
Abstract
Amphibole is one of the most important hydrous minerals of the middle and lower continental crust and plays a key role in the formation of intermediate to silica-rich magmas. This study reports a consistent set of amphibole trace element partition coefficients derived from fractional crystallization experiments at 0.7 GPa in a piston cylinder apparatus. Starting materials were doped with trace elements on the 20–40 ppm level and measured using laser ablation (LA)-ICP-MS. Amphibole is stable from 1010 to 730 °C and systematically changes its composition from pargasite to magnesiohornblende to cummingtonite, while coexisting liquids vary from andesite to dacite and rhyolite. Amphibole–liquid partition coefficients increase systematically with decreasing temperature and increasing SiO2 in the liquid. Potassium displays an inverse behavior and partitioning decreases with decreasing temperature. Rare earth element (REE) partition coefficients, assumed to occupy the M4 site within the amphibole structure, increase continuously up to one order of magnitude. The calculated lattice parameters, ideal cation radius (r 0) and Young’s modulus (E) remain nearly constant with decreasing temperature. The high-field strength elements Zr and Hf that occupy the M2 site of the amphibole structure reveal a fivefold increase in partition coefficients with decreasing temperature and constant lattice parameters r 0 and E. Partition coefficients correlate with edenite, tschermaks and cummingtonite exchange vectors indicating that the maximum partition coefficient (D 0) for an ideal cation radius increases with decreasing edenite component, while the latter decreases linearly with temperature. Regressing Amph/L D Ca against trace elements results in fair to excellent correlations (r 2 0.55–0.99) providing a predictive tool to implement the trace element partition coefficients in numerical geochemical modeling. Our data result in positive correlations between Amph/L D Nb/Ta and Amph/L D Ca, and Mg#. Spoon-shaped REE patterns and subchondritic Nb/Ta ratios in tonalitic to granodioritic plutonic rocks and andesitic to rhyolitic magmas directly constrained by measured trace element compositions of coexisting liquids are consistent with hornblende gabbro fractionation in the middle to lower crust. The systematic change of the measured trace element composition of fractionating calc-alkaline liquids indicates that hornblende gabbro formation in the middle to lower crust also exerts an important control on some commonly used trace element ratios such as Sr/Y, Sr/Ba or Nb/Zr.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amphibole plays a key role in our understanding of geochemical processes in the Earth crust and mantle. In addition to being a primary host for H2O and halogens in many igneous and high-grade metamorphic rocks, amphibole is characterized by a flexible crystal structure characterized by a large number of sites with variable size accommodating a wide spectrum of trace elements as a function of pressure, temperature, oxygen fugacitiy and melt or fluid composition (Adam and Green 1994; Sisson 1994; Hilyard et al. 2000; Klein et al. 1997; Tiepolo et al. 2007). Amphibole crystallization and melting exerts an important control on magmatic differentiation processes at intermediate to lower crustal levels (Cawthorn et al. 1976; Hilyard et al. 2000; Davidson et al. 2007). Many volcanic rocks ranging from (alkali) basalt to basaltic andesite and dacite contain amphibole phenocrysts indicating crystallization near the liquidus. Thus, an appropriate set of partition coefficients (D’s) permits calculation of the trace element concentration of coexisting liquids for a wide variety of arc volcanic and plutonic compositions. Such partition coefficients are derived either from experimental studies (Adam and Green 1994; Hilyard et al. 2000; Tiepolo et al. 2007 and references therein) or from natural rocks by measuring compositions of amphibole phenocrysts and interstitial volcanic glass (Sisson 1994; Ewart and Griffin 1994). These studies revealed that partition coefficients between amphibole and melt primarily depend on melt composition and crystal structure, while oxygen fugacity, pressure and temperature are of secondary importance (Tiepolo et al. 2007). However, most of the bulk compositions used to determine amphibole–liquid trace element partitioning were alkali basaltic to basanitic (e.g., Adam and Green 1994; La Tourrette et al. 1995; Dalpé and Baker 2000; Tiepolo et al. 2000a, b), a few andesitic (e.g., Brenan et al. 1995; Klein et al. 1997; Tiepolo et al. 2000b), and rarely dacitic (Hilyard et al. 2000) or rhyolitic (Sisson 1994). In addition, due to the similar chemical behavior of element groups in particular sites of the amphibole lattice, some elements were measured, while others with similar chemical behavior were calculated using the lattice strain model of Blundy and Wood (1994).
As a prerequisite for predicting partition coefficients, the partition coefficient of a cation with similar chemical behavior, the ideal radius and the elasticity of the respective lattice site (Young’s modulus) must be known. Therefore, several attempts have been undertaken to model these parameters using compositional and structural information of natural amphiboles and silicate liquids (Nielsen 1985; Hilyard et al. 2000; Tiepolo et al. 2007); the latter being proposed to exert the dominant control on amphibole trace element partitioning (Tiepolo et al. 2007).
The present study provides laser ablation (LA)-ICP-MS amphibole–melt partitioning data along the experimentally determined liquid line of descent of a calc-alkaline basalt at 0.7 GPa. These new experimental data on amphibole–liquid pairs cover the temperature range from 1010 to 780 °C and are relevant for intermediate to lower crustal magma reservoirs from which most of the silicic magmas are extracted. We evaluate the systematic compositional changes of amphibole and coexisting silicate liquid as a function of temperature and compositions and discuss the relevance of our new set of partition coefficients for the genesis of silicic magmas in island and continental arc settings.
Methods
Trace element doping of experimental starting materials
A trace element-bearing diopside glass was employed as a source of trace elements in the experiments. A mixture of fired oxides (SiO2 and MgO, 1000 °C) and dried carbonate (CaCO3, 400 °C) was weighed, homogenized and vitrified at 1460 °C at ambient pressure in a vertical quench furnace to obtain a diopside glass. The glass was ground under alcohol prior to the addition of 32 trace elements at the 2000 ppm level, except U and Th, which were added at the 1000 ppm level. Most of the trace elements were added as oxide powders except Li, Sr and Ba, which were added as titanate, K as carbonate, Mo and W as metal complexes and Rb (HCl), Th and U (HNO3) as solutions. This mixture was subsequently vitrified in a platinum crucible at 1460 °C for 3.5 h. The composition of the recovered glass was determined using electron microprobe and laser ablation (LA)-ICP-MS analysis. The concentrations are generally within 10 % of the target value of 1000 and 2000 ppm (Table 1), with Mo and U being substantially lower (1629 and 765 ppm, respectively). Rb, Cs and Pb concentrations are very low and were most probably lost by volatilization during the melting process (Rb, Cs), while Pb most likely alloyed with the Pt crucible during synthesis.
High-pressure experiments
Fractional crystallization experiments were performed using a piston cylinder apparatus and approached in a stepwise manner. The measured glass composition of an experiment was determined and remixed as new starting material for a subsequent experiment at lower temperatures (for details of the experimental procedures, see Nandedkar et al. 2014). Starting from a synthetic chemical equivalent of a primitive olivine tholeiite with 3 wt% H2O, the liquid composition was remixed from oxides, hydroxides, phosphates and silicates and used as starting composition for the following experiment. Compounds used were fired and dried reagent grade oxides and silicates (fired at 1000 °C, SiO2, TiO2, Fe2O3, MgO, CaSiO3, dried at 220–350 °C; γ-Al2O3, MnO, Fe2SiO4, Na2SiO3, K2SiO3, Ca5(PO4)3(OH)), and dried mixtures of AlOOH (200 °C) and Al(OH)3 (100 °C) to adjust the desired H2O-content provided the alumina concentration of the target composition was high enough. In the last fractionation steps where Al2O3 concentration was low(er) and H2O concentrations high, distilled water was added by a microsyringe to an anhydrous powder mixture during preparation of the capsules. Two wt% of the pre-synthesized diopside glass containing the trace elements was added to each starting material as finely ground glass, and the reported major element compositions for the starting material were calculated accordingly. This resulted in concentrations of trace elements at around 40 ppm (20 ppm for Th and U) in each experiment, well within the range of Henry’s law (<50 ppm, e.g., Klein et al. 2000).
Electron probe microanalysis (EPMA)
Experimental run products were analyzed with wavelength-dispersive spectrometers (WDS) on a JEOL JXA-8200 electron probe micro-analyzer. An acceleration voltage of 15 kV and a variable beam current and beam size (7 nA and 10–20 µm for glasses and 20 nA and 1 µm for minerals) were used to minimize alkali migration during analyses of hydrous glasses. Peak and background counting times were 20 s. All data were corrected with the PRZ model (Goldstein et al. 1992; Korolyuk et al. 2009). For glasses, H2O was used as additional element for PRZ corrections and calculated as difference to 100 wt%. Amphibole compositions have been normalized using charge balance (46 positive charges without OH, F, Cl) and a fixed ferric/total iron ratio (Fe3+/Fetot) of 0.267. This value is slightly higher than 0.22 that was determined for a tschermakite synthesized at NNO in a similar calc-alkaline system (Clowe et al. 1988). As our experiments are close to, or slightly above, the NNO oxygen buffer (Nandedkar et al. 2014). We adopted a value of 0.267 in order to distribute cations in the amphibole structure (Spear and Kimball 1984). Modal proportions of the experimental runs were calculated using non-weighted, least-squares regression (LSR) analyses implemented in Microsoft EXCEL® 2010 balancing the nominal composition of the starting material against the averages of all analyzed phases in the experimental charges. Melt fractions were high in all runs (>75 %), and the modal proportions of amphibole vary between 0.7 and 16.6 % as observed in Nandedkar et al. (2014). A summary of experimental conditions, glass compositions and coexisting phases are given in Table 2.
LA-ICP-MS analysis
Trace element compositions were analyzed by laser ablation inductively coupled mass spectrometry (LA-ICP-MS) with a new wave UP193-FX 193 nm ArF excimer laser coupled to a sector field ICP-MS (Thermo Element XR) installed in the laboratories of the University of Lausanne, Switzerland. The aerosol produced during laser ablation was transported in a He atmosphere from the ablation cell at a flow rate of 1.00 l/min and then mixed with Ar at flux rates of 0.690–0.735 l/min, before entering the ICP-MS. Repetition rates of the laser were between 5 and 10 Hz. The laser energy was measured with an external photometer and varied between 5.2 and 5.8 J/cm2. During acquisition, the background was measured for 120 s and the ablation intervals were between 5 and 60 s. Spot sizes varied between 10 and 20 μm for experiments down to 920 °C and 5 and 10 μm for small crystals in experiments below 900 °C, and 50 μm for experimental glasses. Dwell times varied between 10 and 20 ms. As an external standard, the standard reference material NIST SRM 612 (Pearce et al. 1997 and reference therein), produced by the National Institute of Standards and Technology (NIST), was used maximizing 139La > 3.5 × 106 cps and 232Th > 4.5 × 106 cps and minimizing doubly charged ions Ba2+⁄Ba+ < 2 and ThO+⁄Th+ < 0.5. The NIST SRM 612 was measured twice before and after each series. We note that the revised reference concentrations for NIST SRM 612 (Jochum et al. 2011) for our elements of interest are within 3 % of the values published by Pearce et al. (1997), and we therefore kept the Pearce et al. values for external standardization. Data reduction was performed using the program SILLS (Guillong et al. 2008). As internal standard, CaO concentrations determined with the electron probe micro-analyzer were used. Limits of detection for each signal interval were calculated as count rate plus three times the standard deviation divided by the element sensitivity. Major and trace element compositions of liquids and amphibole are listed in Tables 3 and 4.
Spot size, signals and data treatment
The spot size of the laser beam had to be adjusted, according to the crystal size. This additionally included a reduction in repetition rates and energy density of the laser. Despite these precautions, the signals were short because of the small size of experimentally grown crystals. It is not straightforward to set integration intervals as amphibole incorporates a large number of different trace elements within its crystal structure complicating distinction from the surrounding glass. Therefore, we used the following two criteria to distinguish between pure and mixed signals. First, we checked for the most incompatible element Cs in amphibole and only analyses for which Cs was below detection limit were accepted. Second, we only accepted the lowest Ba and La concentrations in amphibole for further processing, which are known to be fairly incompatible in amphibole (e.g., Tiepolo et al. 2007). The remaining analyses were considered acceptable and are averaged and reported in Table 4, together with their propagated 2 sigma uncertainties. Partition coefficients D’s (amphibole–liquid) were then calculated (Table 5). To further test the coherency of the dataset, we plotted the moderately compatible Yttrium (Y) partition coefficient against more incompatible and compatible elements (Electronic Appendix: EA Fig. 1). Simple linear regression provides r 2 (sums of residual squared) between 0.6 and 0.99, indicating consistent behavior of compatible and incompatible element D’s in our experiments.
Results
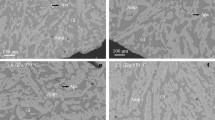
All experiments were near-liquidus runs yielding abundant and homogeneous liquid (>75 %) without any visible quench crystallization. Amphibole in our experiments was stable over a wide temperature range (1010–730 °C) and crystallized together with plagioclase and Ti-magnetite (Table 2). At higher temperatures (1010–950 °C), amphibole coexists with orthopyroxene, and at lower temperatures (860–730 °C), apatite is present as an accessory phase. The compositions of experimentally produced amphiboles range from pargasite at high temperature coexisting with basaltic andesite liquid, to magnesiohornblende, coexisting with andesitic/dacitic liquids, to cummingtonite, coexisting with rhyolite at temperatures below 780 °C. Backscattered electron (BSE) images of the experimental charges illustrate the decreasing crystal size of experimental amphibole with decreasing temperature (Fig. 1). Amphibole crystals are fairly homogeneous, but some amphiboles contain tiny inclusions of melt and/or plagioclase (Fig. 2).

Backscattered electron (BSE) images of experimental charges at various temperature illustrating coexisting minerals and the reduction in crystal size with decreasing temperature

Characteristic X-ray distribution maps of experiment RN10 s at 950 °C illustrating a large amphibole crystal. Amphibole crystals are fairly homogeneous and display only weak zoning, but larger crystals (>30 µm) may contain tiny inclusions of plagioclase crystals
Major element composition and partitioning of amphibole
The Si content of amphibole increases monotonically from 5.9 to 7.8 cations p.f.u. with increasing SiO2 content of the coexisting liquid. The Al concentration in amphibole decreases with decreasing temperature (Nandedkar et al. 2014). This is mostly related to the decrease in fourfold coordinated aluminum (AlIV) with temperature, whereas the calculated sixfold coordinated aluminum (AlVI) remains nearly constant. The titanium contents increase at temperatures of 920 °C and subsequently decrease when Ti-rich Fe oxides appear on the liquidus and co-precipitate with amphibole. The alkalis and calcium decrease with decreasing temperature. Magnesium increases while total iron concentrations remain nearly constant, indicating an increase in Mg# of amphibole with decreasing temperature. This indicates that the edenite (\({\text{NaAl}}^{{({\text{IV}})}}{ \Box_{ - 1}} {\text{Si}}_{ - 1}\)), tschermaks (Al2Mg−1Si−1) and cummingtonite (MgCa−1) exchange components control the composition of amphibole with decreasing temperature.
Figure 3 displays the relationships between the exchange coefficients (amphibole/liquid) and temperature for various oxides: The relationships between the Al2O3, Na2O and K2O exchange coefficients (amphibole/liquid) and temperature all exhibit a negative correlation (Fig. 3a). This reflects trends of decreasing concentrations of these elements in amphibole and increasing (Na, K) or only slightly decreasing (Al) concentrations in the coexisting liquid phase. In contrast, Ca, Fe (total), Mn and Mg (not shown) reveal increasing exchange coefficients with decreasing temperature (Fig. 3b) reflecting the highly polymerized nature of low-temperature rhyodacitic to rhyolitic liquids rejecting incorporation of these elements in the liquid phase and, thus, resulting in increasing Kd values.

Amphibole–melt distribution coefficients (Amph/L D’s) as a function of temperature for a Al2O3, Na2O and K2O; and b CaO, FeOtot and MnO
Trace element partition coefficients
Amphibole has the general structural formula A0−1B2C5T8O22(OH)2 (Leake 1978; Leake et al. 1997). The structural positions can be further split into single sites. The eightfold coordinated M4 sites are surrounded by 8 anions characteristic for large monovalent to divalent cations such as Ca2+ and Na+. The different octahedrally coordinated M1–M3 sites are surrounded by 6 oxygens, which mainly incorporate medium-sized, divalent to trivalent cations (Mg2+, Fe2+, Mn2+, Al3+, Fe3+). The partition coefficient of an element (i) is defined as:
where C denotes the weight concentration of a particular element (i) in either the liquid (Table 3) or in amphibole (Table 4). The partition coefficients and their uncertainties, and some petrogenetically relevant partition coefficient ratios are reported in Table 5. The incorporation of trace elements is intimately linked to the behavior of major elements such as Ca and alkalies (for REE) or Ti (for HFSE) and is, thus, thermodynamically controlled and dependent on temperature, pressure and composition (Blundy and Wood 1994; Klein et al. 1997; Sisson 1994; Wood and Blundy 1997). Partial melting and/or fractional crystallization of amphibole-dominant systems result in tonalitic to dacitic melts with middleREE/heavy REE and middle REE/light REE fractionation producing convex downward (“spoon-shaped”) REE pattern (Dessimoz et al. 2012; Hilyard et al. 2000).
Rare earth elements
The amphibole–liquid partition coefficients for rare earth elements (REE) are illustrated in Fig. 4a. The light rare earth elements (LREE; La-Sm) are incompatible elements at high temperature, but become compatible (with the exception of La) with decreasing temperature. In contrast, medium to heavy rare earth elements (MREE–HREE; Gd–Lu) are more compatible and their partition coefficients are generally >1. The compatibility of HREE is slightly decreasing with increasing atomic number in agreement with previous studies (e.g., Sisson 1994; Klein et al. 1997; Bottazzi et al. 1999). The most compatible rare earth elements in the amphibole structure are dysprosium (Dy) and holmium (Ho). The fractionation between LREE/MREE (Amph/LDLa/Amph/LDSm = 0.12–0.08) and (Amph/LDLa/Amph/LDYb = 0.09–0.06) decreases slightly with temperature for calcic amphiboles (Table 5). The behavior of Y is analogous to that of HREE due to its identical valence and similar cation radius.

a REE partition coefficients color-coded for experimental temperature. Experiment RN8 was performed using a double capsule, and partition coefficients for both (inner and outer) capsules are provided. Open symbols data from Sisson (1994), error bars represent 2 sigma standard deviations from the mean; b Onuma diagram (Onuma et al. 1968) for the M4′ lattice site illustrates REE partition coefficients as a function of cation radius. The color code indicates partition coefficients derived from the same experiment. Lines are fitted to Eq. (2) (Blundy and Wood 1994); lattice parameters are given in Table 7
The trivalent REE are typically eightfold coordinated and form a parabolic trend on an Onuma diagram (Fig. 4b), with the exception of Europium (Eu), which reveals a negative anomaly with respect to samarium (Sm) and gadolinium (Gd). The most common explanation is that Eu occurs as both divalent and trivalent cations (Philpotts 1970; Klein et al. 1997). With decreasing temperature, all REE partition coefficients increase by almost an order of magnitude (Fig. 4a). Applying the lattice strain model of Blundy and Wood (1994), the REE’s and Y (excluding Eu) are fitted to a parabola with lattice parameters reported in Table 6. D 0 increases with decreasing temperature, while r 0 (average: 1.022 Å) remains between the radii of Dy (1.027 Å) and Ho (1.015 Å). The Young’s modulus E does not significantly change with temperature and remains fairly constant with an average value of 348.5 ± 20.8 GPa.
High-field strength elements
High-field strength elements (HFSE) are tetra- or pentavalent cations and are plotted against inverse temperature (10,000/T(K), Fig. 5). Niobium (Nb), tantalum (Ta), zirconium (Zr) and hafnium (Hf) are moderately incompatible to weakly compatible (Hf at low temperature), with the exception of titanium (Ti), which is compatible in the amphibole structure, as also shown in previous studies (e.g., Bottazzi et al. 1999; Hilyard et al. 2000; Tiepolo et al. 2007). Partition coefficient ratios Amph/L D Nb/Amph/L D Ta (0.89–1.41) and Amph/LDZr/Amph/LDHf (0.44–0.75) generally increase with decreasing temperature but no systematic trends are evident for the case of Zr and Hf (Table 5), indicating that other factors are controlling the partition coefficient ratios. This will be discussed below. In analogy to REEs, the lattice parameters (D 0, r 0 and E) can be estimated and are reported in Table 6. However, given the limited number of elements that enter the M2 site and the caveat that the M1–M3 sites in amphibole are not strictly equivalent (e.g., Tiepolo et al. 2000b), the lattice parameters are only rough estimates of trial-and-error fits and are far less robust than the ones derived for REEs (Fig. 4b). Despite these limitations, D0 increases with decreasing temperature and r0 varies within a limit of 0.637–0.655 Å, for an assumed E of 1500 GPa. The higher value of E corresponds to a less elastic (stiffer) behavior of the M1–M3 sites compared to the more flexible M4 site; our values are higher than the compilation of elastic moduli for the amphibole M2 site of Klein et al. (1997) but consistent with the data of Brenan et al. (1995).

Partition coefficients of high-field strength elements (Ti, Nb, Ta, Zr, Hf) plotted against inverse temperature. Experiment RN8 was performed using a double capsule, and partition coefficients for both (inner and outer) are shown. Lines are eye-ball fits to the experimental data. Open symbols are data from Klein et al. (1997) (K97) and Tiepolo et al. (2007) (T07)
Large ion lithophile elements
Large ion lithophile elements (LILE) are incompatible elements with partition coefficients generally well below unity (Fig. 6), with the exception of potassium (K) in the study of Dalpe and Baker (2000). The partition coefficient of the major element potassium (K) indicates an opposite trend relative to all other trace elements (with the exception of Rb, Tiepolo et al. 2007) and decreases with temperature from 0.41 to 0.035 (Fig. 3a). Strontium (Sr) does not show a clear trend and remains almost constant for our dataset, while at the highest temperatures our values are comparable to those (scattered data) from the literature (LaTourrette et al. 1995; Dalpe and Baker 2000). Ba’s partition coefficient increases from <0.2 at 1020 °C to 0.65 at 780 °C.

Partition coefficients for LILE elements K, Ba and Sr plotted against inverse temperature (K). K (and Rb data from the literature) decrease with decreasing temperature and increasing SiO2 content of the melt, Sr is almost independent of temperature lacking a clear behavior with decreasing temperature, while Ba increases with decreasing temperature. Error bars represent 2 sigma standard deviations of the mean. Literature data: LaTourrette et al. (1995) (LT95), Dalpe and Baker (2000) (DB00), Tiepolo et al. (2007) (T07). Shaded field are K partition coefficients from Tiepolo et al. (2000a, b)
Transition metals
The partition coefficients for transition metals are rarely reported in the literature, in particular for molybdenum (Mo) and tungsten (W). While vanadium (V) is a compatible element, with Amph/LDV ranging from 3.9 to 11.7, Mo and W are incompatible elements (Amph/LDMo 0.01 to 0.58; Amph/LDw 0.004 to 0.194 (Table 5; Fig. 7). For Mo and W, there is a notable increase in partition coefficients between 920 and 890 °C. For comparison, we also show Amph/LDSc, which increase with decreasing temperature (4.7–35.0). Our Sc data are in good agreement with the parameterization of Tiepolo et al. (2007).

Partition coefficients of transition metals (V, Mo, W) and Sc from this work and for the available literature data (T07: Tiepolo et al. 2007). Note the significant increase at 890 °C for Mo and W
Actinides
Uranium (U) and thorium (Th) amphibole–liquid partition coefficients are reported in Fig. 8 together with the partition coefficients for Pb. Th and U show increasing partition coefficients with decreasing temperature with D Th varying from 0.015 to 0.275 and D U from 0.01 to 0.188. Similar to Mo and W, U and Th show a notable increase in partition coefficients between 920 and 890 °C. Within error, the Amph/LDU and Amph/LDTh are identical, similar to what has been reported by Tiepolo et al. (2000b). However, our data show substantially higher Amph/LDU and Amph/LDTh than previous studies (LaTourrette et al. 1995; Brenan et al. 1995; Tiepolo et al. 2000a) which is probably related to the expansion of the temperature range down to 780 °C and/or Si-rich compositions.

Partition coefficients for actinides (U, Th) and Pb compared to literature data. Note the significant increase between 890 and 920 °C for all three elements. T07 indicates predicted partition coefficients according to the exponential fits of literature data (Tiepolo et al. 2007)
Discussion
Attainment of equilibrium
An important prerequisite for accurate trace element partitioning coefficients is the attainment or close approach to thermodynamic equilibrium of melts and crystals during the experiments. Although we did not perform reversal experiments, there are several arguments that suggest close approximation to equilibrium. The attainment of equilibrium in these experiments is facilitated by large liquid proportions and evident by consistent Fe–Mg partitioning and consistent trends in the major element chemical evolution of liquids and minerals (see Nandedkar et al. 2014, for a detailed discussion). Determinations of H2O contents of quenched silicate liquids (glasses) by Raman spectroscopy revealed that water was quantitatively kept in the experimental charges (Nandedkar et al. 2014). The H2O-rich nature of the experimental glasses further enhanced equilibration. To facilitate equilibration of doped diopside glass and the starting material, the doped glass was fused twice and ground to grain sizes less than 10 μm for 1 h prior to mixing with the major element oxides. The homogeneous composition of the quenched experimental glasses as indicated by standard deviations of the mean of less than <10 % for most trace elements (Table 3) further support our interpretation that major and trace element equilibrium was achieved. As an additional test, we calculated amphibole–plagioclase temperatures (Holland and Blundy 1994, HB2 94) for those experiments where both phases coexist, with plagioclase compositions as given by Nandedkar et al. (2014). We find that calculated temperatures overestimate experimental temperatures by 2–56 °C, which is acceptable given the rather variable plagioclase compositions.
Another issue is the significant increase in some trace element partition coefficients (e.g., U, Th, Pb, Mo, V) between the experiments performed at 920 and 890 °C (Figs. 7, 8). One possibility is that once equilibrium between amphibole and liquid has been attained, oxide phases crystallize later during the experiments, without complete reequilibration of the experimental charge and, therefore, resulting in an increase in the mineral–melt partition coefficients. If this process were indeed significant, one would expect a significant drop of the trace elements under consideration in the liquid phase. Inspection of Table 3 clearly indicates that the liquid concentrations of U, Th, Pb, Mo and V are not notably different for the various experiments, indicating that late-stage crystallization of oxides and incomplete equilibration between liquid and amphibole cannot explain the increase in partition coefficients. Instead, the increase in partition coefficients is related to an increase in these elements in amphibole (Table 4). An alternative possibility is that small submicron inclusions of melt, or apatite or spinel affected the analyses below 900 °C, yet major element analysis or phosphorus does not provide clear evidence that this was the case. On the other hand, experimental amphibole–liquid partition coefficients for a complete series of trace elements below 900 °C are extremely rare and the power law fits for U, Th and Pb (Tiepolo et al. 2007) as a function of melt compositions are in reasonable agreement with our new data. Whether or not the substantial increase in amphibole–liquid D’s for U, Th, Pb, Mo and V is related to changes of the melt structure is beyond the scope of this study.
Amphibole exchange vectors, REE partition coefficients and regression models
Our new set of experimental data over a temperature range of 230 °C can be used to evaluate the relationship between trace element partitioning and amphibole exchange vectors, such as the edenite (\({\text{NaAl}}^{{({\text{IV}})}}\Box_{ - 1} {\text{Si}}_{ - 1}\)), Tschermak’s (AlIVAlVI Si−1 (Mg, Fe2+, Mn2+)−1) and the cummingtonite (Ca (Mg, Fe2+, Mn2+)−1) exchanges. The former two are intimately related to AlIV; both the exchange vectors and AlIV monotonically decrease with temperature (Fig. 9). The temperature dependence of the edenite exchange in calcic amphiboles has previously been recognized (Blundy and Holland 1990; Holland and Blundy 1994) and is the basis of the amphibole–plagioclase geothermometer. The decreasing edenite and Tschermak’s components correlate linearly with increasing D 0 [M4] (Fig. 9). The cummingtonite component increases from 0.25 at 1010 °C to 0.6 at 780 °C. All three amphibole exchange vectors display R 2 correlation coefficients exceeding 0.8, whereas the ideal radius r 0 remains constant.

a Edenite, Tschermak’s and cummingtonite components of amphiboles as a function of temperature. Solid black lines represent weighted linear fits. b Edenite, Tschermak’s and cummingtonite components of amphiboles as a function of the ideal cation radius for REE (D 0). Solid black lines represent weighted linear fits
Figure 10 shows that the maximum REE partition coefficient D 0 increases exponentially with increasing silica content of the coexisting melt. It is, however, not straightforward to identify the dominant factor(s) controlling the partitioning behavior between amphibole and an evolving liquid because both composition of amphiboles and the composition/polymerization of the melt change concomitantly with decreasing temperature and they are evidently highly correlated. It is, however, remarkable that the ideal radius r0 of M4 is constant and, thus, independent of temperature and amphibole composition, indicating that melt composition/polymerization exerts the dominant control on the incorporation of trace elements on the M4 site.

D 0 for REE as a function of melt silica content (H2O-free basis) for our new experimental data, compared to similar data from calc-alkaline systems of Klein et al. (1997)
To evaluate this further, we calculated simple regression models taking into account both melt composition and amphibole compositional variations. We first tested correlations of Amph/LD’s with X nf/X total, where X nf is the molar fraction of network-forming cations calculated as the sum of network-forming cations (Si + Al fraction balanced by Na + K) divided by the total cations in the melt (Nielsen 1985). Given the excellent correlations of amphibole exchange vectors and trace element partitioning, empirical models relating the Ca distribution coefficient Amph/L D ca to the trace element partition coefficients have been calculated (Table 7) as previously proposed by Sisson (1994). These models take both mineral and melt compositions (and also temperature) into account. We applied linear and York-type regressions (York 1966) with uncorrelated errors between Amph/L D Ca and Amph/L D trace element; the results including errors are listed in Table 7. The R 2 for the fitted trace element partition coefficients ranges from 0.55 to 0.99. The REE’s substituting for Ca on the M4 site display excellent correlations with R 2 > 0.9, except for Sm and Nd that are between 0.85 and 0.9. We also evaluated the influence of Mg# and total Ti in amphibole but these parameters did not improve the overall results. This indicates that for phenocryst–melt pairs, Amph/LDREE partitioning can be estimated from the Ca distribution alone providing an easy-to-use algorithm to implement amphibole–liquid partitioning coefficients into geochemical models in the absence of tight temperature constraints that would alternatively allow the implementation using the lattice strain model (at least for REE and HFSE, see below).
Lattice strain modeling and incorporation of REE in amphibole
The REE partition coefficients are plotted against their ionic radii as an Onuma-type plot (Fig. 4b), (Onuma et al. 1968). Applying the elastic strain model of crystals developed by Brice (1975), Beattie (1994) and Blundy and Wood (1994) and the equation of Blundy and Wood (1994),
(with r 0 ideal radius; E Young’s modulus; R ideal gas constant; N A Avogadro constant; T temperature in [K] and D 0 maximum partition coefficient at r 0), we obtain a coherent set of parabolas with nearly identical ideal radius r 0 for the M4 site in amphibole, with similar Young’s moduli and increasing D 0 (Fig. 4b). Lattice strain parameters for tonalitic amphiboles and literature data for basanites (Adam and Green 1994; LaTourrette et al. 1995) and andesites (Brenan et al. 1995; Green and Pearson 1985) were compiled by Klein et al. (1997). R 0’s of these studies vary between 1.03 and 1.04 Å with the exception of Brenan et al. (1995) who reported a significantly smaller r 0 of 0.99 Å for the M4 site. The r 0’s of this study are within the reported errors of Klein et al. (1997) and indicate that the D 0 of amphibole increases with decreasing temperature and that r 0 remains roughly constant. Young’s moduli of the compiled studies range from 110 to 390 GPa (Adam and Green 1994; Brenan et al. 1995; Green and Pearson 1985; LaTourrette et al. 1995). Our data constrain a rather narrow interval between 310 and 370 GPa, at the upper bound of previous studies. Most likely, considerably lower values reported in previous studies could be affected by melt contamination most evident for the most incompatible elements (La, Ce) resulting in calculated Young’s moduli that are too low.
The details of the incorporation mechanisms of REE into the amphibole M4 structure are still a matter of debate (Bottazzi et al. 1999; Dalpé and Baker 2000; Tiepolo et al. 2000a, b, 2007). Due to their chemical behavior similar to Ca, the REEs are commonly assumed to enter the M4 site in the amphibole structure (e.g., Brenan et al. 1995; Hilyard et al. 2000; Klein et al. 1997), although HREE and Y may also enter the octahedral M2 site in amphibole with an elevated richterite component (Bottazzi et al. 1999). Bottazzi et al. (1999) argue that the cation radius of Ca is 1.12 Å, but the calculated r 0’s derived by lattice strain modeling of our data are between 1.019 and 1.026 Å, in excellent agreement with 1.037 Å for the kaersutite investigated by Bottazzi et al. (1999). To resolve this apparent discrepancy between the dominating Ca cation on M4, with a radius substantially larger than our calculated r0, it has been argued that one of the two M4 sites of the amphibole structure is smaller (M4′ = 1.03 Å; M4 = 1.10–1.11 Å). This is referred to as a cummingtonite-like component, where the M4 site is smaller because of the distortion of the adjacent tetrahedral polyhedrons. This smaller site results in a higher compatibility for HREEs (centred on Dy). Although our study lacks single-crystal-site refinement data, we do not have any indication that the HREE would prefer octahedral sites, similar to observations of Dalpé and Baker (2000). While the cummingtonite component of amphibole in this study increases with decreasing synthesis temperature, r0′s calculated with the lattice strain model remain constant, even for the lowest temperature, cummingtonite-rich amphiboles (Table 6). This indicates that REEs in amphiboles in fractionating calc-alkaline magmas are incorporated in the M4′ site, as a consequence of ordering of the M-HREE on the M4′ site similar to that occupied by Fe–Mg but substantially smaller than the conventional M4 position occupied by Ca. Alternatively, Ca on the M4 site might not strictly be eightfold coordinated. Ionic radii for VIICa or VICa are 1.06 and 1.00 Å, respectively (Shannon 1976). Our calculated r0 on the M4 site is 1.023 Å and 1.037 Å can be calculated for kaersutite from the data from Bottazzi et al. (1999) assuming an oxygen ionic radius of 1.38 Å. A slightly distorted M4 site in some amphiboles could therefore be an alternative explanation for the discrepancy between the r0 calculated from lattice strain modeling and the ionic radius of eightfold coordinated Ca. The ionic radii of VIICa or VICa would be in better agreement with our D0 modeled from the REE.
The calculated parabolas for the trivalent REE indicate that Eu partition coefficients deviate from the fitted parabola. This anomalous behavior is commonly attributed to significant amounts of Eu2+ (Philpotts 1970; Klein et al. 1997). The Eu anomaly increases with decreasing temperature (Table 8). We first calculated the amount of Eu3+ by using the lattice parameters for trivalent REEs (Table 6) and the cation radius for Eu3+ (1.066 Å). The partition coefficients calculated for Eu3+ are reported in Table 8. In order to calculate the partition coefficients for Eu2+, we adopt the approach of Klein et al. (1997) assuming that all Eu2+ with a radius of 1.25 Å occupy the M4 site. The corrected partition coefficients of Eu2+ must then be plotted on a parabola fitting divalent cations, such as Ca, having the same r 0 as the corresponding REEs on the same M4 site. As outlined above, the fitted Young’s modulus for trivalent REE remains approximately constant for the given M4′ site, independent of temperature and amphibole composition. Therefore, we applied the Young’s modulus reported by Brenan et al. (1995) to calculate the partition coefficients of Eu2+ using partition coefficients for Ca reported in Table 5. With the partition coefficients for Eu2+ and Eu3+, we can estimate the fraction of Eu2+ and Eu3+ by mass balance (Table 8). The measured Eu partition coefficients are closer to the partition coefficients calculated for Eu3+ (xEu3+ ranging between 0.58 and 0.78, Table 8), which points to an overall high oxidation state for the experiments (NNO or higher, see above and Nandedkar et al. 2014). The Eu2+ partition coefficients are systematically increasing (0.32–0.72) and are in the same range as Sr partition coefficients (0.34–0.44, except the last experiment with 0.57).
High-field strength elements part I: Ti, Zr, Hf
HFSE reveal an increasing compatibility with decreasing temperature similar to the behavior of the REE. Ti is always compatible, and at lower temperatures Zr and Hf also become compatible. Although Klein et al. (1997) performed their experiments at 1 GPa, partition coefficients match the high-temperature results of this study, indicating that the pressure difference of 0.3 GPa to our study has no significant influence on the high-field strength element partition coefficients. Partition coefficients in calc-alkaline systems determined at higher pressures (1.5–2.5 GPa, Dalpé and Baker 2000) are lower than those of this study. For crystal chemical reasons, it is assumed that the larger Zr and Hf prefer the sixfold coordinated M2 site of the amphibole (Dalpé and Baker 2000; Klein et al. 1997), while Ti can be incorporated into the M1 and M2 sites if amphibole is dehydrogenated (Oberti et al. (2000). The estimated lattice parameter r 0 for a fixed E is similar to the values (0.64 Å) determined for amphibole from andesites (Brenan et al. 1995), while smaller r 0 (0.61–0.63 Å) and lower Young’s moduli (530–960 GPa) were reported for amphibole crystallized from tonalitic melts (Klein et al. 1997) and basanites (LaTourrette et al. 1995). This variation in the data reflects the large uncertainty in the accurate calculation of lattice parameters using three elements only (Ti, Zr and Hf). However, our data provide a robust constraint on the general increase in Zr and Hf partitioning with decreasing temperature and increasing SiO2 content in the liquids.
High-field strength elements part II: Nb and Ta incorporation in amphibole: fractionation
Tiepolo et al. (2000b) present an exhaustive dataset on Nb–Ta partitioning between amphibole and liquid, where they could show that (a) Amph/L D Nb/Amph/L D Ta are inversely correlated with Mg# of amphibole and (b) Amph/L D Nb/Amph/L D Ta are positively correlated with the M1–O bond length of the amphibole lattice. For their dataset of alkaline systems, they fitted the M1–O bond length to amphibole composition and derived the following equation:
We plotted our data and those from the literature and observed an intriguing relationship between Amph/L D Nb/Amph/L D Ta and Mg# (Fig. 11a). While some of the data are negatively correlated, our data and some of the literature data (Hilyard et al. 2000) show a positive correlation, supporting the hypothesis of Tiepolo et al. (2000a, b) that the ionic radii of Nb and Ta are not strictly identical and might differ by 0.01–0.02 Å. There must exist, however, additional or alternative parameters that control Nb and Ta fractionation in amphibole. Liquid SiO2 content and, therefore, the polymerization of silicate liquids has often been shown to be a crucial parameter for increasing partition coefficients of trace elements, yet Fig. 11b shows that melt X nf/X (or SiO2, not shown) is poorly correlated with Amph/L D Nb/Amph/L D Ta and that the fractionation of Nb and Ta must be controlled by crystal chemistry instead. In order to test how the size of the M1–M3 sites in amphibole influence Amph/L D Nb/Amph/L D Ta, we calculated the size of an average M2 site for our experimental amphiboles and those of Tiepolo et al. (2000b), by simply multiplying the cation fraction of M2 cations with their respective ionic radii for octahedrally coordinated cations (Shannon 1976). The results are listed in Table 9, compared to the measured rM1 and the average r M1–M3 (the mean of M1–M3 of Tiepolo et al. 2000b), and illustrated in Fig. 12. We find that simply subtracting 1.38 Å, the ionic radius for oxygen, from the listed M1–O and the average M1–3–O provides a surprisingly good fit with both the measured M1–O and the average M1–3–O of Tiepolo et al. (2000b) (Fig. 12) by using ionic radii for high-spin Fe (Fe2+: 0.78 Å, Fe3+: 0.645 Å, Shannon 1976), but poor fits if we assume an ionic radius of low-spin Fe (Fe2+: 0.61 Å, Fe3+: 0.55 Å, Shannon 1976). Mössbauer spectroscopic studies on amphiboles indicate that both Fe2+ and Fe3+ are in the high-spin state (e.g., Della Ventura et al. 2005). Our data and the data of Tiepolo et al. (2000a, b) thus indicate that the M1–M3 sites in amphibole are significantly larger than the Shannon radii of octahedrally coordinated pentavalent Nb and Ta (0.64 Å), and the positive correlation of Amph/L D Nb/Amph/L D Ta with ionic radii of M1–M3 sites confirms that (a) Nb–Ta fractionation in amphibole is controlled by the lattice parameters and (b) Nb is slightly larger than Ta (see also Blundy and Wood 2003). We thus argue that Amph/L D Nb/Amph/L D Ta is controlled by the bulk site occupancy of cations entering the M1–M3 site in amphibole, and not only by Mg# and Ti, as indicated by the equation of Tiepolo et al. (2000a, b), which was developed for amphiboles in alkaline systems, but provides poor fits for our new set of amphiboles crystallized from calc-alkaline magmas. The most coherent correlation for calc-alkaline and alkaline systems is shown by Amph/L D ca versus Amph/L D Nb/Amph/L D Ta (Fig. 11c). Good linear fits with R 2 between 0.66 (our data) and 0.68 (Tiepolo et al. 200b) for calcic amphibole with cummingonite <0.4 are obtained, indicating that the combined effects of amphibole and melt composition (and thus temperature) controls Nb–Ta amphibole–melt partitioning. An ancillary result from our analysis of amphibole composition and Amph/L D Nb/Amph/L D Ta is that Nb–Ta fractionation is not restricted to amphiboles with Mg# < 0.7 (cf. Foley et al. 2002), thus limiting the importance of Fe-rich amphibolite melting in explaining the low Nb/Ta ratio of Archean Tonalite-Trondhjemite-Granodiorite series (TTG's).

a Mg# versus Amph/L D Nb/Amph/L D Ta, for our new experimental data, compared to data from the literature. Note the positive correlation for our new data and the negative correlation for some of the literature data. b Amph/L D Nb/Amph/L D Ta versus X nf/X of coexisting liquids, on an anhydrous basis. c Amph/L D Nb/Amph/L D Ta versus Amph/L D Ca indicating the combined effects of liquid and mineral composition adequately explains the data. Regression lines are calculated as weighted linear regressions for our new data (red) and the data of Tiepolo et al. (2000a, b) (blue). Data sources: Tiepolo et al. 2000b (T00), Dalpe and Baker (2000) (DB00), Hilyard et al. 2000 (HY00)

Calculated ionic radius for the amphibole M2 site of our new experimental data (red symbols) and the data of Tiepolo et al. (2000b) (T00). The data are compared to measured values of the M1 site, to the average M1–M3 site, both from Tiepolo et al. 2000b (their Table 2), and calculated M2 sites (with data from Tiepolo et al. 2000a, b) for low-spin and high-spin Fe, with ionic radii tabulated by Shannon (1976). Note that the measured single-crystal-site refinement data from Tiepolo et al. (2000b) are fully consistent with Fe in the high-spin state
Implications of the experimental data on derivative Si-rich liquids
Significant amphibole fractionation and delayed plagioclase crystallization have been identified as important phase equilibria controls on derivative magma composition in hydrous, arc-related, calk-alkaline systems leading to (1) corundum-normative intermediate to acidic differentiates (Cawthorn et al. 1976; Müntener et al. 2001; Davidson et al. 2007; Nandedkar et al. 2014) and (2) high Sr/Y ratios comparable to “adakites” inferred to originate from partial melting of mafic crust (slab and/or sub-arc crust melting, e.g., Defant and Drummond 1990; Moyen 2009). Figure 13 depicts the evolution of REE using an andesitic composition as a starting material (NH 116, Hürlimann et al. 2016). For each experimental liquid, “enrichment factors” have been calculated based on the measured trace element compositions of the experimental glasses (Table 3) assuming an effective constant trace element composition of the starting material (EA Table 1), and the results are shown in Fig. 13. Despite some uncertainties in the assumption of a constant starting material, our calculations use liquid compositions only and are thus independent of (highly variable) partition coefficients from the literature (e.g., plagioclase, Bedard 2006). The liquid REE evolution shows that amphibole–plagioclase–Fe-Ti oxide ± apatite-saturated melts evolve trough-like REE patterns with increasing depletion of MREE, and in the last steps below 800 °C significant depletions in Eu and LREE (Fig. 13). Interestingly, the derivative liquids calculated in this way display increasing Sr/Y ratios from 40 (starting composition of the andesite) to almost 140 over the temperature interval between 1010 and 830 °C, before decreasing in the last steps below 800 °C, presumably related to significant apatite crystallization in rhyodacitic to rhyolitic differentiates (Fig. 14). On the other hand, Sr/Ba ratios remain almost constant, which can be explained by our relatively low-K starting material (see Nandedkar et al. 2014, for a discussion) and late saturation in biotite at 700 °C only. Relative to literature data for the Archean (Tang et al. 2015) and the southern Cascade volcanic products (Grove et al. 2002; Clynne et al. 2008; Wende et al. 2015; Sisson and Calvert, personal communication), the combined effects of amphibole–plagioclase–Fe-Ti oxide ± apatite fractionation as simulated by our experiments are more consistent with the Archean dataset, while the Cascades field shows higher Ba/Sr at a given Sr/Y. Other trace element ratios such as Nb/Zr calculated in the same way decrease until zircon saturation is reached. Combined apatite and zircon crystallization is most consistent with our experimental data. Our new experimental data illustrate possible evolutionary trends of trace element ratios for crystallizing (or melting) middle to lower crustal rocks.

Evolution of REE of crystallizing calc-alkaline liquids. Starting composition is an andesite dyke from the Adamello massif. Enrichment factors were calculated for each experiment from measured liquid compositions and the respective starting material (Electronic Appendix EA 1). The REE develop from a LREE enriched to a MREE-depleted “trough-like” pattern. During the last two experiments, LREE and Eu significantly decrease. NH116 from Hürlimann et al. (2016)

Sr/Y vs Sr/Ba (a) and Sr/Y vs Nb/Zr (b) from experimental liquids compared to Archean rocks and the southern Cascades. Archean data compiled by Tang et al. (2015), Cascades data from Clynne et al. (2008), Grove et al. (2002) and Wende et al. (2015) and Sisson and Calvert (pers. comm). Trace element ratios are calculated from the model developed in Fig. 13. Note that for the last two experiments at 730 and 700 °C zircon saturation values (calculated after Gervasoni et al. 2016) were used. Saturation of accessory phases (zircon, apatite: Zr, Sr, Y) and biotite/k-feldspar (Ba) has an important control on trace element ratios
Combining both major (Nandedkar et al. 2014) and trace element (this work) constraints on the evolution of near-primary, mantle-derived hydrous calc-alkaline magmas by fractional crystallization at lower to middle crustal levels provides limits on some major and trace element indicators to infer geological processes such as the ASI (corundum-normative magmas) and high Sr/Y (“adakitic”) signatures. Given the difficulty of preserving slab source trace element ratios during magma transport to the eruptive or plutonic products, we infer that most trace element signatures and ratios are probably modified by deep crustal differentiation processes and/or plagioclase accumulation effects (e.g., Macpherson et al. 2006; Dessimoz et al. 2012). This relaxes the requirement of partial melting of a garnet-bearing mafic source either at the slab–mantle interface in the subducted plate or by melting of the lower arc crust. Amphibole-dominated fractionation in andesitic to dacitic compositions in intermediate to deep magma reservoirs might also be able to fractionate Nb from Ta and explain the subchondritic Nb/Ta ratios of many evolved silicic plutonic rocks, which has been attributed mainly to the effects of mica (e.g., Stepanov et al. 2014).
Conclusions
Amphibole compositions analyzed in this study span a large compositional range from pargasite to magnesiohornblende to cummingtonite. The corresponding (coexisting) liquids range from andesite to rhyolite, at temperatures from 1010 to 730 °C at 0.7 GPa. The consistency of the experimentally derived amphibole–liquid partition coefficients derived by LA-ICP-MS along the liquid line of descent can be utilized to calculate their equilibrium liquid compositions in calc-alkaline systems. Trace element groups (such as REE, HFSE, LLE, transition metals and actinides) reveal increasing partition coefficients with decreasing temperature and increasing polymerization of the liquid. The only exception is K displaying an opposite trend with decreasing partition coefficients with temperature. Trivalent REEs occupy the M4′ site in the amphibole structure, while important lattice parameters (r 0 and E) remain constant over a temperature interval of 230 °C and can be well modeled with the lattice strain model of Blundy and Wood (1994). The D 0 for trivalent elements correlate linearly with the edenite, tschermaks and cummingtonite component of the amphibole. We derived simple expressions relating Ca partitioning between amphibole and liquid and trace element partition coefficients that can be implemented in geochemical modeling of crystallization/melting processes of intermediate to acidic calc-alkaline systems. The increasing Amph/L D Nb/Amph/L D Ta with decreasing temperature indicates that amphibole crystallization in the middle to lower crust contributes to the development of subchondritic Nb/Ta ratios in evolved Si-rich arc magmas. Our set of new amphibole partition coefficients can be applied to calculate equilibrium liquid compositions for H2O-rich intermediate andesites to high-silica rhyolite, where amphibole is a major phase-determining phase equilibria and calc-alkaline differentiation trends. Finally, based on directly measured liquid compositions from 1010 to 700 °C we infer that fractional crystallization develops a remarkable middle REE trough in chondrite-normalized andesitic to rhyolitic liquids, an increase followed by a decrease in the Sr/Y ratio, and other trace element ratios (e.g., Sr/Ba, Nb/Zr).
References
Adam J, Green TH (1994) The effects of pressure and temperature on the partitioning of Ti, Sr and REE between amphibole, clinopyroxene and basanitic melts. Chem Geol 117(1–4):219–233
Beattie P (1994) Systematics and energetics of trace-element partitioning between olivine and silicate melts: implications for the nature of mineral/melt partitioning. Chem Geol 117:57–71
Bedard J (2006) Trace element partitioning of plagioclase feldspar. Geochim Cosmochim Acta 70(14):3717–3742
Blundy JD, Holland TJB (1990) Calcic amphibole equilibria and a new amphibole-plagioclase geothermometer. Contrib Mineral Petrol 104(2):208–224
Blundy JD, Wood BJ (1994) Prediction of crystal-melt partition coefficients from elastic moduli. Nature 372:452–454
Blundy JD, Wood BJ (2003) Mineral-melt partitioning of U, Th and their daughters. Rev Mineral 52:59–123
Bottazzi P, Tiepolo M, Vannucci R, Zanetti A, Brumm R, Foley SF, Oberti R (1999) Distinct site preferences for heavy and light REE in amphibole and the prediction of D-Amph/L(REE). Contrib Mineral Petrol 137(1–2):36–45
Brenan JM, Shaw HF, Ryerson FJ, Phinney DL (1995) Experimental determination of trace-element partitioning between pargasite and a synthetic hydrous andesitic melt. Earth Planet Sci Lett 135(1–4):1–11
Brice JC (1975) Some thermodynamic aspects of the growth of strained crystals. J Cryst Growth 28:249–253
Cawthorn RG, Strong DF, Brown PA (1976) Origin of corundum-normative intrusive and extrusive magmas. Nature 259(5539):102–104
Clowe CA, Popp RK, Fritz JJ (1988) Experimental investigation of the effect of oxygen fugacity on ferric-ferrous ratios and unit-cell parameters of 4 natural clinoamphiboles. Am Mineral 74:487–499
Clynne MA, Muffler LJP, Siems, DF, Taggart JE, Bruggman P (2008) Major and EDXRF trace element chemical analyses of Volcanic rocks from Lassen Volcanic National Park and Vicinity, California. USGS open-file report 2008–109, U.S. Geological Survey, Reston, VA
Dalpé C, Baker DR (2000) Experimental investigation of large-ion-lithophile-element-, high-field-strength-element- and rare-earth-element-partitioning between calcic amphibole and basaltic melt: the effects of pressure and oxygen fugacity. Contrib Mineral Petrol 140(2):233–250
Davidson J, Turner S, Handley H, MacPherson C, Dosseto A (2007) Amphibole ‘sponge’ in arc crust? Geology 35:787–790
Defant MJ, Drummond MS (1990) Derivation of some modern arc magmas by melting of young subducted lithosphere. Nature 347:662–665
Della Ventura G, Iezzi G, Redhammer GJ, Hawthorne FC, Scaillet B, Novembre D (2005) Synthesis and crystal-chemistry of alkali amphiboles in the system Na2O–MgO–FeO–Fe2O3–SiO2–H2O as a function of fO2. Am Mineral 90:1375–1383
Dessimoz M, Müntener O, Ulmer P (2012) A case for hornblende dominated fractionation of arc magmas: the Chelan Complex (Washington Cascades). Contrib Mineral Petrol 163(4):567–589
Ewart A, Griffin WL (1994) Application of proton-microprobe data to trace element partitioning in volcanic rocks. Chem Geol 117:251–284
Foley SF, Tiepolo M, Vannucci R (2002) Growth of early continental crust controlled by melting of amphibolites in subduction zones. Nature 417:837–840
Gervasoni F, Klemme S, Rocha-Junior ERV, Berndt J (2016) Zircon saturation in silicate melts: a new and improved model for aluminous and alkaline melts. Contrib Mineral Petrol 171:21
Goldstein JI, Newbury DE, Echlin P, Joy DC, Fiori C, Lifshin E (1992) Scanning electron microscopy and X-ray microanalysis. Plenum Press, New York
Green TH, Pearson NJ (1985) Experimental-determination of REE partition coefficients between amphibole and basaltic to andesitic liquids at high-pressure. Geochim Cosmochim Acta 49(6):1465–1468
Grove TL, Parman SW, Bowring SA, Price RC, Baker MB (2002) The role of an H2O-rich fluid component in the generation of primitive basaltic andesites and andesites from the Mt. Shasta region, California. Contrib Mineral Petrol 142:375–396
Guillong M, Meier DL, Allan MM, Heinrich CA, Yardley BWD (2008) SILLS: matlab-based program for the reduction of laser ablation ICP-MS data of homogeneous materials and inclusions. Mineral Ass Can Short Course 40:328–333
Hilyard M, Nielsen RL, Beard JS, Patino-Douce A, Blencoe J (2000) Experimental determination of the partitioning behavior of rare earth and high field strength elements between pargasitic amphibole and natural silicate melts. Geochim Cosmochim Acta 64(6):1103–1120
Holland T, Blundy J (1994) Non-ideal interactions in calcic amphiboles and their bearing on amphibole-plagioclase thermometry. Contrib Mineral Petrol 116(4):433–447
Hürlimann N, Müntener O, Ulmer P, Nandedkar R, Chiaradia M, Ovtcharova M (2016) Primary magmas in continental arcs and their differentiated products: petrology of a post-plutonic dyke suite in the Tertiary Adamello batholith (Alps). J Petrol 57:495–534
Jochum KP, Weis U, Stoll B, Kuzmin D, Yang Q, Raczek I, Jacob DE, Stracke A, Birbaum K, Frick DA, Günther D, Enzweiler J (2011) Determination of reference values for NIST SRM 610–617 glasses following ISO guidelines. Geostand Geoanal Res 35(4):397–429
Klein M, Stosch HG, Seck HA (1997) Partitioning of high field-strength and rare-earth elements between amphibole and quartz-dioritic to tonalitic melts: an experimental study. Chem Geol 138(3–4):257–271
Klein M, Stosch HG, Seck HA, Shimizu N (2000) Partitioning of high field-strength and rare-earth elements between clinopyroxene and garnet in andesitic to tonalitic systems. Geochim Cosmochim Acta 64:99–115
Korolyuk YN, Usova LV, Nigmatulina EN (2009) Accuracy in the determination of the compositions of main rock forming silicates and oxides on a JXA-8100 microanalyer. J Anal Chem 64(10):1042–1046
LaTourrette T, Hervig RL, Holloway JR (1995) Trace element partitioning between amphibole, phlogopite, and basanite melt. Earth Planet Sci Lett 135(1–4):13–30
Leake BE (1978) Nomenclature of amphiboles. Am Mineral 63(11):1023–1052
Leake BE, Woolley AR, Arps CES, Birch WD, Gilbert MC, Grice JD, Hawthorne FC, Kato A, Kisch HJ, Krivovichev VG, Linthout K, Laird J, Mandarino JA, Maresch WV, Nickel EH, Rock NMS, Schumacher JC, Smith DC, Stephenson NCN, Ungaretti L, Whittaker EJW, Guo YZ (1997) Nomenclature of amphiboles: report of the subcommittee on amphiboles of the International Mineralogical Association, commission on new minerals and mineral names. Am Mineral 82(9–10):1019–1037
Macpherson CG, Dreher ST, Thirlwall MF (2006) Adakites without slab melting: high pressure differentiation of island arc magma, Mindanao, the Philippines. Earth Planet Sci Lett 243:581–593
Moyen JF (2009) High Sr/Y and La/Yb ratios: the meaning of the ‘adakitic signature’. Lithos 112(3–4):556–574
Müntener O, Kelemen PB, Grove TL (2001) The role of H2O during crystallization of primitive arc magmas under uppermost mantle conditions and genesis of igneous pyroxenites: an experimental study. Contrib Mineral Petrol 141:643–658
Nandedkar RH, Ulmer P, Müntener O (2014) Fractional crystallization of primitive hydrous arc magmas: an experimental study at 0.7 GPa. Contrib Mineral Petrol 167(6):1015
Nielsen RL (1985) A method for the elimination of the compositional dependence of trace-element distribution coefficients. Geochim Cosmochim Acta 49(1):1575–1779
Oberti R, Vannucci R, Zanetti A, Tiepolo M, Brumm RC (2000) A crystal chemical re-evaluation of amphibole/melt and amphibole/clinopyroxene D-Ti values in petrogenetic studies. Am Mineral 85(3–4):407–419
Onuma N, Higuchi H, Wakita H, Nagasama H (1968) Trace element partition between 2 pyroxenes and host lava. Earth Planet Sci Lett 5:47–51
Pearce NJG, Perkins WT, Westgate JA, Gorton MP, Jackson SE, Neal CR, Chenery SP (1997) A compilation of new and published major and trace element data for NIST SRM 610 and NIST SRM 612 glass reference materials. Geostand Newsl 21(1):115–144
Philpotts JA (1970) Redox estimation from a calculation of Eu2+ and Eu3+ concentrations in natural phases. Earth Planet Sci Lett 9:257–268
Shannon RD (1976) Revised effective ionic-radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst 32:751–767
Sisson TW (1994) Hornblende-melt trace-element partitioning measured by ion microprobe. Chem Geol 117(1–4):331–344
Spear FS, Kimball KL (1984) RECAMP—a FORTRAN IV program for estimating Fe3+ contents in amphiboles. Comput Geosci 10(2–3):317–325
Stepanov A, Mavrogenes JA, Meffre S, Davidson P (2014) The key role of mica during igneous concentration of tantalum. Contrib Mineral Petrol 167:1009–1016
Tang M, Rudnick RL, McDonough WF, Gaschnig RM, Huang Y (2015) Europium anomalies constrain the mass of recycled lower continental crust. Geology 43:703–706
Tiepolo M, Vannucci R, Bottazzi P, Oberti R, Zanetti A, Foley S (2000a) Partitioning of rare earth elements, Y, Th, U, and Pb between pargasite, kaersutite, and basanite to trachyte melts: implications for percolated and veined mantle. Geochem Geophys Geosyst 1:2000GC000064
Tiepolo M, Vannucci R, Oberti R, Foley S, Bottazzi P, Zanetti A (2000b) Nb and Ta incorporation and fractionation in titanian pargasite and kaersutite: crystal-chemical constraints and implications for natural systems. Earth Planet Sci Lett 176(2):185–201
Tiepolo M, Oberti R, Zanetti A, Vannucci R, Foley SF (2007) Trace-element partitioning between amphibole and silicate melt in amphiboles: crystal chemistry, occurrence, and health issues. Rev Mineral 67:417–451
Wende AM, Johnson CM, Beard BL (2015) Tracing changes in mantle and crustal influences in individual cone-building stages at Mt. Shasta using U-Th and Sr isotopes. Earth Planet Sci Lett 428:11–21
Wood BJ, Blundy JD (1997) A predictive model for rare earth element partitioning between clinopyroxene and anhydrous silicate melt. Contrib Mineral Petrol 129(2–3):166–181
York D (1966) Least-squares fitting of a straight line. Can J Phys 44(5):1079–1086
Acknowledgments
Alexey Ulianov is thanked for expertise and discussion about the LA-ICP-MS data acquisition. Markus Wälle’s help during post-processing of LA-ICP-MS data with SILLS was very valuable. The work is part of the ProDoc program “4-D Adamello” and was supported by the Swiss National Science Foundation (grants PDFMP2-123097/1 and PDAMP2-123074). Comments by Jon Blundy, Massimo Tiepolo, Roland Stalder and Stefan Klemme on an early version were very helpful. Reviews by Tom Sisson and Roger Nielsen are gratefully acknowledged and helped to improve the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Timothy L. Grove.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

{kind=link}
Cite this article
Nandedkar, R.H., Hürlimann, N., Ulmer, P. et al. Amphibole–melt trace element partitioning of fractionating calc-alkaline magmas in the lower crust: an experimental study. Contrib Mineral Petrol 171, 71 (2016). https://doi.org/10.1007/s00410-016-1278-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-016-1278-0