
Abstract
Experiments at high pressures and temperatures were carried out (1) to investigate the crystal-chemical behaviour of Fe4O5–Mg2Fe2O5 solid solutions and (2) to explore the phase relations involving (Mg,Fe)2Fe2O5 (denoted as O5-phase) and Mg–Fe silicates. Multi-anvil experiments were performed at 11–20 GPa and 1100–1600 °C using different starting compositions including two that were Si-bearing. In Si-free experiments the O5-phase coexists with Fe2O3, hp-(Mg,Fe)Fe2O4, (Mg,Fe)3Fe4O9 or an unquenchable phase of different stoichiometry. Si-bearing experiments yielded phase assemblages consisting of the O5-phase together with olivine, wadsleyite or ringwoodite, majoritic garnet or Fe3+-bearing phase B. However, (Mg,Fe)2Fe2O5 does not incorporate Si. Electron microprobe analyses revealed that phase B incorporates significant amounts of Fe2+ and Fe3+ (at least ~ 1.0 cations Fe per formula unit). Fe-L2,3-edge energy-loss near-edge structure spectra confirm the presence of ferric iron [Fe3+/Fetot = ~ 0.41(4)] and indicate substitution according to the following charge-balanced exchange: [4]Si4+ + [6]Mg2+ = 2Fe3+. The ability to accommodate Fe2+ and Fe3+ makes this potential “water-storing” mineral interesting since such substitutions should enlarge its stability field. The thermodynamic properties of Mg2Fe2O5 have been refined, yielding H°1bar,298 = − 1981.5 kJ mol− 1. Solid solution is complete across the Fe4O5–Mg2Fe2O5 binary. Molar volume decreases essentially linearly with increasing Mg content, consistent with ideal mixing behaviour. The partitioning of Mg and Fe2+ with silicates indicates that (Mg,Fe)2Fe2O5 has a strong preference for Fe2+. Modelling of partitioning with olivine is consistent with the O5-phase exhibiting ideal mixing behaviour. Mg–Fe2+ partitioning between (Mg,Fe)2Fe2O5 and ringwoodite or wadsleyite is influenced by the presence of Fe3+ and OH incorporation in the silicate phases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the past few years, post-spinel phases have attracted more and more attention due to the discovery of several high-pressure (hp) oxide phases with stoichiometries other than M3O4. Phases with M4O5, M5O6 and M7O9 stoichiometry have now been added to the list of potential post-spinel phases that might be present in the Earth’s deep upper mantle and transition zone (e.g., Enomoto et al. 2009; Lavina et al. 2011; Lavina and Meng 2015; Woodland et al. 2012, 2013, 2015; Guignard and Crichton 2014; Ishii et al. 2014, 2015; Myhill et al. 2016; Sinmyo et al. 2016; Uenver-Thiele et al. 2017a, b). Phases with an M4O5 stoichiometry (O5-phase) were found in a number of simple chemical systems, including those containing Cr, Al, Fe3+ as trivalent cations, and Mg and Fe2+ as divalent cations (e.g., Enomoto et al. 2009; Woodland et al. 2012, 2013; Ishii et al. 2014, 2015; Uenver-Thiele et al. 2017a, b). Often the O5-phase occurs in an M4O5 + M2O3 post-spinel assemblage, as in the case of Fe4O5 + Fe2O3, FeMgFe2O5 + Fe2O3, Fe2Cr2O5 + Cr2O3 or Fe2Cr2O5 + Cr2O3 becoming stable at the expense of the spinel-structured phase at conditions of the deep upper mantle (Woodland et al. 2012; Ishii et al. 2014, 2015; Uenver-Thiele et al. 2017a, b).
Solid solutions involving the Fe4O5 component are of particular interest because the ability to contain both Fe2+ and Fe3+ makes their stability sensitive to redox processes. The incorporation of Mg into Fe4O5 was first reported by Woodland et al. (2013); Boffa Ballaran et al. (2015) subsequently demonstrated that Mg can completely substitute for Fe2+, producing an Mg endmember, Mg2Fe2O5. Both endmember compositions share the same space group Cmcm with a CaFe3O5-type structure (Lavina et al. 2011; Boffa Ballaran et al. 2015), suggesting that the entire Mg–Fe2+ solid solution series is stable at high pressures and temperatures. Myhill et al. (2016) questioned whether Fe4O5 could occur in a peridotitic upper mantle or transition zone environment since their preliminary calculations implied that its stability required an oxygen fugacity that exceeded the expected mantle range (e.g., Frost and McCammon 2008). However, such a conclusion was based upon incomplete knowledge of potential phase assemblages, particularly those with Mg-rich silicates. In addition, even if such a situation were generally true, this does not rule out the stability of an O5-phase in localized environments, such as those responsible for diamond formation where olivine and pyroxene do not coexist. In fact the microtexture of magnetite in an inclusion in diamond described by Jacob et al. (2016) led Uenver-Thiele et al. (2017b) to conclude that this magnetite was originally Fe4O5 at the conditions of diamond formation.
The recent investigations of the phase relations for MgFe2O4 and Mg0.5Fe0.5Fe2O4 by Uenver-Thiele et al. (2017a, b) also place important constraints on the stability of the Mg2Fe2O5 endmember and the MgFeFe2O5 composition. However, knowledge of the properties of (Mg,Fe)2Fe2O5 solid solutions and the phases with which they can coexist under mantle-relevant conditions remains incomplete. Thus, the purpose of this study is twofold: (1) to address the crystal-chemical behaviour of the Fe4O5–Mg2Fe2O5 join and (2) to further investigate the phase assemblages in which an O5-phase is stable, including those involving high-pressure Mg–Fe silicates.
Methods
High-pressure experiments
Five different starting compositions were employed in this study (Tables 1, 2). A starting mixture of MgO + MgFe2O4 in a 1:1 molar ratio as well as pre-synthesized MgFe2O4 and Mg0.5Fe0.5Fe2O4 were employed to investigate the crystal-chemical behaviour of solid solutions along the Fe4O5–Mg2Fe2O5 join (Table 1). The synthesis of magnesioferrite and Mg0.5Fe0.5Fe2O4 is reported in Uenver-Thiele et al. (2017a, b), respectively. A mixture of 70 mol% MgFe2O4 and 30 mol% Mg2SiO4 was used to investigate equilibria involving Mg2Fe2O5 and high-pressure Si-bearing phases (Table 2). Another starting mixture of 75 mol% MgSiO3 and 25 mol% Fe2O3 (Table 2), giving a composition of Mg3Fe2Si3O12, was produced by combining sintered hematite with enstatite glass. The glass was produced by melting a stoichiometric mixture of MgO and SiO2 in a Pt-crucible at 1660 °C using a high-temperature furnace for 1 h. It was quenched in ice water and re-grounded for a second cycle of melting at 1650 °C for a further 20 min. This procedure yielded a clear glass with no microscopic quench crystals.
Experiments up to 13 GPa were performed with a 800-t Walker-type multi-anvil press at the University of Frankfurt. Pressure calibration and pressure cell design are given in Brey et al. (2008). Temperature was monitored by W5Re95–W26Re74 thermocouples with the electromotive force uncorrected for pressure. Capsule designs follow those described in Uenver-Thiele et al. (2017a). For experiments at pressures ≥ 14 GPa, 5000-t and 1000-t multi-anvil presses were employed at the Bayerisches Geoinstitut, Bayreuth. The experimental setup and pressure calibration are described in Keppler and Frost (2005). High-pressure assemblies with a LaCrO3 heater were employed with the temperature monitored by W3Re97–W25Re75 thermocouples with the electromotive force uncorrected for pressure. Uncertainties in pressure and temperature are ± 0.5 GPa and ± 30–50 °C, respectively (Keppler and Frost 2005). Samples were packed into Pt-foil capsules, except for experiment Z1507o that employed Ag-foil as capsule material. Several experiments were conducted with small amounts of PtO2 to avoid reduction of ferric iron. In others, PtO2 was omitted since we intended to produce mixed-valence Fe3+–Fe2+-bearing solid solutions. The high oxygen fugacity of the experiments as imposed by the high Fe3+ contents of the starting materials meant that Fe-loss to the Pt capsule was negligible. The experiments were first cold pressurized, followed by heating to the desired temperature. They were terminated by turning off the power to the heater, but maintaining the pressure. Decompression was then immediately initiated. The resulting run products, as well as experimental run conditions are reported in Tables 1 and 2.
Analytical techniques
Recovered samples were characterized primarily by electron microprobe (EPMA) and powder X-ray diffraction, with selected samples also investigated by transmission electron microscopy (TEM) and micro-Raman spectrometry. Fragments of sample material were embedded in epoxy and polished for EPMA analysis. Measurements were performed at the University of Frankfurt using a five-spectrometer JEOL JXA-8900 superprobe operating in the wavelength-dispersive mode (at 15 kV, 20 nA, 1 µm spot size). Pure MgO, Fe2O3, Pt and CaSiO3 were used as primary standards. Integration times for Si and Mg were 40 s on the peak and background, while for Fe and Pt an integration time of 40 s on the peak and 20 s on the background was employed. For experiment Z1507o, forsterite (Mg), fayalite (Fe) and wollastonite (Si) were employed as standards. Integration times for Mg, Fe and Si were the same as described above. A CITZAF algorithm was employed for matrix correction (Armstrong 1993).
Powder X-ray diffraction patterns were obtained on a STOE Stadi P diffractometer operating at 45 kV and 35 mA with monochromatic Mo Kα (λ = 0.70926 Å) radiation, equipped with a linear PSD or a Mythen detector at the University of Frankfurt. An internal Si standard was added to the sample material and mounted in a 0.5-mm-diameter capillary. Diffraction patterns were collected in transmission mode between 1°–100° 2θ. Several samples were analysed using a Philips X´Pert PRO diffractometer at the Bayerisches Geoinstitut. Co Kα1 (λ = 1.78897 Å) radiation, monochromated with a symmetrically cut curved Johansson Ge (111) crystal and a Philips X’celerator detector were employed. A small amount of Si powder was added to the sample as an internal standard and mounted on a silicon wafer. Data collection was performed in reflection mode at 40 kV and 40 mA and between 10° and 120° 2θ. The diffraction patterns were analysed with the General Structure Analysis System (GSAS, Larson and van Dreele 1994) software package with the EXPGUI interface of Toby (2001) for phase identification and determination of the unit-cell parameters for the (Mg,Fe)2Fe2O5 solid solutions.
For selected samples, further phase identification was performed using a Renishaw micro-Raman spectrometer (RM-1000) equipped with a 633 nm HeNe laser at the University of Frankfurt. The 519 cm− 1 peak of a Si wafer was used for calibration (Temple and Hathaway 1973). Unpolarized confocal Raman spectra were collected with a 50x microscope objective and a laser power of ~ 10 mW. The wavenumber accuracy was about ± 2 cm− 1. The focused laser spot on the sample was approximately ~ 2 µm in diameter.
Transmission electron microscopic characterizations were carried out using a 200 kV Philips CM-20FEG TEM equipped with an energy-dispersive X-ray spectrometer (NO-RAN Ge detector) and a parallel electron energy-loss spectrometer (Gatan 666 PEELS). A small chip from sample Z1586u used in the X-ray diffraction measurement was crushed under ethanol and the dispersion was placed on a Lacey carbon film on Cu-grid. For textural as well as crystallographic investigations, conventional TEM techniques such as bright field (BF) and dark field (DF) imaging, as well as selected area electron diffraction (SAED) were employed. Chemical compositions were cross-checked by energy-dispersive X-ray (EDX) analyses. The Fe-L2,3-edge electron energy-loss near-edge structure (ELNES) of the selected sample was also examined using spectra collected in diffraction mode with a collection semi-angle of β = 2.7 mrad, an energy-dispersion of 0.1 eV per channel and 10–20 s integration time per read-out. The energy resolution is 0.8–0.9 eV, measured as the full width of the zero-loss peak at half-maximum. Quantification of the Fe-L2,3 ELNES followed the procedure described by van Aken et al. (1998) and van Aken and Liebscher (2002), using an empirically calibrated universal curve.
Results and discussion
Phase relations of (Fe,Mg)2Fe2O5 solid solutions
In order to investigate the crystal-chemical behaviour of Fe4O5–Mg2Fe2O5 solid solutions, six additional experiments were performed in the pressure–temperature range of 12–20 GPa and 1300–1600 °C to augment the data of Woodland et al. (2013) and Uenver-Thiele et al. (2017a, b). The conditions and run products from the experiments are summarized in Table 1, along with those experiments from Uenver-Thiele et al. (2017a, b) for which unit-cell parameters and the molar volumes for the O5-phase are presented here for the first time. Representative electron microprobe analyses of each phase synthesized in the six new experiments are listed in Table 3.
In most experiments, the O5-phase occurs with hematite, except for two cases where an “O5” bulk composition was used (Table 1; samples Z1584u and Z1234u). Some samples contain a coexisting unquenchable phase (UQ) that exhibits a mottled texture in backscattered electron (BSE) images (Fig. 1a–c), indicating breakdown during decompression. However, the UQ-phase is marked by straight grain boundaries of larger crystals with 120° triple junctions implying that equilibrium was attained during the experiment (see Fig. 1a-c). The bulk composition of the UQ-phase was determined from 4 µm diameter spot analyses, revealing homogenous compositions with only minor variations for each sample (Table 3). Different stoichiometries were obtained (Tables 1, 3) depending on the experimental run conditions. For example, in experiment M519 (Mg,Fe)2Fe2O5 and hematite occur up to 30 µm in size and coexist together with an interstitial UQ-phase (Table 1; Fig. 1b, c). However, the mottled texture indicative of decomposition is not complete and relict domains of the original phase are apparent in BSE images (e.g., Fig. 1c). Microprobe analyses were performed in both parts of these grains; a “normal” spot size (1 µm) for the un-mottled part and a larger 4 µm spot for the mottled region (Fig. 1c). The compositions of these two regions are in perfect agreement with each other, yielding 38.97(25) wt% MgO and 52.88(44) FeO for the relict parts and 39.35(4) wt% MgO and 52.85(23) wt% FeO, for the mottled parts, respectively (Table 3). Reflections in the X-ray powder diffraction pattern also confirm the presence of a phase in addition to (Mg,Fe)2Fe2O5, hematite, (Mg,Fe)O and magnesioferrite, the latter two of which are decomposition products (see Uenver-Thiele et al. 2017a). Extra reflections, corresponding to d-values of about 2.967 and 1.713 Å are observed and do not match any of the known Mg–Fe-oxide phases. It is likely that these reflections belong to the relict phase identified in the BSE images. Unfortunately, further analysis of the crystal structure of this phase is currently hampered by its apparent instability at ambient pressure and by overlapping peaks in the powder diffraction pattern. Assessing the stoichiometry of this relict phase is also ambiguous since no PtO2 was added to the experiment and the composition of the coexisting O5-phase of Mg1.53(1)Fe0.47(1)Fe2O5 indicates that some reduction of ferric to ferrous iron occurred during the experiment. Thus, a number of stoichiometries, each with different Fe3+/Fe2+ would be consistent with the observed bulk composition from EPMA. However, combining the robust measure of Mg/Fetot for this phase along with possible phase field topologies with a coexisting O5-phase and hematite place some constraints on its stoichiometry (Fig. 2). For example, an O9-stoichiometry with composition Mg4.55Fe1.45Fe2O9 would be essentially collinear with the O5-phase and hematite in the system MgO–FeO–FeO1.5, indicating that the three phases cannot be in equilibrium (Fig. 2). In addition, any phases containing more Fe2+ than the A 6 B 2 O 9 stoichiometry should not be in equilibrium with hematite. This leaves Mg2.85Fe0.15Fe2O6, Mg3.42Fe0.58Fe2O7 or Mg3.99Fe1.01Fe2O8 as potential compositions (Fig. 2), all stoichiometries that have already been suggested to have narrow stability fields in the Fe2+-free MgO–FeO1.5 binary system at pressures of 10–13 GPa and temperatures ≥ 1200 °C (Uenver-Thiele et al. 2017a). On the other hand, such an UQ-phase was not found in experiments conducted with a (Mg0.5Fe0.5)Fe2O5 bulk composition (Uenver-Thiele et al. 2017b). Thus, the phase assemblage observed in sample M519 is important in that it indicates the possibility that a variety of Fe3+-bearing oxide stoichiometries may be stable at high pressures and temperatures, even if Fe2+ apparently acts to destabilize some of these structures (like observed for wadsleyite; e.g., Fig. 3).

Back scatter electron (BSE) images of the run products from experiment Z1584u (a), exhibiting well developed crystals of (Mg,Fe)2Fe2O5 together with an unquenchable phase (UQ). Here, (Mg,Fe)2Fe2O5 is the most abundant phase exhibiting straight grain boundaries and triple junctions between individual grains. Hematite is present as small interstitial grains or has the same dimension as (Mg,Fe)2Fe2O5. The run products produced in experiment M519, b, c indicate the assemblage of (Mg,Fe)2Fe2O5 + UQ phase + hematite. The unquenchable phase also exhibits straight grain boundaries and triple junctions that are preserved from before the quench (b). However, mottled textures in the inner part of the grains point to partial decomposition during decompression of the experiment (c). The four different circles indicated in c show the position and the spot size used for microprobe analyses within mottled and relict domains of the unquenchable phase

Ternary diagram for the MgO–FeO–FeO1.5 system illustrating the possible phase relations for sample M519. Without explicitly knowing the nature of the unquenchable phase (UQ), a number of stoichiometries with different Fe3+/Fe2+, but constant Mg/Fetot are consistent with the microprobe analyses. However, the topologies of the coexisting phase fields place some constraint on the stoichiometry of the UQ phase (see text)

Experimental data plotted along with calculated stability fields for olivine (ol), wadsleyite (wds) and ringwoodite (rgw) (after Katsura and Ito 1989) in the Mg2SiO4–Fe2SiO4 system
Mg–Fe silicates coexisting with (Mg,Fe)2Fe2O5
Five experiments were performed to further explore phase relations involving the O5-phase and Mg–Fe silicates at high pressures and temperatures (Table 2). The combination of BSE imaging and EPMA analyses reveals at least three phases coexisting in these experiments (Tables 2, 4). The most abundant phase is (Mg,Fe)2Fe2O5, which does not incorporate Si in its structure, in agreement with previous observations (e.g., Woodland et al. 2013; Bindi et al. 2016). In the X-ray powder diffraction patterns, (Mg,Fe)2Fe2O5 could be indexed using the crystal structure model reported by Boffa Ballaran et al. (2015). Raman spectra of the O5-phase exhibit two characteristic peaks at ~ 315 and ~ 673 cm− 1, along with several other overlapping peaks lying between 450 and 550 cm− 1 (Electronic Supplementary Material S.1).
As anticipated, an additional phase having (Mg,Fe)2SiO4 stoichiometry was identified by EPMA (see Table 4) and characterized by means of X-ray diffraction and/or Raman spectroscopy. Considering that many diffraction peaks of the three (Mg,Fe)2SiO4 polymorphs: olivine, wadsleyite and ringwoodite overlap with those of the O5-phase, Raman spectra often proved to be particularly useful in phase identification by comparing them with spectra from McMillan and Akaogi (1987) and Guyot et al. (1986).
The occurrence of olivine, wadsleyite or ringwoodite is consistent with the phase relations of Mg2SiO4–Fe2SiO4 solid solutions at high pressure and temperature (Katsura and Ito 1989, see Fig. 3). One exception is the presence of wadsleyite in experiment Z1531o conducted at 1350 °C and 20 GPa (Fig. 3). Considering the study of Mrosko et al. (2015), the occurrence of wadsleyite may be due to the incorporation of Fe3+ and OH. They reported that this combination significantly expands the wadsleyite stability field to higher pressures and higher Fe2+ contents at the expense of ringwoodite. In fact, Raman spectra of wadsleyite from this experiment reveal small peaks at ~ 3340 and 3580 cm− 1, which can be attributed to OH-stretching modes (Kleppe et al. 2005). These two bands were also observed in sample Z1532u, although their intensities were significantly lower. The origin of small amounts of H2O in Z1531o and Z1532u could lie with the fact that small amounts of PtO2 were added to both experiments. It is known that the very fine-grained PtO2 powder can contain adsorbed atmospheric CO2 as indicated by the presence of traces of magnesite in such experiments (Table 2; Uenver-Thiele et al. 2017a), and it is conceivable that small amounts of H2O could also have been adsorbed on the PtO2 as well.
Our new experiments are richer in Mg than those previously reported by Woodland et al. (2013) and demonstrate that (Mg,Fe)2Fe2O5 can coexist with silicates having mantle-relevant compositions (i.e., with XMg = ~ 0.9). In two experiments (Z1531o, Z1532u), traces of magnesite were detected in the X-ray powder diffraction patterns. As suggested by Uenver-Thiele et al. (2017a), its presence is likely due to CO2 adsorbed onto the PtO2 powder (see above). The coexistence of (Mg,Fe)2Fe2O5 solid solutions with several silicate phases (olivine, wadsleyite, ringwoodite, clinopyroxene) or oxides (spinel and spinelloid V) was previously reported by Woodland et al. (2013) for rather Fe-rich bulk compositions. From the results of experiment Z1507o (Table 3), we can now add majoritic garnet to the list of phases with which O5-phase can coexist. Microprobe analyses (Table 4) indicate a composition of 82 mol% (Mg,Fe)4Si4O12 and 18 mol% (Mg,Fe)3Fe2Si3O12. In the absence of other constraints, the actual apportioning of Fe2+ between the two garnet components is currently not possible.
Compared to other samples that have a homogenous distribution of phases, run products from Z1587o and Z1586u are marked by distinctly different textures (Fig. 4a, b). The O5-phase forms large crystals and is most abundant, but there are two other phases with a lower mean atomic number \(\stackrel{-}{Z}\) illustrated by a lower contrast in BSE images (dark and middle grey). Olivine/wadsleyite (Z1586u) and ringwoodite (Z1587o) (dark grey in Fig. 4a, dark-middle grey in Fig. 4b) are almost entirely enclosed by another phase with a slightly different grey scale that contains distinctly less SiO2 than the (Mg,Fe)2SiO4 polymorphs (Table 3). Investigation of sample Z1586u by TEM revealed nicely developed crystals (Fig. 5b) and selected area electron diffraction (SAED) patterns (Fig. 5a, c, d) could be indexed on the basis of a monoclinic cell (space group P2 1 /c) with lattice parameters a = 10.6249(17), b = 14.1332(28), c = 10.1015(18) Å and β = 104.08(1)°. This is consistent with phase B reported by Finger et al. (1989), which is surprising since no water was added to the experiment.

BSE images of run products of experiments: a Z1586u and b Z1587o. In both cases, olivine or ringwoodite are enclosed by Fe3+-bearing phase B. The sample consists of ~ 50% large crystals of (Mg,Fe)2Fe2O5. In sample Z1586u, Fe3+-bearing phase B is much more abundant than olivine and is not only associated with olivine. In this case, the Fe content of phase B is ~ five times higher than in olivine. In sample Z1587o, phase B is as abundant as ringwoodite. However, both phases differ in their FeO content by only ~ 2 wt%, which explains the only slightly different contrast in the BSE image

a Selected area electron diffraction (SAED) pattern and b high-resolution TEM lattice image along the [010] zone axis of Fe3+-bearing phase B from experiment Z1586u. Some stacking faults parallel to the (100) plane are visible. c SAED patterns along the [001] and d the [100] zone axes in the phase B structure, respectively
Raman spectra with peaks characteristic for phase B (McMillan et al. 1991; Liu et al. 1998) were subsequently obtained from samples Z1586u and Z1587o (Fig. 6). A number of the peaks are quite broad, which can be explained by the rather high Fe content of our samples (Table 4). Small peaks at ~ 3360 and 3412 cm− 1 confirm the presence of OH groups in the structure.

Ambient Raman spectrum of Fe3+-bearing phase B in sample Z1586u, with some structural OH revealed by small peaks at ~ 3360 and 3412 cm− 1
EPMA measurements for the Fe-bearing phase B found in sample Z1586u gave a composition of Mg11.17(4)Fe1.04(6)Si3.79(3)O21H2, which, from the deviation from 4 Si per formula unit, implies ~ 0.43(5) Fe3+ cations have entered the structure via the charge-balanced substitution: [4]Si4+ + [6]Mg2+ = 2Fe3+. Fe-L2,3-edge energy-loss near-egde structure (ELNES) spectra based on EELS measurements were taken from this phase (sample Z1586u) to evaluate the Fe3+/Fetot ratio more quantitatively. The mixed valance state of iron was confirmed by the splitting in the white-lines, showing two separate Fe-L2,3 maxima for ferrous and ferric iron (Fig. 7). The Fe3+/Fetot ratios determined from the spectra vary from 0.35(4) to 0.46(4) in agreement with the stoichiometric calculation from EPMA analyses. To our knowledge, this is the first report of phase B incorporating significant amounts of Fe3+ and it is found to coexist with (Mg,Fe)2Fe2O5 as well as with either olivine or ringwoodite.

Representative Fe-L2,3-edge ELNES spectrum taken from phase B (sample Z1586u). The spectrum has been gain-normalized, background-subtracted and deconvoluted using the corresponding low-loss spectrum (not shown). The black dotted lines represent the position of the Fe-L2,3 white-line maxima for Fe2+ (at ~ 707.8 eV, fixed for the energy calibration) and Fe3+ (~ 710 eV), respectively. The grey dashed line below the spectrum represents the double arctan background function. The Fe3+/ƩFe quantification was made following the method of van Aken et al. (1998)
Crystal-chemical behaviour of (Mg,Fe)2Fe2O5 solid solutions
The range in composition obtained for Fe4O5–Mg2Fe2O5 solid solutions in this study, combined with literature data indicates that a complete solid solution exists across the binary join (see Table 5). Molar volumes decrease linearly with increasing Mg content (Fig. 8a). Application of symmetric or asymmetric solution models led to no statistical improvement in the fit to the data and the uncertainties in the interaction parameters were roughly equal to the derived values. Thus, we conclude that with the present uncertainties, the molar volumes vary essentially ideally across the binary. Extrapolation to the Mg2Fe2O5 endmember composition yields a molar volume of 52.99(1) cm3 (Fig. 8a), which is in agreement with the values reported from the single-crystal refinements of Boffa Ballaran et al. (2015) and Siersch et al. (2017).

Variations in a molar volume and lattice parameters (b–d), as a function of XMg in the O5-phase. Error bars represent uncertainties obtained from refinement of the powder diffraction patterns using GSAS (Toby et al. 2001). Most errors are the same size or smaller than the symbol
The change in molar volume along the solid solution is primarily due to a near linear decrease of ~ 0.08 Å in the b-parameter with increasing Mg content (Fig. 8c), consistent with the observations of Woodland et al. (2013). Moreover, our more extensive dataset clearly indicates additional, more subtle changes in both the a- and c-parameters with composition (Fig. 8b, d), which could not be resolved by Woodland et al. (2013). Single-crystal structural refinement of the Mg2Fe2O5 endmember reveals that Mg occupies all three crystallographic sites to various degrees, with a preference over Fe3+ for the M3 site (Boffa Ballaran et al. 2015). It is very likely that the large Fe2+ atom exhibits a similar site preference as Mg. Therefore the small changes in the a-parameter, which is controlled by the height of the M3 triangular prism, may be mainly due to Fe2+–Mg substitution at the M3 site with little or no influence from Fe3+. Even though the c-parameter is by far the largest in the unit-cell, it undergoes only ~ 25% of the shortening across the join compared to the b-parameter (Fig. 8b, d). These systematics can be understood by considering the following two effects of Mg–Fe2+ substitution on the structure of the Fe4O5–Mg2Fe2O5 solid solutions: (1) the M2–O1–M2 angle formed by the only corner-sharing bonds in the structure can change (i.e., rotation of M2 octahedra) to accommodate some of the geometric effects of substituting a smaller Mg cation for Fe2+, thereby compensating for some of the necessary shortening along the c-axis; (2) significant occupancy of M1 by Mg will have a strong influence on decreasing the M1–O2 bond distance, which principally leads to shortening of the b-parameter (Boffa Ballaran et al. 2015; Woodland et al. 2013). The similarity in unit-cell parameters for (Mg,Fe)2Fe2O5 solid solutions synthesized at different temperatures (see Tables 1, 2) suggests that the observed order–disorder behaviour is more a crystal-chemical phenomenon than being thermally induced or related to the quench process of the experiments.
Thermodynamic properties of Mg2Fe2O5
In their study of high-pressure Fe-oxides, including Fe4O5, Myhill et al. (2016) provided an estimate of the thermodynamic properties of the Mg2Fe2O5 endmember primarily based on Mg–Fe2+ partitioning between (Mg,Fe)2Fe2O5 and olivine solid solutions reported by Woodland et al. (2013). Experiment Z1461o, conducted at 1300 °C and 16 GPa, produced a three-phase assemblage of MgFe2O5 coexisting with Fe2O3 and MgO, indicating that it lies essentially on phase boundary defined by the equilibrium Mg2Fe2O5 = 2 MgO + Fe2O3 (Table 1). This P–T point, along with further constraints on the position of this phase boundary as depicted in Fig. 3 of Uenver-Thiele et al. (2017a) allow us to refine the standard enthalpy (H°298) of Mg2Fe2O5 in combination with new data for the equation of state of this material from Siersch et al. (2017) and molar volume reported here. The thermodynamic properties of MgO and Fe2O3 were taken from the dataset of Holland and Powell (2011), as suggested by Myhill et al. (2016). The properties of Mg2Fe2O5 are given in Table 6, along with their sources. The heat capacity was derived from the molar sum of the heat capacities of MgO and MgFe2O4, as compiled by Holland and Powell (2011). The study of Uenver-Thiele et al. (2017a) demonstrated that their data for these two phases (along with that of hematite) reproduce the experimentally determined position of the breakdown reaction MgFe2O4 = MgO + Fe2O3 reasonably well over a temperature range of ~ 400 °C. From our analysis, we obtain H°298 = − 1981.5 kJ mol− 1 for Mg2Fe2O5. This result is ~ 5 kJ mol− 1 less negative than that previously estimated by Myhill et al. (2016). The small amount of Mg present in the coexisting hematite [0.0618(9) Mg cations on a three-oxygen basis], as reported by Uenver-Thiele et al. (2017a) in experiment Z1461o translates to a XFe2O3 = 0.979. Assuming ideal mixing, this slight deviation from pure hematite contributes an uncertainty of only ~ 300 J at the conditions of the experiment.
Mg–Fe partitioning between (Mg,Fe)2Fe2O5 and silicate phases
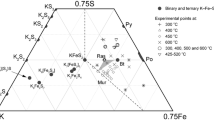
The partitioning of Mg and Fe2+ indicates that (Mg,Fe)2Fe2O5 has a strong preference for Fe2+ compared to coexisting silicate phases (Fig. 9). This behaviour was also observed by Woodland et al. (2013), but we can now extend this to majoritic garnet and phase B. Defining \({K_{\text{D}}}={{X_{{{\text{F}}{{\text{e}}^{2+}}}}^{{{\text{silicate}}}} \times X_{{{\text{Mg}}}}^{{{{\text{O}}_5}}}} \mathord{\left/ {\vphantom {{X_{{{\text{F}}{{\text{e}}^{2+}}}}^{{{\text{silicate}}}} \times X_{{{\text{Mg}}}}^{{{{\text{O}}_5}}}} {X_{{{\text{Mg}}}}^{{{\text{silicate}}}} \times X_{{{\text{F}}{{\text{e}}^{2+}}}}^{{{{\text{O}}_5}}}}}} \right. \kern-0pt} {X_{{{\text{Mg}}}}^{{{\text{silicate}}}} \times X_{{{\text{F}}{{\text{e}}^{2+}}}}^{{{{\text{O}}_5}}}}}\), we obtain \(K_{{\text{D}}}^{{{{{\text{grt}}} \mathord{\left/ {\vphantom {{{\text{grt}}} {{{\text{O}}_5}}}} \right. \kern-0pt} {{{\text{O}}_5}}}}}=\sim 0.09\) and \(K_{{\text{D}}}^{{{{{\text{phB}}} \mathord{\left/ {\vphantom {{{\text{phB}}} {{{\text{O}}_5}}}} \right. \kern-0pt} {{{\text{O}}_5}}}}}=\sim 0.05\), respectively, where Fe2+ was calculated from stoichiometry (Table 7).

Mg–Fe2+ partitioning between the (Fe,Mg)2Fe2O5 and coexisting Si-bearing phases plotted as tie lines in terms of the molar Mg/(Mg + Fe2+) of each phase. Data from Woodland et al. (2013) are plotted as dashed tie lines for comparison
For partitioning with the Mg–Fe-silicate polymorphs olivine, wadsleyite and ringwoodite, an analogous treatment can be expressed in terms of the exchange reaction:
The standard state Gibbs free energy of this equilibrium can be expressed as:
where KD is defined as above and Kγ accounts for non-ideal behaviour. Assuming the observed ideal volume of mixing for (Mg,Fe)2Fe2O5 solid solutions is representative of general ideal mixing behaviour for this phase, Eq. (2) becomes:
when taking into account the known non-ideal behaviour of olivine, wadsleyite or ringwoodite, here modelled as symmetric solid solutions. Values for KD at the P and T of our experiments can be computed by combining standard thermodynamic data for the Fe and Mg-endmembers of silicate phases from Holland and Powell (2011) with data from Myhill et al. (2016, 2017) for Fe4O5 and our results for Mg2Fe2O5 (Table 6). The interaction parameters, \(W_{{{\text{Fe}} - {\text{Mg}}}}^{{{\text{silicate}}}}\), for olivine (2000 kJ), wadsleyite (7500 kJ) and ringwoodite (4160 kJ) on a single-site basis were taken from Frost (2003). The results of these calculations are presented in Table 7, along with the KD values obtained from our five new Si-bearing experiments and several samples from Woodland et al. (2013). There is reasonable agreement for partitioning with olivine, supporting our assumption that (Mg,Fe)2Fe2O5 solid solutions behave ideally. On the other hand, there are discrepancies in KD values for Mg–Fe2+ partitioning with wadsleyite and ringwoodite, implying that these silicate phases, particularly wadsleyite, should have even higher Mg contents than those measured in the experimental run products (Table 7). For ringwoodite, the differences in KD are fairly small, on the order of ~ 0.04–0.05 and a likely explanation is that the presence of Fe3+ influences Mg–Fe2+ partitioning. This effect is not expected for olivine due to the lack of significant Fe3+ incorporation into this phase (e.g., Koch et al. 2004). Similar small differences are also observed for wadsleyite-bearing experiments of Woodland et al. (2013). However, samples Z1531o and Z1532u from this study both exhibit much larger differences in calculated and observed KD (Table 7). These differences are even much larger than can be accounted for by considering the effect of strongly non-ideal interactions between the Mg2SiO4 and \({\text{(Fe}}_{{5/3}}^{{3+}}{\square _{1/3}})\)Fe3+O4 components in wadsleyite (\(W_{{{\text{Fe}}^{3+} - {\text{Mg}}}}^{{{\text{wadsleyite}}}}\) = 20 kJ) as described by Frost and McCammon (2009) for oxidizing conditions (Re–ReO2 buffer). However, the work of Mrosko et al. (2015) suggests that our results may be due to the combination of Fe3+ and OH incorporation in wadsleyite. Thus, we attribute the differences in calculated and observed KD to the presence of Fe3+ and OH in wadsleyite. However, it is clear from the KD values presented in Table 7 that the O5-phase will always have the highest Fe content in mantle-relevant assemblages.
Redox stability of (Mg,Fe)2Fe2O5 solid solutions
A number of experiments reported in Table 1 contain a (Mg,Fe)2Fe2O5 solid solution coexisting with hematite, which allows us to estimate the oxygen fugacity (ƒO2) of equilibration via the equilibrium:
where
From the very limited deviation in hematite composition from the pure Fe2O3 as described above, we can reasonably assign unit activity of Fe2O3 for this phase. This, along with the assumption of ideal mixing on sites in (Mg,Fe)2Fe2O5 (i.e., a = X2) and rearranging yields the following simplification to Eq. 5:
The ƒO2 can then be computed by combining compositional data from this study and from Uenver-Thiele et al. (2017b) with standard state thermodynamic data for hematite, Fe4O5 and O2 from Holland and Powell (2011), Myhill et al. (2016) and Stagno and Frost (2010), respectively. The systematics of equilibrium (4) relative to the Re–ReO2 and the metastable fayalite–magnetite quartz (FMQ) oxygen buffers are illustrated in Fig. 10 as a function of pressure at 1300 and 1500 °C. Also plotted are the results of the few samples that were equilibrated at these two temperatures. As expected, the experiments employing PtO2 to minimize the formation of Fe2+ record the highest ƒO2, lying about 5 log units above FMQ. Somewhat lower ƒO2 values are computed for the two experiments where no PtO2 was added and partial reduction of Fe3+ occurred (M519, M566; Fig. 10; Table 2). Unsurprisingly the lowest ƒO2 values are calculated for the experiments with the Mg0.5Fe0.5Fe2O4 starting composition, where Fe3+/∑Fe = 0.8. However, our experiments with coexisting (Mg,Fe)2Fe2O5 and hematite were clearly performed under strongly oxidizing conditions, as was intended for investigating the behaviour of Fe3+ under deep mantle conditions. Although such conditions are much more oxidizing than generally expected for the deep upper mantle and transition zone (e.g., Frost and McCammon 2009), this does not exclude their appearance due to local excursions to high oxygen fugacities through interactions with subducted material or migrating carbonatite melts. These regions, where redox gradients occur, may also be important for the crystallization of diamond (Stachel et al. 1998).

logƒO2 of the equilibrium between Fe2 + 2Fe3 + 2O5 and hematite plotted against pressure at 1300 and 1500 °C along with values calculated for several samples run at the same temperatures. The arrows indicate the shift in logƒO2 related to Mg incorporation in Fe4O5. Also shown for reference are the positions of the metastable FMQ and Re–ReO2 redox buffer equilibria (Ballhaus et al. 1991; Myhill et al. 2016, 2017)
Concluding remarks
Combining the results on the crystal-chemical behaviour of Fe4O5–Mg2Fe2O5 solid solutions of this study with those of Uenver-Thiele et al. (2017a, b) and Woodland et al. (2013), it is apparent that complete solid solution exists across the binary join. A steady decrease in molar volume with increasing Mg content indicates ideal mixing behaviour. The variation in molar volume arises primarily from shortening along the b-axis with increasing Mg content. Although our results are in good agreement with the observations of Woodland et al. (2013), our wider compositional range also reveals more subtle changes in the a- and c-parameters with composition.
The stability of the O5-phase depends on the phase assemblage and bulk composition. For example, the study of Si-free bulk compositions (Uenver-Thiele et al. 2017a, b) demonstrates that the minimum pressure for (Mg,Fe)2Fe2O5 coexisting with hematite is ~ 10–11 GPa at ~ 1500–1600 °C. However, the maximum stability of the O5-phase in these Si-free systems depends on the redox conditions, and thus, on the Fe2+/Mg ratio of (Mg,Fe)2Fe2O5 (compare results of Uenver-Thiele et al. 2017a with b). The endmember assemblage Mg2Fe2O5 + Fe2O3 remains stable up to 20 GPa, whereas the maximum stability of Mg0.5Fe0.5Fe2O5 + Fe2O3 lies at ~ 14–16 GPa at ~ 1000–1300 °C and decreases to higher temperatures. This contrasts with the results of the Si-bearing experiments where hematite is absent. Here, (Mg,Fe)2Fe2O5 can be stabilized down to depths corresponding to the lower transition zone (~ 20 GPa) under more reducing conditions, consistent with the results of Myhill et al. (2016) who found Fe4O5 to be stable to at least 22 GPa.
In Si-bearing bulk compositions, (Mg,Fe)2Fe2O5 can occur, even though it does not incorporate Si into its structure, consistent with previous studies (e.g., Woodland et al. 2013; Bindi et al. 2016). The new experiments reported here demonstrate that the list of known Si-phases with which (Mg,Fe)2Fe2O5 can coexist (olivine, wadsleyite, ringwoodite, spinelloid V or clinopyroxene; Woodland et al. 2013) can now be extended to include majoritic garnet and Fe3+-bearing phase B. Chemical analyses of the garnet (Z1507o) indicate a composition of 82 mol% (Mg,Fe)4Si4O12 and 18 mol% (Mg,Fe)3Fe2Si3O12. Since phase B with the chemical formula Mg12Si4O19(OH)2 is known to become stable under hydrous conditions, its presence in our “dry” experiments was surprising, particularly since no PtO2 was added to these experiments that could have been a source of adsorbed H2O. Raman spectra obtained from phase B confirmed the presence of some structural water as OH; however, further quantification of the exact amount of OH by infrared spectroscopy or secondary ion mass spectrometry (SIMS) is required. A possible explanation for the stabilization of phase B might be the incorporation of Fe3+, which is reported here for the first time. EPMA and EDX measurements in the TEM gave consistent results with a composition of Mg11.17(4)Fe1.04(6)Si3.79(3)O21H2. Additional Fe-L ELNES analysis yielded a \({\text{F}}{{\text{e}}^{{\text{3}}+}}/\sum {{\text{Fe}}=\sim 0.{\text{41}}\left( {\text{4}} \right)}\), confirming that Fe3+ entered the structure via the charge-balanced substitution: [4]Si4+ + [6]Mg2+ = 2Fe3+. Based on the fact that phase B can not only store water, but also can accommodate Fe3+ and Fe2+ makes this hydrous magnesium silicate even more interesting as a possible mantle phase. From only two experiments, we have little constraint on the P–T stability of Fe3+-bearing phase B, except that it appears to be stable between at least 14 and 20 GPa at fairly low temperature (Table 3). An analogous substitution involving Cr3+ was recently reported for anhydrous phase B (Bindi et al. 2016; Sirotkina et al. 2017). Assuming the crystal chemistry of phase B and anhydrous phase B is similar with respect to substitution of trivalent cations, the incorporation of Cr3+ and Fe3+ together has the potential for expanding the stability field of phase B, particularly in terms of redox conditions. The same situation also holds for (Mg,Fe)2Fe2O5 in that the substitution of other trivalent cations (i.e., Cr and Al) will act to stabilize this phase to lower oxygen fugacities and also to a potentially wider range of pressures and temperatures, making it even more relevant for the mineralogy of the deep upper mantle and transition zone. The incorporation of such cations will also expand the range of bulk compositions in which the O5-phase can be stable. Investigation of such solid solutions is currently underway.
References
Armstrong JT (1993) Matrix correction program CITZAF, Version 3.5. California Institute of Technology
Ballhaus C, Berry RF, Green DH (1991) High pressure experimental calibration of the olivine–orthopyroxene–spinel oxygen geobarometer: implications for the oxidation state of the upper mantle. Contrib Mineral Petrol 107:27–40
Bindi L, Sirotkina EA, Bobrov A, Nestola F (2016) Chromium solubility in anhydrous phase B. Phys Chem Miner 43:103–110
Boffa Ballaran T, Uenver-Thiele L, Woodland AB, Frost DJ (2015) Complete substitution of Fe2+ by Mg in Fe4O5: the crystal structure of the Mg2Fe2O5 end-member. Am Mineral 100:628–632
Brey GP, Bulatov V, Girnis A (2008) Geobarometry for peridotites: experiments in simple and natural systems from 6 to 10 GPa. J Petrol 49:3–24
Enomoto A, Kojitani H, Akaogi M, Miura H, Yusa H (2009) High-pressure transitions in MgAl2O4 and a new high-pressure phase of Mg2Al2O5. J Solid State Chem 182:389–395
Finger LW, Ko J, Hazen RM, Gasparik T, Hemley RJ, Prewitt CT, Weidner DJ (1989) Crystal chemistry of phase B and an anhydrous analogue: implications for water storage in the mantle. Nature 341:140–142
Frost DJ (2003) The structure and sharpness of (Mg,Fe)2SiO4 phase transformations in the transition zone. Earth Planet Sci Lett 216:313–328
Frost DJ, McCammon CA (2008) The redox state of Earth’s mantle. Annu Rev Earth Planet Sci 36:389–420
Frost DJ, McCammon CA (2009) The effect of oxygen fugacity on the olivine to wadsleyite transformation: Implications for remote sensing of mantle redox state at the 410 km seismic discontinuity. Am Mineral 94:872–882
Guignard J, Crichton WA (2014) Synthesis and recovery of bulk Fe4O5 from magnetite, Fe3O4. A member of a self-similar series of structures for the lower mantle and transition zone. Mineral Mag 78:361–371
Guyot F, Boyer H, Madon M, Velde B, Poirier JP (1986) Comparison of the Raman microprobe spectra of (Mg,Fe)2SiO4 and Mg2GeO4 with olivine and spinel structures. Phys Chem Miner 13:91–95
Holland TJB, Powell R (2011) An improved and extended internally consistent thermodynamic dataset for phases of petrological interest, involving a new equation of state for solids. J Metamorph Geol 29:333–383
Ishii T, Kojitani H, Tsukamoto S, Fujino K, Mori D, Inaguma Y, Tsujino N, Yoshino T, Yamazaki D, Higo Y, Funakoshi K, Akaogi M (2014) High-pressure phase transitions in FeCr2O4 and structure analysis of new post-spinel FeCr2O4 and Fe2Cr2O5 phases with meteoritical and petrological implications. Am Mineral 99:1788–1797
Ishii T, Kojitani H, Fujino K, Yusa H, Mori D, Inaguma Y, Matsushita Y, Yamaura K, Akaogi M (2015) High-pressure high-temperature transitions in MgCr2O4 and crystal structures of new Mg2Cr2O5 and post-spinel MgCr2O4 phases with implications for ultrahigh-pressure chromitites in ophiolites. Am Mineral 100:59–65
Jacob DE, Piazolo S, Schreiber A, Trimby P (2016) Redox-freezing and nucleation of diamond via magnetite formation in the Earth’s mantle. Nat Commun 7:11891
Katsura T, Ito E (1989) The system Mg2SiO4-Fe2SiO4 at high pressures and temperatures: precise determination of stabilities of olivine, modified spinel, and spinel. J Geophys Res 94:15663–15670
Keppler H, Frost DJ (2005) Introduction to minerals under extreme conditions. In: Miletich R (ed) Mineral behaviour at extreme conditions, vol 7. EMU Notes in Mineralogy, pp 1–30
Koch M, Woodland AB, Angel RJ (2004) Stability of spinelloid phases in the system Mg2SiO4–Fe2SiO4–Fe3O4 at 1100 °C and up to 10.5 GPa. Phys Earth Planet Inter 143:171–183
Larson AC, Van Dreele RB (1994) GSAS general structure analysis system. Los Alamo National Laboratory, New Mexico
Lavina B, Meng Y (2015) Unraveling the complexity of iron oxides at high pressure and temperature: synthesis of Fe5O6. Sci Adv 1(5):e1400260
Lavina B, Dera P, Kim E, Meng Y, Downs RT, Weck PF, Sutton SR, Zhao Y (2011) Discovery of the recoverable high-pressure iron oxide Fe4O5. Proc Natl Acad Sci 108:17281–17285
Liu L-G, Lin C-C, Mernagh TP, Irfune T (1998) Raman spectra of phase B at various pressures and temperatures. J Phys Chem Solids 59:871–877
McMillan P, Akaogi M (1987) Raman spectra of β-Mg2SiO4 (modified spinel) and ɣ-Mg2SiO4 (spinel). Am Mineral 72:361–364
McMillan PF, Akaogi M, Sato RK, Poe B, Foley F (1991) Hydroxyl groups in β-Mg2SiO4. Am Mineral 76:354–360
Mrosko M, Koch-Müller M, McCammon CA, Rhede D, Smyth JR, Wirth R (2015) Water, iron, redox environment: effects on the wadsleyite–ringwoodite phase transition. Contrib Mineral Petrol 170:9
Myhill B, Ojwang DO, Ziberna L, Frost D, Boffa Ballaran T, Miyajima N (2016) On the P–T–fO2 stability of Fe4O5 and Fe5O6-rich phases: a thermodynamic and experimental study. Contrib Mineral Petrol 171:1–11
Myhill B, Ojwang DO, Ziberna L, Frost D, Boffa Ballaran T, Miyajima N (2017) Correction to: on the P–T–fO2 stability of Fe4O5 and Fe5O6-rich phases: a thermodynamic and experimental study. Contrib Mineral Petrol 171:51
Siersch NC, Boffa Ballaran T, Uenver-Thiele L, Woodland AB (2017) Compressibility and high-pressure structural behavior of Mg2Fe2O5. Am Mineral 102:845–850
Sinmyo R, Bykova E, Ovsyannikov SV, McCammon C, Kupenko I, Ismailova L, Dubrovinsky L (2016) Discovery of Fe7O9: a new iron oxide with a complex monoclinic structure. Sci Rep 6:32852
Sirotkina EA, Bobrov AV, Bindi L, Irifune T (2017) Chromium-bearing phases in the Earth’s mantle: evidence from experiments in the Mg2SiO4–MgCr2O4 system at 10–24 GPa and 1600 °C. Am Mineral 103:151–160
Stachel T, Harris JW, Brey GP (1998) Rare and unusual mineral inclusions in diamond from Mwadui, Tanzania. Contrib Mineral Petrol 132:34–47
Stagno V, Frost DJ (2010) Carbon speciation in the asthenosphere: experimental measurements of the redox conditions at which carbonate-bearing melts coexist with graphite or diamond in peridotite assemblages. Earth Planet Sci Lett 300:72–84
Temple PA, Hathaway CE (1973) Multiphonon spectrum of silicon. Phys Rev B 7:3685
Toby BH (2001) EXPGUI, a graphical user interface for GSAS. J Appl Crystallogr 34:210–213
Uenver-Thiele L, Woodland AB, Boffa Ballaran T, Miyajima N, Frost DJ (2017a) Phase relations of MgFe2O4 at conditions of the deep upper mantle and transition zone. Am Mineral 102:632–642
Uenver-Thiele L, Woodland AB, Boffa Ballaran T, Miyajima N, Frost DJ (2017b) Phase relations of Fe–Mg spinels including new high-pressure post spinel phases and implications for natural samples. Am Mineral 102:2054–2064
Van Aken PA, Liebscher B (2002) Quantification of ferrous/ferric ratios in minerals: new evaluation schemes of Fe L23 electron energy-loss near-edge spectra. Phys Chem Miner 29:188–200
Van Aken PA, Liebscher B, Styrsa VJ (1998) Quantitative determination of iron oxidation states in minerals using Fe L2,3-edge electron energy-loss near edge structure spectroscopy. Phys Chem Miner 25:323–327
Woodland AB, Frost DJ, Trots DM, Klimm K, Mezouar M (2012) In situ observation of the breakdown of magnetite (Fe3O4) to Fe4O5 and hematite at high pressures and temperatures. Am Mineral 97:1808–1811
Woodland AB, Schollenbruch K, Koch M, Boffa Ballaran T, Angel RJ, Frost DJ (2013) Fe4O5 and its solid solutions in several simple systems. Contrib Mineral Petrol 166:1677–1686
Woodland AB, Uenver-Thiele L, Boffa Ballaran T (2015) Synthesis of Fe5O6 and the high-pressure stability of Fe2+–Fe3+-oxides related to Fe4O5. Goldschm Abstr 2015:3446
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft through grants WO 652/20-1 and BO 2550/7-1 to ABW and TBB, respectively. E. Alig is thanked for helping with obtaining the X-ray powder diffraction patterns. We are grateful to Thomas Kautz, Nicki Siersch and Svyatoslav Shcheka for their help with the multi-anvil experiments at the University of Frankfurt and the Bayerisches Geoinstitut. Heidi Höfer is thanked for her help with the microprobe analysis. We also acknowledge the comments by Bob Myhill and two anonymous reviewers which helped to improve the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Jochen Hoefs.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Uenver-Thiele, L., Woodland, A.B., Miyajima, N. et al. Behaviour of Fe4O5–Mg2Fe2O5 solid solutions and their relation to coexisting Mg–Fe silicates and oxide phases. Contrib Mineral Petrol 173, 20 (2018). https://doi.org/10.1007/s00410-018-1443-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-018-1443-8