113/gsecongeo.97.6.1273
Large RR, Maslennikov VV, Robert F, Danyushevsky LV, Chang Z (2007) Multistage sedimentary and metamorphic origin of pyrite and gold in the giant Sukhoi Log deposit, Lena gold province, Russia. Econ Geol 102:1233–1267. https://doi.org/10.2113/gsecongeo.102.7.1233
Large RR, Halpin JA, Danyushevsky LV, Maslennikov VV, Bull SW, Long JA, Gregory DD, Lounejeva E, Lyons TW, Sack PJ, McGoldrick PJ, Calver CR (2014) Trace element content of sedimentary pyrite as a new proxy for deep-time ocean–atmosphere evolution. Earth Planet Sci Lett 389:209–220. https://doi.org/10.1016/j.epsl.2013.12.020
Lasaga AC (1998) Kinetic theory in the earth sciences, 1st edn. Princeton Univ. Press, Princeton
Lennie AR, Vaughan DJ (1992) Kinetics of the marcasite–pyrite transformation: an infrared spectroscopic study. Am Mineral 77:1166–1171
Li T, Guo Z, Li X, Wu Z, Zhang K, Liu H, Sun H, Liu Y, Zhang H (2015) Colloidal synthesis of marcasite FeS2 nanoparticles with improved electrochemical performance. RSC Adv 5(120):98967–98970. https://doi.org/10.1039/C5RA22610D
Li K, Etschmann BE, Rae N, Reith F, Ryan CG, Kirkham R, Howard D, Rosa D, Zammit C, Pring A, Ngothai Y, Hooker A, Brugger J (2016) Ore petrography using megapixel X-ray imaging: rapid insights into element distribution and mobilisation in complex Pt and U–Ge–Cu ores. Econ Geol 111:487–501. https://doi.org/10.2113/econgeo.111.2.487
Lipin SV, Uskov VS, Klokman VR (1942) Heat of iron disulphide formation. J Appl Chem (Russian) 15:411–421
Liu YG, Lv XB, Zhang ZJ, You GJ, Cao XF (2011) Genesis of Daqiao gold deposit in Xihe County, Gansu province. Min Deposit 6:1085–1099 (in Chinese with English abs.)
Masslennikov VV, Masslennikova SP, Large RR, Danyushevsky LV, Herrington RJ, Stanley CJ (2013) Tellurium-bearing minerals in zoned sulfide chimneys from Cu–Zn massive sulfide deposits of the Urals, Russia. Miner Petrol 107:67–99. https://doi.org/10.1007/s00710-012-0230-x
Milke R, Neusser G, Kolzer K, Wunder B (2013) Very little water is necessary to make a dry solid silicate system wet. Geology 41(2):247–250. https://doi.org/10.1130/G33674.1
Murowchick JB (1992) Marcasite inversion and the petrographic determination of pyrite ancestry. Econ Geol 87(4):1141–1152. https://doi.org/10.2113/gsecongeo.87.4.1141
Murowchick JB, Barnes HL (1986) Marcasite precipitation from hydrothermal solutions. Geochim Cosmochim Acta 50:2615–2629. https://doi.org/10.1016/0016-7037(86)90214-0
Murray JW, Jannasch HW, Honjo S, Anderson RF, Reeburgh WS, Top Z, Friederich GE, Codispoti LA, Izdar E (1989) Unexpected changes in the oxic/anoxic interface in the Black Sea. Nature 338(6214):411. https://doi.org/10.1038/338411a0
Nickel EH, Ross JR, Thornber MR (1974) The supergene alteration of pyrrhotite-pentlandite ore at Kambalda Western Australia. Econ Geol 69(1):93–107. https://doi.org/10.2113/gsecongeo.69.1.93
Paterson D, De Jonge M, Howard D, Lewis W, McKinlay J, Starritt A, Kusel M, Ryan C, Kirkham R, Moorhead G (2011) The X-ray fluorescence microscopy beamline at the Australian synchrotron. AIP Conf Ser. https://doi.org/10.1063/1.3625343
Pearce MA (2015) EBSDinterp 1.0: a MATLAB® program to perform microstructurally constrained interpolation of EBSD data. Microsc Microanal 21(4):985–993. https://doi.org/10.4225/08/5510090C6E620
Pecharsky VK, Zavalij PY (2003) Fundamentals of powder diffraction and structural characterization of materials. Kluwere Academic Publishers, Boston, p 713
Petersen U (1965) Regional geology and major ore deposits of central Peru. Econ Geol 60(3):407–476. https://doi.org/10.2113/gsecongeo.60.3.407
Peterson KM, Heaney PJ, Post JE (2016) A kinetic analysis of the transformation from akaganeite to hematite: an in situ time-resolved X-ray diffraction study. Chem Geol 444:27–36. https://doi.org/10.1016/j.chemgeo.2016.09.017
Prior DJ, Mariani E, Wheeler J (2009) EBSD in the earth sciences: applications, common practice, and challenges. Springer, Boston, pp 345–360
Putnis A (2009) Mineral replacement reactions. Rev Mineral Geochem 70:87–124. https://doi.org/10.2138/rmg.2009.70.3
Putnis CV, Mezger K (2004) A mechanism of mineral replacement: Isotope tracing in the model system KCl–KBr–H2O. Geochim Cosmochim Acta 68:2839–2848. https://doi.org/10.1016/j.gca.2003.12.009
Qian G, Xia F, Brugger J, Skinner WM, Bei J, Chen G, Pring A (2011) Replacement of pyrrhotite by pyrite and marcasite under hydrothermal conditions up to 220 ºC: an experimental study of reaction textures and mechanisms. Am Mineral 96(11–12):1878–1893. https://doi.org/10.2138/am.2011.3691
Reich M, Utsunomiya S, Kesler SE, Wang L, Ewing RC, Becker U (2006) Thermal behavior of metal nanoparticles in geologic materials. Geology 34:1033–1036. https://doi.org/10.1130/G22829A
Rickard D, Luther GW (2007) Chemistry of iron sulfides. Chem Rev 107:514–562. https://doi.org/10.1021/cr0503658
Rieder M, Crelling JC, Šustai O, Drábek M, Weiss Z, Klementová M (2007) Arsenic in iron disulfides in a brown coal from the North Bohemian Basin Czech Republic. Int J Coal Geol 71(2–3):115–121. https://doi.org/10.1016/j.coal.2006.07.003
Rising BA (1973) Phase relations among pyrite, marcasite and pyrrhotite below 300 °C. Dissertation, Pennsylvania State University, pp 192
Ruppert LF, Hower JC, Eble CF (2005) Arsenic-bearing pyrite and marcasite in the Fire Clay coal bed, Middle Pennsylvanian Breathitt Formation, eastern Kentucky. Int J Coal Geol 63(1–2):27–35. https://doi.org/10.1016/j.coal.2005.02.003
Ryan C (2000) Quantitative trace element imaging using PIXE and the nuclear microprobe. Int J Imag Syst Tech 11:219–230. https://doi.org/10.1002/ima.1007
Ryan CG, Etschmann B, Vogt S, Maser J, Harland C, Van Achterbergh E, Legnini D (2005) Nuclear microprobe-synchrotron synergy: towards integrated quantitative real-time elemental imaging using PIXE and SXRF. Nucl Instrum Methods Phys Res B 231:183–188. https://doi.org/10.1016/j.nimb.2005.01.054
Ryan C, Siddons D, Moorhead G, Kirkham R, De Geronimo G, Etschmann B, Dragone A, Dunn P, Kuczewski A, Davey P (2009) High-throughput X-ray fluorescence imaging using a massively parallel detector array, integrated scanning and real-time spectral deconvolution. J Phys Conf Ser 186:012–013. https://doi.org/10.1088/1742-6596/186/1/012013
Ryan C, Kirkham R, Hough R, Moorhead G, Siddons D, De Jonge M, Paterson D, De Geronimo G, Howard D, Cleverley J (2010a) Elemental X-ray imaging using the Maia detector array: the benefits and challenges of large solid-angle. Nucl Instrum Methods Phys Res A 619:37–43. https://doi.org/10.1016/j.nima.2009.11.035
Ryan C, Siddons D, Kirkham R, Dunn P, Kuczewski A, Moorhead G, De Geronimo G, Paterson D, De Jonge M, Hough R, Lintern M, Howard D, Kappen P, Cleverley J (2010b) The new MAIA detector system: methods for high definition trace element imaging of natural material. Am Inst Phys Conf Ser 1221:9–17. https://doi.org/10.1063/1.3399266
Ryan C, Siddons D, Kirkham R, Li Z, De Jonge M, Paterson D, Kuczewski A, Howard D, Dunn P, Falkenberg G (2014) MAIA X-ray fluorescence imaging: capturing detail in complex natural samples. J Phys Conf Ser. https://doi.org/10.1088/1742-6596/499/1/012002
Schieber J (2007) Oxidation of detrital pyrite as a cause for Marcasite formation in marine lag deposits from the devonian of the Eastern US. Deep Sea Res 54:1312–1326. https://doi.org/10.1016/j.dsr2.2007.04.005
Schieber J (2011) Marcasite in black shales—a mineral proxy for oxygenated bottom waters and intermittent oxidation of carbonaceous muds. J Sediment Res 81(7):447–458. https://doi.org/10.2110/jsr.2011.41
Schoonen MAA, Barnes HL (1991) Mechanisms of pyrite and marcasite formation from solution: III. Hydrothermal processes. Geochim Cosmochim Acta 55(12):3491–3504. https://doi.org/10.1016/0016-7037(91)90050-F
Schoonen MAA, Barnes HL (1991b) Reactions forming pyrite and marcasite from solution: I. Nucleation of FeS2 below 100 °C. Geochim Cosmochim Acta 55(6):1495–1504. https://doi.org/10.1016/0016-7037(91)90122-L
Schoonen MAA, Barnes HL (1991c) Reactions forming pyrite and marcasite from solution: II. Via FeS precursors below 100 °C. Geochim Cosmochim Acta 55(6):1505–1514. https://doi.org/10.1016/0016-7037(91)90123-M
Shannon RD (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst A32:751–767. https://doi.org/10.1107/S0567739476001551
Song J, Xia F, Zhao M, Zhong YL, Li W, Loh KP, Bao Q (2015) Solvothermal growth of bismuth chalcogenide nanoplatelets by the oriented attachment mechanism: an in situ PXRD study. Chem Mater 27:3471–3482. https://doi.org/10.1021/acs.chemmater.5b00903
Spagnoli D, Refson K, Wright K, Gale JD (2010) Density functional theory study of the relative stability of the iron disulfide polymorphs pyrite and marcasite. Phys Rev B 81:094–106. https://doi.org/10.1103/PhysRevB.81.094106
Storey CD, Prior DJ (2005) Plastic deformation and recrystallization of garnet: a mechanism to facilitate diffusion creep. J Petrol 46(12):2593–2613. https://doi.org/10.1093/petrology/egi067
Su W, Heinrich CA, Pettke T, Zhang X, Hu R, Xia B (2009) Sediment-hosted gold deposits in Guizhou, China: products of wall-rock sulfidation by deep crustal fluids. Econ Geol 104(1):73–93. https://doi.org/10.2113/gsecongeo.104.1.73
Sung Y-H, Brugger J, Ciobanu CL, Pring A, SkinnerW NM (2009) Invisible gold in arsenian pyrite and arsenopyrite from a multistage Archean gold deposit: Sunrise Dam Eastern Goldfields Province, Western Australia. Mineralium Dep 44:765–791. https://doi.org/10.1007/s00126-009-0244-4
Tessalina SG, Jurdan F, Belogub EV (2017) Significance of Late Devonian—lower carboniferous ages of hydrothermal suphides and sericites from the Urals Volcanic-Hosted Massive Sulphide deposits. Ore Geol Rev 85:131–139. https://doi.org/10.1016/j.oregeorev.2016.08.005
Vokes FM (1993) The metamorphism of pyrite and pyritic ores: an overview. Mineral Mag 57:3–18. https://doi.org/10.1180/minmag.1993.057.386.02
Wang H, Pring A, Ngothai Y, O'Neill B (2005) A low-temperature kinetic study of the exsolution of pentlandite from the monosulfide solid solution using a refined Avrami method. Geochim Cosmochim Acta 69:415–425. https://doi.org/10.1016/j.gca.2004.06.031
Webster NAS, Pownceby MI, Madsen IC, Kimpton JA (2013) Effect of oxygen partial pressure on the formation mechanisms of complex Ca-rich ferrites. ISIJ Int 53(5):774–781. https://doi.org/10.2355/isijinternational.53.774
Williams-Jones AE, Heinrich CA (2005) Vapor transport of metals and the formation of magmatic-hydrothermal ore deposits. Economic Geology 100th Anniversary Volume 1905–2005 100:1287–1312
Wu R, Zheng YF, Zhang XG, Sun YF, Xu JB, Jian JK (2004) Hydrothermal synthesis and crystal structure of pyrite. J Cryst Growth 266(4):523–527. https://doi.org/10.1016/j.jcrysgro.2004.02.020
Wu YF, Li JW, Evans K, Koenig AE, Li ZK, O’Brien H, Lahaye Y, Rempel K, Hu SY, Zhang ZP, Yu JP (2018) Ore-forming processes of the Daqiao epizonal orogenic gold deposit, West Qinling orogen, China: constraints from textures, trace elements, and sulfur isotopes of pyrite and marcasite, and raman spectroscopy of carbonaceous material. Econ Geol 113:1093–1132. https://doi.org/10.5382/econgeo.2018.4583
Wu YF, Evans K, Li JW, Fougerouse D, Large RR, Guagliardo P (2019) Metal remobilization and ore-fluid perturbation during episodic replacement of auriferous pyrite from an epizonal orogenic gold deposit. Geochim Cosmochim Acta 245:98–117. https://doi.org/10.1016/j.gca.2018.10.031
Xia F, Zhou J, Pring A, Ngothai Y, O’Neill B, Brugger J, Chen G, Colby C (2007) The role of pyrrhotite (Fe7S8) and the sample texture in the hydrothermal transformation of pentlandite ((Fe, Ni)9S8) to violarite ((Ni, Fe)3S4). React Kinet Catal L 92(2):257–266. https://doi.org/10.1007/s11144-007-5197-y
Xia F, Zhou J, Brugger J, Ngothai Y, O’Neill B, Chen G, Pring A (2008) Novel route to synthesize complex metal sulfides: hydrothermal coupled dissolution−reprecipitation replacement reactions. Chem Mater 20(8):2809–2817. https://doi.org/10.1021/cm7033883
Xia F, Brugger J, Chen G, Ngothai Y, O’Neill B, Putnis A, Pring A (2009) Mechanism and kinetics of pseudomorphic mineral replacement reactions: a case study of the replacement of pentlandite by violarite. Geochim Cosmochim Acta 73(7):1945–1969. https://doi.org/10.1016/j.gca.2009.01.007
Xia F, Pring A, Brugger J (2012) Understanding the mechanism and kinetics of pentlandite oxidation in extractive pyrometallurgy of nickel. Miner Eng 27:11–19. https://doi.org/10.1016/j.mineng.2011.12.001
Xia F, Chen D, Scarlett NV, Madsen IC, Lau D, Leoni M, Caruso RA (2014) Understanding solvothermal crystallization of mesoporous anatase beads by in situ synchrotron PXRD and SAXS. Chem Mater 26:4563–4571. https://doi.org/10.1021/cm501810x
Xing Y, Brugger J, Tomkins A, Shvarov Y (2019) Arsenic evolution as a tool for understanding formation of pyritic gold ores. Geology 47(4):335–338. https://doi.org/10.1130/G45708.1
Xu L, Wu BX, Wang YL, Wang ZX, Wang G, Sun ZP (2015) Fluid inclusion characteristics and geological significance at Daqiao gold deposit. J Jinlin Univ (Earth Sci Ed) 45:568–569 (in Chinese with English abs.)
Zhao J, Brugger J, Grguric BA, Ngothai Y, Pring A (2017) Fluid-Enhanced coarsening of mineral microstructures in hydrothermally synthesized bornite–digenite solid solution. ACS Earth Space Chem 1:465–474. https://doi.org/10.1021/acsearthspacechem.7b00034
Acknowledgements
This work was funded by the Australian Research Council through a discovery project (DP170101893). The authors acknowledge the Australian Microscopy & Microanalysis Research Facility (AMMRF) at the Centre for Microscopy, Characterization and Analysis, The University of Western Australia, for access to SEM and EPMA, and The Australian Synchrotron (part of ANSTO) for access to the Powder Diffraction and X-ray Florescence beamlines. The authors are also grateful to Alexandra Suvorova and Malcolm Roberts for technical assistance on SEM and EPMA and wish to thank Fatemah Nikkhou and Rahul Ram for helping with synchrotron power diffraction data collection, and Australian Synchrotron staff Justin Kimpton and Daryl Howard for setting up the beamlines. The authors are also indebted to Associate Editor Prof. Steve Reddy, reviewer Dr. Jeff Steadman, and one anonymous reviewer for constructive comments that improved the manuscript. Xizhi Yao acknowledges Murdoch University for providing a Murdoch University Strategic Scholarship (MUSS).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Steven Reddy.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yao, X., Xia, F., Deditius, A.P. et al. The mechanism and kinetics of the transformation from marcasite to pyrite: in situ and ex situ experiments and geological implications. Contrib Mineral Petrol 175, 27 (2020). https://doi.org/10.1007/s00410-020-1665-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-020-1665-4