
Abstract
Osteopontin (OPN) is a multifunctional phosphorylated protein that is involved in physiological and pathological events. Emerging evidence suggests that OPN also plays a critical role in the pathogenesis of respiratory diseases. OPN can be produced and secreted by various cell types in lungs and overexpression of OPN has been found in acute lung injury/acute respiratory distress syndrome (ALI/ARDS), pulmonary hypertension (PH), pulmonary fibrosis diseases, lung cancer, lung infection, chronic obstructive pulmonary disease (COPD), and asthma. OPN exerts diverse effects on the inflammatory response, immune cell activation, fibrosis and tissue remodeling, and tumorigenesis of these respiratory diseases, and genetic and pharmacological moudulation of OPN exerts therapeutic effects in the treatment of respiratory diseases. In this review, we summarize the recent evidence of multifaceted roles and underlying mechanisms of OPN in these respiratory diseases, and targeting OPN appears to be a potential therapeutic intervention for these diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Respiratory diseases, including chronic obstructive pulmonary disease (COPD), asthma, lung cancer, pulmonary hypertension (PH), idiopathic pulmonary fibrosis (IPF), pulmonary infection, and other respiratory diseases, are multifactorial diseases that result in morbidity and mortality [1]. The pathogenesis of these respiratory diseases is tremendously complex, and the underlying mechanisms remain largely unknown. Osteopontin (OPN), also known as secreted phosphoprotein 1 (SPP1) or early T-lymphocyte activation-1 (ETA-1), is a member of the small integrin-binding ligand N-linked glycoprotein (SIBLING), which functions both as a matricellular protein (when bound to the matrix) and as an inflammatory cytokine when secreted in a soluble form [2]. OPN can be produced by various cell types (e.g., immune cells) and has been reported to exert pleiotropic effects on bone remodeling, tumorigenesis, the inflammatory response, cell proliferation, and migration and is involved in various diseases (e.g., rheumatoid arthritis, cancers, and cardiovascular diseases) [3,4,5].
Accumulating evidence suggests that OPN also plays a critical role in the pathogenesis of respiratory diseases [6,7,8,9,10]. Under physiological conditions, OPN expression in the plasma and lung tissues (e.g., alveolar macrophages) is relatively weak; however, OPN is significantly upregulated in several inflammatory, fibrotic, malignant, and vascular respiratory diseases, and targeting OPN exerts therapeutic effects in the treatment of respiratory diseases [6,7,8,9,10]. In this review, we summarize the structure and function of OPN, the roles and underlying mechanisms of OPN in respiratory disease pathologies and recent advances in the pharmacological and molecular modulation of OPN in the treatment of PH. According to the current literature, targeting OPN appears to be a potential therapeutic strategy for the treatment of respiratory diseases.
OPN
OPN Structure
OPN is a 41–75 kDa multifunctional matricellular protein encoded by the human SPP1 gene and its murine counterpart Spp1. The OPN-encoding gene has 7 exons and is located on chromosome 4 region 22 (4q1322.1) in the human genome and on murine chromosome 5 [2]. Several OPN isoforms are generated by alternative translation, alternative splicing, and posttranslational modifications (PTMs). Alternative translation of full-length SPP1 mRNA can generate 2 OPN isoforms: secreted OPN (sOPN) and intracellular OPN (iOPN). The 2 isoforms exhibit different expression levels in various cell types and exert differential effects [11]. Furthermore, alternative splicing of the human OPN transcript results in 5 isoforms, OPN-a (full length), OPN-b (missing exon 5), OPN-c (missing exon 4), OPN-4 (also known as OPN-d, missing exons 4 and 5), and OPN-5 (the longest isoform, with an extra exon located between exons 3 and 4). All human splicing isoforms contain the following functional domains: (1) Arg-Gly-Asp (RGD) domain, (2) SVVYGLR domain (SLAYGLR in murine), (3) ELVTDFTDLPAT domain, (4) calcium-binding domain, and (5) heparin-binding domain [2]. OPN has thrombin and matrix metalloproteinase (MMP) cleavage sites [11].
OPN Functions
OPN plays multifarious roles in a variety of physiological and pathological events. Under physiological conditions, circulating and tissue OPN expression levels are relatively low, and OPN is suggested to play a key role in regulating biomineralization, wound healing, and developmental processes [11, 12]. Under pathological conditions, OPN is often significantly upregulated and exerts pleiotropic effects in several inflammatory, autoimmune, degenerative, fibrosis, and oncologic diseases, such as diabetes, stroke, kidney injury, cardiac fibrosis, and cancers [3,4,5, 13]. The pleiotropic effects of sOPN are partly attributed to its interaction with several cell surface receptors (e.g., integrins and CD44 variants), calcium, and heparin. Binding of OPN to its receptor (e.g., integrins, CD44, and ICOSL) activates multiple signaling pathways, including the PI3K/AKT, MEK/ERK, NF-κB, and JAK/STAT pathways, which regulate various cellular functions, including cell-mediated immunity, cell proliferation, invasion, migration, and fibrosis [14, 15]. However, information regarding the function of iOPN is limited, and iOPN was suggested to function as an adaptor or scaffold protein in several signal transduction pathways [e.g., the Toll-like receptor 9 (TLR9) pathway] and play a key role in regulating the function of immune cells, antiviral response, and tumorigenesis [16,17,18,19]. Furthermore, posttranslational modification (PTM) is another crucial mechanism that controls the functions of OPN, which may also affect the roles of OPN in health and diseases [20, 21].
OPN Receptors
Integrins are the most important cell surface receptor of OPN; the RGD domain allows interaction with several integrins, including αv (β1, β3, β5, β6, β8) and (α5, α8) β1 and/or αIIbβ3; the SVVYGLR domain binds to α4β1, α4β7, and α9β; and α4β1 is suspected to bind to the ELVTDFTDLPAT domain [22, 23]. Additionally, CD44 is another important receptor of OPN, and OPN has also been shown to interact with CD44, specifically CD44v3 and CD44v6-7 variants, via the C-terminal calcium domain [24,25,26]. Recently, ICOSL (inducible T-cell costimulator ligand) has been identified as a novel receptor of OPN, and OPN can promote tumor metastasis by binding ICOSL [27]. Moreover, OPN also interacts with various ECM proteins, such as fibronectin and collagen types I, II, III, IV, and V [21, 28].
Posttranslational Modifications (PTMs)
The human OPN protein is subjected to extensive PTMs, including phosphorylation, O-glycosylation, sulfation, and transglutamination, which allow for a monomeric molecular weight ranging from 42 to 75 kDa. These PTMs can be tissue-specific and influence both OPN structure and function. To date, 36 phosphoresidues, including serine, threonine, and tyrosine residues, have been localized in the OPN sequence and the phosphorylation of OPN is mainly mediated by Golgi FAM20C [29, 30]. Furthermore, the degree of OPN phosphorylation can be cell-type specific, and deferentially phosphorylated OPN may exhibit different biological effects [31]. Furthermore, 5 O-glycosylation sites have also been identified in exon 6 of the human OPN sequence [20, 29]. O-glycosylation can also influence the functional properties of OPN. For instance, in vitro, overexpression of O-glycosylation mutant OPN can significantly inhibit the proliferation and migration of lung cancer cells [32]. OPN also serves as a substrate for the enzyme transglutaminase 2 (TG2), which catalyzes the formation of cross-linking protein aggregates [33]. TG2-mediated OPN polymerization induces a more ordered conformational structure, and polymeric OPN displays higher collagen-binding activity and promotes enhanced cell adhesion and migration compared with monomeric OPN [21, 34, 35]. However, further in-depth investigations are needed to elucidate the functional properties of these various forms of OPN.
OPN in Respiratory Diseases
OPN in ALI/ARDS
Acute lung injury/acute respiratory distress syndrome (ALI/ARDS) is a life-threatening respiratory disease with high rates of mortality and morbidity. The pathogenesis of ALI/ARDS is complex and mainly characterized by alveolar epithelial and endothelial barrier dysfunction, severe inflammatory response within the lung, alveolar edema, impaired surfactant synthesis, and significant hypoxemia. An excessive inflammatory response within the lung is a key pathophysiological characteristic of ALI/ARDS [36]. As a potent inflammatory mediator, OPN has been shown to be involved in the inflammatory response of ALI/ARDS [37,38,39,40,41].
Intrapulmonary causes (e.g., influenza virus and coronavirus) can significantly increase OPN expression in plasma and/or lung tissues, and upregulated OPN is suggested to play a detrimental role in the progression of ALI/ARDS (Fig. 1A) [37,38,39]. Higher serum OPN levels were observed in patients with severe coronavirus disease 2019 (COVID-19)-related ARDS than in healthy controls, and higher circulating OPN was associated with higher odds of death and mechanical ventilation [38, 39]. In addition, in influenza virus-induced ALI/ARDS, significant upregulation of OPN expression is observed in the plasma and/or lung tissue of patients and mice with influenza virus infection, and OPN exacerbates the progression of ALI/ARDS by promoting influenza virus-induced macrophage necroptosis, increasing alveolar epithelial cell apoptosis, downregulating epithelial sodium channel (ENaC) expression and extracellular matrix (ECM) destruction [37, 42]. OPN ablation in mice reduces viral replication and inhibits lung inflammation and injury [37, 40]. Similarly, various extrapulmonary causes, including sepsis, acute kidney injury (AKI), intestinal ischemia‒reperfusion, and transfusion, also result in significant upregulation of OPN in lung tissues and/or plasma [41, 43]. Notably, OPN can be excessively produced and released by lung tissues, immune cells (e.g., macrophages), and other tissues (e.g., injured kidney tissue), and upregulated OPN has been demonstrated to exacerbate cell necroptosis, induce lung endothelial hyperpermeability, facilitate immune cell infiltration (e.g., neutrophils), and increase cytokine and chemokine production, which may synergistically aggravate lung injury [6, 41, 43,44,45]. Consistently, the progression of ALI/ARDS was exacerbated by the administration of recombinant OPN (rOPN) and attenuated in OPN knockout (OPN−/−) mice or anti-OPN antibody-treated mice [6, 41, 43,44,45]. In addition, increased ratio of T helper 17 cells (Th17)/regulatory T cells (Treg) has been identified as a risk indicator for early ALI/ARDS [46]. Macrophages can be polarized into proinflammatory M1 macrophages and anti-inflammatory M2 macrophages, and macrophage M1/M2 imbalance is implicated in ALI/ARDS at various stages. A recent study has shown that OPN increases Th17/Treg ratio and promotes macrophage polarize to proinflammatory M1 phenotype by downregulating von Hippel–lindau (VHL) expression and ubiquitination-dependent hypoxia-inducible factor-1α (HIF-1α) degradation, thus exacerbating ALI/ARDS [47].

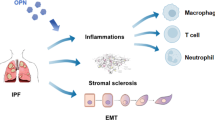
Roles and underlying mechanisms of OPN in ALI/ARDS, PH, and Pulmonary fibrosis diseases. A Detrimental role and underlying mechanisms of OPN in ALI/ARDS. B Beneficial role and underlying mechanisms of OPN in ALI/ARDS. C Detrimental role and underlying mechanisms of OPN in PH. D Detrimental role and underlying mechanisms of OPN in pulmonary fibrosis diseases. OPN, osteopontin; ALI/ARDS, acute lung injury/acute respiratory distress syndrome; PH, pulmonary hypertension; Th17/Treg, T helper 17 cells (Th17)/regulatory T cells (Treg); ENaC, epithelial sodium channel; ERK1/2, extracellular regulated protein kinases 1/2; Akt, PASMC, pulmonary artery smooth muscle cell; PAF, pulmonary adventitial fibroblasts; ECM, extracellular matrix; TGF-β1, transforming growth factor-β1; NETs, neutrophil extracellular traps; EMT, epithelial-to-mesenchymal transition (Figures are created using Biorender. com).
However, conflicting data suggest that OPN plays a protective role in several types of ALI/ARDS. For example, OPN expression was upregulated in the bronchoalveolar lavage fluid (BALF) of patients with ARDS and histone- and LPS-induced ALI/ARDS mice [40]. OPN, particularly phosphorylated OPN, can bind with high affinity to extracellular histones, which act as key danger-associated molecular pattern (DAMP) proteins that promote inflammation and cell death. The interaction between OPN and extracellular histones subsequently inhibits the proinflammatory and cytotoxic effects of extracellular histones by suppressing the formation of tissue-damaging neutrophil extracellular traps (NETs) (Fig. 1B) [40]. Increased proinflammatory mediator (e.g., IL-6) production and aggravated lung injury were observed in OPN−/− mice. Thus, further investigations are warranted to further illustrate the exact roles and underlying mechanisms of OPN in the pathogenesis of ALI/ARDS.
OPN in Pulmonary Hypertension
Pulmonary hypertension (PH) is a progressive and fetal respiratory disease characterized by remodeling of resistance pulmonary arteries, leading to increases in pulmonary artery pressure and right heart failure. Recent studies have demonstrated that OPN plays a key role in the pathogenesis of PH (Fig. 1C) [48]. Expression of OPN in the plasma and/or lung tissues is markedly upregulated in patients with idiopathic pulmonary arterial hypertension (iPAH), connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH), chronic thromboembolic pulmonary hypertension (CTEPH), and congenital heart disease-associated PAH (CHD-PAH), and the plasma concentration of OPN shows a positive correlation with PH severity [10, 49,50,51,52,53]. Similarly, the expression of OPN was also found to be upregulated in the lung tissues of murine models of PAH [hypoxia-, monocrotaline (MCT)-, and systemic-to-pulmonary shunt-induced PAH] [10, 52, 54, 55]. In in vivo experiments, knockout of OPN substantially alleviated the development of PAH [10]. Treatment with rOPN can significantly promote the proliferation and migration of pulmonary artery adventitial fibroblasts from PH animals [54].
Pulmonary vascular remodeling in three-layered structure (i.e., intima, media, and adventitia) is the most important structural alteration in PH and involves various cell types (i.e., endothelial cells, smooth muscle cells, and fibroblasts, respectively). Overproliferation and resistance to apoptosis of pulmonary artery smooth muscle cells (PASMCs) are important hallmark features of PH-pulmonary vascular remodeling [48]. As previously reported, OPN acts as a key mediator for promoting vascular smooth muscle cell (VSMC) proliferation and migration, and upregulation of OPN is suggested to contribute to pathological vascular remodeling associated with various vascular disorders (e.g., arteriosclerosis) [56]. Similarly, OPN is upregulated in proliferating PASMCs compared with quiescent PASMCs, which may further enhance the proliferation and migration of PASMCs by activating integrin αvβ3-mediated Akt and ERK1/2 signaling pathways [52]. Furthermore, PASMC senescence is a key pathogenic mechanism for pulmonary vascular remodeling. OPN is upregulated in senescent PASMCs, and senescent PASMC-derived OPN can also stimulate the proliferation and migration of normal PASMCs [10]. In addition, during the pathological vascular remodeling process, pulmonary adventitial fibroblasts (PAFs) are aberrantly activated and show excessive proliferation, migration, and differentiation. The expression levels of OPN and its cognate receptors (αvβ3, CD44) are markedly increased in the pulmonary vascular adventitia and PAFs of PH patients and animal PH models. OPN is suggested to contribute to the constitutively activated (highly proliferative, migratory, and proinvasive) phenotypes of PAFs by activating the ERK1/2 and Akt pathways, which may further exacerbate the pulmonary vascular remodeling process [54]. In addition, OPN has been reported to promote the differentiation and angiogenesis of endothelial cells in systemic vasculature by activating various pathways (e.g., PI3K/Akt and ERK1/2 pathways) [15]. However, the exact roles and underlying mechanism of OPN in pulmonary vascular endothelial cells remain unknown and require further investigation.
OPN in Pulmonary Fibrosis Diseases
Pulmonary fibrosis diseases are progressive and lethal respiratory diseases that occur as a consequence of various chronic pulmonary diseases. During the pathological process of pulmonary fibrosis, the overpopulation and aberrant activation of fibroblasts and myofibroblasts induce excessive ECM deposition, aberrant lung repair, tissue scar formation, disruption of the lung parenchymal architecture, and irreversible lung function impairment [57]. OPN has been reported to function as a profibrotic cytokine in many fibrotic diseases such as skin, heart, and kidney fibrosis [5, 58, 59]. Emerging evidence suggests that OPN also plays a pivotal role in several pulmonary fibrosis diseases, including idiopathic pulmonary fibrosis (IPF), cystic fibrosis, silicosis, and smoking and asbestos-induced pulmonary fibrosis (Fig. 1D) [7, 60].
IPF is the most common fatal fibrotic respiratory disease of unknown cause, with a median survival of approximately 3–5 years since diagnosis, and is characterized by alveolar epithelial cell injury, fibroblast-to-myofibroblast differentiation, ECM accumulation, and epithelial-to-mesenchymal transition (EMT) [61]. OPN is markedly upregulated in the lung tissue and BALF of IPF patients compared with healthy controls and is negatively correlated with lung function [7, 62]. Likewise, upregulation of OPN was also observed in animals with bleomycin-induced pulmonary fibrosis [62,63,64]. In IPF lungs, macrophages, and epithelial cells are suggested to be the major sources of OPN production [7, 63, 65]. The proliferation, migration, and adhesion of fibroblasts followed by ECM (e.g., collage) accumulation are significantly enhanced by OPN treatment [7, 63, 65]. OPN can also promote the proliferation and migration of alveolar epithelial cells [65]. Moreover, transforming growth factor-β1 (TGF-β1)-mediated EMT plays an important role in the pathogenesis of IPF [61]. OPN has been reported to enhance TGF-β1-mediated EMT by activating Smad2/3 signaling [64]. Furthermore, the predominant infiltration of alternatively activated macrophages (M2) in the lungs acts as a key regulator of pulmonary fibrogenesis. Injured type II alveolar epithelial cell-released Sonic hedgehog (Shh) can stimulate the secretion of OPN in macrophages, and secreted OPN promotes M2 macrophage polarization by enhancing JAK2/STAT3 activity, which may subsequently exacerbate pulmonary fibrogenesis [66]. Recently, a macrophage subpopulation that highly expressed OPN was identified in the fibrotic lower lobes of IPF patient lungs [7]. Given that OPN exerts a pro-proliferation effect on monocytes/macrophages, it can be hypothesized that macrophage-derived OPN may also further enhance macrophage proliferation in IPF [67]. Reduced inflammatory cell infiltration and lung ECM (e.g., collage) accumulation, attenuated EMT, and improved lung function have been observed in OPN−/− mice or OPN inhibitor (e.g., OPN siRNA)-treated mice [64, 65]. Taken together, these findings suggest that OPN also acts as a profibrotic cytokine in IPF.
Moreover, OPN has been implicated in other types of pulmonary fibrosis diseases, such as silicosis and carbon nanotube-induced pulmonary fibrosis. Upregulation of OPN has been observed in the lung tissues of patients with silicosis and mice with silica-induced pulmonary fibrosis [68, 69]. A recent study reported that OPN derived from silica-exposed macrophage exosomes triggers fibroblast-to-myofibroblast differentiation, which may exacerbate the progression of silicosis [60]. Similarly, OPN expression is also upregulated in the lungs of mice with multi-walled or single-walled carbon nanotube-induced pulmonary fibrosis [70, 71]. Elevated OPN was suggested to promote fibroblast-to-myofibroblast differentiation and enhance ECM deposition by activating the TGF-β1/Smad pathway, leading to pulmonary fibrosis [70]. More investigations are needed to further illustrate the role of OPN in the development of pulmonary fibrosis diseases.
OPN in Lung Cancer
Lung cancer is the leading cause of cancer-related mortality worldwide. Despite improvements in surveillance and treatment, the 5-year survival rate of lung cancer is still low [72]. It is necessary to explore novel therapeutic targets and understand their role and underlying mechanism in the pathogenesis of lung cancer. Upregulation of OPN has been observed in various types of cancer, and OPN has been considered an oncogenic driver that promotes tumor growth, metastasis, angiogenesis, and chemoresistance [73, 74]. Numerous studies have shown that the expression of OPN is also significantly upregulated in tumor tissues and/or plasma of patients with non-small cell lung cancer (NSCLC) or small cell lung cancer (SCLC), and cigarette smoking, the main risk factor for lung cancer, has been shown to increase OPN expression in lung cancer cells [75,76,77]. Upregulation of OPN is associated with tumor staging, lymph node involvement, and poor prognosis in patients with lung cancer [75, 78, 79]. In advanced NSCLC, elevated plasma OPN levels are correlated with a decreased response to chemotherapy and poor prognosis [80, 81]. Similarly, plasma OPN levels are also markedly increased in early-stage NSCLC, reduced after resection and increased with recurrence [82]. Plasma OPN may serve as a useful biomarker for predicting cancer prognosis and survival.
Among the three OPN splice variants (OPN-a, OPN-b, and OPN-c), OPN-a is identified as the most highly expressed isoform in lung cancers and several cancer cell lines [83, 84]. It is noteworthy that different OPN splice variants may have distinct characteristics in tumor growth and metastasis. For example, overexpression of OPN-a is reported to enhance the proliferation, migration, and invasion of lung cancer cells, whereas OPN-c exerts the opposite effect [84]. Irrespective of OPN isoforms, most data suggest that OPN overexpression is implicated in tumor growth, metastasis, angiogenesis, and therapy resistance of lung cancer by regulating integrin αvβ3 and CD44-mediated cellular functions [85]. OPN-a has been reported to enhance cell proliferation by activating the CD44/NF-κB pathway in lung cancer cells (e.g., A549 cells) with low integrin αvβ3 levels and inhibit the proliferation of lung cancer cells (e.g., CL1-5 cells) with high integrin αvβ3 levels, indicating that OPN pathological roles in lung cancers may be cell-specific due to the interaction of OPN with different receptors [83]. OPN can also promote lung cancer cell migration and invasion by activating the integrin αvβ3/FAK/Akt and NF-κB-dependent pathways [14, 75]. Furthermore, angiogenesis is essential for the growth and metastasis of solid tumors, OPN, especially OPN-a, has been demonstrated to promote angiogenesis in lung cancer by binding to integrin αvβ3 and/or indirectly by increasing VEGF production [79, 86, 87]. Moreover, OPN, especially OPN-a, is reported to promote the EMT process by enhancing the OPN-PI3K and OPN-MEK pathways [88]. In addition, OPN also promotes acquired cisplatin resistance in SCLC by increasing the expression of the antiapoptotic protein Bcl-2 and enhances acquired epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) resistance in NSCLC by activating the integrin αvβ3/FAK pathways [8, 76]. Interestingly, cigarette smoke is the primary risk factor associated with lung cancer development. Cigarette smoke exposure can increase OPN expression in lung cancer cells through the JAK/STAT3 pathway, and elevated OPN was suggested to attract mesenchymal stem cell (MSC) recruitment and facilitate lung cancer metastasis (Fig. 2A) [77].

Roles and underlying mechanisms of OPN in lung cancer, pneumonia, pulmonary tuberculosis, COPD, and asthma. A Role and underlying mechanisms of OPN in ALI/ARDS. B Role and underlying mechanisms of OPN in pneumonia. C Beneficial role and underlying mechanisms of OPN in pulmonary tuberculosis. D Detrimental role and underlying mechanisms of OPN in COPD. E Detrimental role and underlying mechanisms of OPN in asthma. OPN, osteopontin; COPD, chronic obstructive pulmonary disease; JAK2/STAT3, Janus kinase-signal transducer 2/activator of transcription-3; NF-κB, nuclear factor kappa B; FAK, focal adhesion kinase; VEGF, vascular endothelial growth factor; PI3K, phosphoinositide 3-kinase; MEK mitogen-activated protein kinase kinase; EMT, epithelial-to-mesenchymal transition; IL-12, interleukin-12; IL-17, interleukin-17; IFN-γ, interferon-γ; Irf7, IFN regulatory Factor 7 (Figures are created using Biorender. com).
Most studies have focused specifically on OPN expression in lung cancer cells. However, in addition to cancer cells, OPN can also be synthesized and secreted by a variety of non-tumor cells in the tumor microenvironment, such as tumor-associated macrophages (TAMs) and cancer-associated fibroblasts (CAFs). Recent evidence has also shown that TAM-derived and CAF-derived OPN can also influence tumor formation, progression, and metastasis [89,90,91]. OPN is reported to be highly expressed in intratumoral TAMs, and TAM-derived OPN is suggested to contribute to chemoresistance and is associated with a worse clinical course in patients with lung cancers [92, 93]. Additionally, high expression of OPN in CAFs has been shown to facilitate tumor growth, metastasis, and therapy resistance in many cancer types [90, 91], but whether CAF-derived OPN exerts similar effects on lung cancer remains unclear.
OPN in Lung Infection
OPN in Pneumonia
Pneumonia is a common and serious respiratory illness with a high rate of morbidity and mortality in humans that is often caused by a bacterial, viral, or fungal infection [94]. Inflammation is an important hallmark of pneumonia, and OPN is significantly upregulated and suggested to play a key role in regulating the inflammatory response in various types of pneumonia, including COVID-19 pneumonia, Klebsiella pneumoniae-induced pneumonia, pneumococcal pneumonia, and eosinophilic pneumonia (Fig. 2B) [95,96,97,98].
Plasma OPN levels in patients with COVID-19 pneumonia and community-acquired pneumonia (CAP) are significantly higher than those in healthy controls and are significantly correlated with the severity of pneumonia [38, 95, 99]. OPN is highly expressed in the plasma, lungs, and alveolar macrophages in patients with severe COVID-19. Higher circulating OPN levels are associated with increased odds of mortality and mechanical ventilation in patients with COVID-19 pneumonia [38]. Elevated OPN drives proinflammatory activation of CD14+ classical monocytes and the differentiation of neutrophils toward a proinflammatory PD-L1+ phenotype, indicating that OPN plays a proinflammatory role in the progression of COVID-19 pneumonia [100]. However, there is conflicting evidence regarding the roles of OPN in different types of bacteria-induced pneumonia. For example, Klebsiella pneumoniae is a frequently isolated causative pathogen in hospital-acquired pneumonia (HAP) and ventilator-associated pneumonia (VAP). The circulating levels of OPN and lung OPN are significantly higher in mice with Klebsiella pneumoniae infection than in healthy controls. OPN was suggested to function as a chemotactic cytokine that facilitates early neutrophil recruitment to clear bacteria, and OPN−/− mice displayed impaired antibacterial defense, as reflected by higher bacterial loads in the lungs and a higher mortality rate [96]. However, another study found that OPN may impair host defense during Streptococcus pneumoniae-induced pneumonia. OPN is also markedly upregulated in plasma and lung tissues and was suggested to facilitate the growth of Streptococcus pneumoniae. OPN−/− mice displayed enhanced antibacterial defense, as reflected by lower bacterial loads in the lungs, less pulmonary inflammation, and a lower mortality rate [97]. These conflicting results indicate that the roles of OPN in pneumonia vary among different pathogens.
OPN in Pulmonary Tuberculosis
Pulmonary tuberculosis, caused by Mycobacterium tuberculosis, is a respiratory infectious disease that poses a threat to global health and is characterized by a severe inflammatory response, lung granulomatous lesion formation with caseation, fibrosis, and cavitation. Th1/Th2 imbalance to Th2 in the peripheral blood is a significant factor in the development of pulmonary tuberculosis [101]. Ample evidence suggests that OPN is a critical mediator in Th1-mediated immunity and tuberculosis granuloma formation and plays a beneficial role in protecting against pulmonary tuberculosis (Fig. 2C) [102, 103]. Higher OPN expression in lung tissues, particularly in alveolar macrophages and lymphocytes, has been observed in patients and mice with pulmonary tuberculosis compared to their healthy controls, and circulating OPN levels are positively correlated with the severity of pulmonary tuberculosis [104,105,106]. Elevated OPN was suggested to polarize the immune response to a Th1-type response by stimulating the secretion of IL-12 and IFN-γ [104]. Moreover, OPN can also stimulate Th17 cytokine (IL-17) secretion, which enhances tuberculosis containment [103]. OPN is highly expressed in tuberculosis granulomas and facilitates granuloma formation by acting as a macrophage chemoattractant [68, 107]. OPN−/− mice displayed impaired host defense against Mycobacterium tuberculosis, as reflected by higher bacterial loads in the lungs, reduced clearance of mycobacteria, and greater granuloma burdens [108]. However, other studies reported an inconsequential role of OPN in protecting against pulmonary tuberculosis. There were no significant differences in bacterial loads and lung inflammation during the early phase of pulmonary tuberculosis between OPN−/− mice and wild-type mice. However, OPN−/− mice showed lower bacterial loads in the lungs, less Th1 cytokine (IFN-γ) production, and a modest higher survival rate during the late phase of pulmonary tuberculosis, and more in-depth investigations are needed to explain the conflicting results [105].
OPN in COPD
Chronic obstructive pulmonary disease (COPD), including chronic bronchitis and emphysema, is a destructive lung disorder characterized by persistent inflammation, progressive and irreversible airway obstruction, and tissue remodeling [109]. Increased OPN expression has been observed in the plasma, lungs, and alveolar macrophages and dendritic cells of patients with COPD and COPD exacerbation, and lung levels of OPN show a positive correlation with COPD severity as assessed by forced expiratory volume in 1 s (FEV1) [110,111,112]. Furthermore, cigarette smoke is the most important risk factor associated with COPD development. Cigarette smoke exposure can increase OPN expression in dendritic cells, and increased OPN was suggested to promote Th17-mediated inflammation, IL-17A production, and emphysema formation, in part through its inhibition of the transcription factor IFN regulatory Factor 7 (Irf7). OPN−/− mice develop significantly less smoke-induced emphysema than wild-type mice (Fig. 2D) [112]. In addition, OPN was reported to bind and impair the bacterial activity of several antimicrobial proteins (e.g., lactoferrin) that are expressed in the airways during COPD, which may increase the vulnerability to acquire bacterial infection during COPD [113].
OPN in Asthma
Asthma is a heterogeneous respiratory disease characterized by airway inflammation, mucus hyperproduction, airway hyperresponsiveness (AHR), and airway remodeling, including fibrotic changes [9, 114, 115]. Asthma heterogeneity is expressed in various phenotypes (clinical presentation) and distinctive endotypes (pathophysiological mechanism). The main phenotypes of asthma have been identified, including allergic asthma, non-allergic asthma, adult-onset (late-onset) asthma, and childhood asthma. Based on the Th2 cytokine profile, asthma can be broadly classified into 2 major endotypes, Th2-high (eosinophilic) and Th2-low (non-eosinophilic) asthma [114, 115]. Th2-high asthma is the most common endotype and is characterized by high levels of Th2 cytokines (e.g., IL-4, IL-5, and IL-13) and eosinophilic and Th2-derived airway inflammation. However, Th2-low asthma is more complex, including low levels of Th2 cytokines, neutrophilic or paucigranulocytic airway inflammation, and involvement of Th1 and Th17 cells [115]. Growing evidence has revealed a strong association between OPN and asthma, and OPN has been suggested to play a pivotal role in the development and progression of several types of asthma [116,117,118,119,120].
OPN is widely expressed in the airway, including bronchial epithelial cells, airway glandular endothelial cells, and immune cells. Compared to healthy controls, increased OPN expression has been observed in the bronchial tissue, BALF, and sputum of asthmatic patients [116,117,118]. Several studies have reported that serum OPN levels are also higher in asthmatic patients than in healthy controls and are positively correlated with age [119, 120]. OPN expression, particularly sub-epithelial OPN expression, showed a positive association with asthma severity [117]. Interestingly, sputum OPN levels are higher in smoking asthma patients than in non-smoking asthma patients, and patients with asthma who smoke often exhibit more severe symptoms than patients with asthma who smoke [121]. Consistently, in a house dust mite (HDM) and ovalbumin (OVA)-induced asthma mouse model, OPN expression was also upregulated in BALF and lung tissues [118, 119].
Upregulation of OPN is suggested to contribute to airway inflammation and remodeling in asthma. In asthmatic patients, OPN expression, particularly sub-epithelial OPN expression, is positively correlated with airway remodeling changes, including thickened reticular basement membrane (RBM) and lung function [117, 122]. In an OVA-induced asthma model, OPN−/− mice displayed reduced airway inflammation, less Th2 cytokine (e.g., IL-4 and IL-13) production, AHR, and attenuated airway remodeling, which can be reversed by administration of rOPN [123, 124]. OPN produced by eosinophils plays a pivotal role in allergic inflammation and tissue remodeling [125]. Similarly, recent studies have shown that eosinophils are an important source of OPN in the lungs of allergic asthma. In allergic asthma, amphiregulin-producing memory Th2 cells can stimulate the secretion of OPN in eosinophils, and eosinophil-derived OPN was shown to contribute to airway fibrosis and vascular remodeling in allergic asthma [9, 126]. In vitro, OPN induces murine lung fibroblasts to switch to a profibrogenic myofibroblast phenotype, as indicated by enhanced proliferation and migration and increased collage deposition [118, 123]. Additionally, sOPN was reported to function as a dual-regulatory cytokine in experimental allergic asthma. In the sensitization phase, sOPN suppresses the recruitment of anti-inflammatory plasmacytoid dendritic cells (pDCs), resulting in an enhanced Th2 response. In the challenge phase, sOPN inhibits the recruitment of proinflammatory conventional dendritic cells (cDCs), resulting in a reduced Th2 response. Different roles of sOPN in the different phases of asthma indicate that the function of sOPN may be regulated by different signaling pathways [127]. Moreover, OPN can bind eosinophil-recruiting chemokines (e.g., CCL11) and impair their defense-like antibacterial activities without affecting their eosinophil-recruiting properties in vitro, which may increase the vulnerability to acquire pneumococcal infection in allergic asthma [128]. However, another study found that OPN promotes the host defense against pneumococcal infection, and OPN−/− mice displayed increased vulnerability to pneumococcal infection in a murine model of allergic asthma [128, 129]. Thus, more investigation is warranted to explain the conflicting results.
In addition to allergic asthma, upregulated OPN is also suggested to contribute to the development of adult-onset (late-onset) asthma, which is more heterogeneous and severe and less associated with allergy than child-onset asthma. Several risk factors (e.g., aging and viral infection) of late-onset asthma can further upregulate OPN expression and increase the polymerization of OPN by TG2, which exacerbates airway fibrosis by activating the TGF-β1/Smad3 pathways [119]. Interestingly, corticosteroids are the first-line drugs for the treatment of asthma, and corticosteroids (e.g., dexamethasone) were reported to downregulate OPN expression in lung tissues, which may partly explain the beneficial effects of corticosteroids in asthma (Fig. 2E) [130]. However, information concerning OPN in other types of asthma (e.g., exercise-induced asthma) and in other effector cells (e.g., mast cells) of asthma remains scarce and needs further investigation.
Outlook and Conclusion
Growing evidence demonstrates the significance of OPN in the pathogenesis of various respiratory diseases. Upregulation of OPN has been observed in ALI/ARDS, pulmonary hypertension, IPF, lung cancers, pneumonia, COPD, and asthma. OPN plays a pleiotropic role in the inflammatory response, immune cell activation, fibrosis and tissue remodeling, and tumorigenesis of these respiratory diseases (summarized in Table 1), and targeting OPN exerts therapeutic effects in the treatment of various respiratory diseases [6,7,8,9,10]. However, our current understanding of the roles and underlying mechanisms of OPN in respiration remains largely unknown, and several gaps remain in the knowledge of OPN and the pathogenesis of respiratory diseases. (1) In respiratory diseases (e.g., lung cancer), OPN mediates diverse functions through interactions with integrins and CD44 variants. Recently, ICOSL, another important OPN receptor, was implicated in the pathogenesis of several respiratory diseases (e.g., asthma, pulmonary hypertension, and pulmonary fibrosis) [131,132,133]. Whether ICOSL also contributes to the pleiotropic effects of OPN in these respiratory diseases requires additional studies. (2) Most studies regarding the roles of OPN in respiratory diseases primarily focus on total OPN expression and do not distinguish any specific OPN isoform expression; however, different OPN isoforms may have distinct functions [84]. Therefore, the expression of different OPN isoforms should be taken into account in further studies. (3) In most current studies, the roles of OPN have been mainly attributed to sOPN, and information concerning the roles of iOPN in respiratory diseases remains scarce. Given that iOPN plays a critical role in regulating immune cell (e.g., NK and dendritic cell) functions and tumorigenesis, which may also be involved in the pathogenesis of several respiratory diseases, more in-depth investigations are needed to elucidate the roles and underlying mechanisms of iOPN in respiratory diseases [16, 134, 135]. (4) To date, clinical evidence regarding the therapeutic effects of OPN-specific inhibitors and/or monoantibodies in respiratory diseases is lacking; therefore, additional high-quality clinical trials are needed to confirm the therapeutic effects of these drugs in patients with respiratory diseases.
In summary, a growing body of evidence shows that OPN is significantly upregulated in various inflammatory, fibrotic, malignant, and vascular respiratory diseases. OPN exerts diverse effects on the inflammatory response, immune cell activation, fibrosis and tissue remodeling, and tumorigenesis of these respiratory diseases. Targeting OPN might be a very promising therapeutic approach in the treatment of respiratory diseases.
References
Sécher T, Guilleminault L, Reckamp K et al (2018) Therapeutic antibodies: a new era in the treatment of respiratory diseases? Pharmacol Ther 189:149–172
Ashkar S, Weber GF, Panoutsakopoulou V et al (2000) Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science 287(5454):860–864
Chen G, Zhang X, Li R et al (2010) Role of osteopontin in synovial Th17 differentiation in rheumatoid arthritis. Arthritis Rheum 62(10):2900–2908
Sawaki D, Czibik G, Pini M et al (2018) Visceral adipose tissue drives cardiac aging through modulation of fibroblast senescence by osteopontin production. Circulation 138(8):809–822
Szalay G, Sauter M, Haberland M et al (2009) Osteopontin: a fibrosis-related marker molecule in cardiac remodeling of enterovirus myocarditis in the susceptible host. Cir Res 104(7):851–859
Khamissi FZ, Ning L, Kefaloyianni E et al (2022) Identification of kidney injury released circulating osteopontin as causal agent of respiratory failure. Sci Adv 8(8):eabm5900
Morse C, Tabib T, Sembrat J et al (2019) Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J 54(2):1802441
Fu Y, Zhang Y, Lei Z et al (2020) Abnormally activated OPN/integrin αVβ3/FAK signalling is responsible for EGFR-TKI resistance in EGFR mutant non-small-cell lung cancer. J Hematol Oncol 13(1):169
Morimoto Y, Hirahara K, Kiuchi M et al (2018) Amphiregulin-producing pathogenic memory T helper 2 cells instruct Eosinophils to secrete osteopontin and facilitate airway fibrosis. Immunity 49(1):134-150.e136
Saker M, Lipskaia L, Marcos E et al (2016) Osteopontin, a key mediator expressed by senescent pulmonary vascular cells in pulmonary hypertension. Arterioscler Thromb Vasc Biol 36(9):1879–1890
Singh A, Gill G, Kaur H et al (2018) Role of osteopontin in bone remodeling and orthodontic tooth movement: a review. Prog Orthod 19(1):18
Liaw L, Birk DE, Ballas CB et al (1998) Altered wound healing in mice lacking a functional osteopontin gene (spp1). J Clin Invest 101(7):1468–1478
Shi L, Sun Z, Su W et al (2021) Treg cell-derived osteopontin promotes microglia-mediated white matter repair after ischemic stroke. Immunity 54(7):1527-1542.e1528
Fong YC, Liu SC, Huang CY et al (2009) Osteopontin increases lung cancer cells migration via activation of the alphavbeta3 integrin/FAK/Akt and NF-kappaB-dependent pathway. Lung Cancer 64(3):263–270
Dai J, Peng L, Fan K et al (2009) Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene 28(38):3412–3422
Leavenworth JW, Verbinnen B, Wang Q et al (2015) Intracellular osteopontin regulates homeostasis and function of natural killer cells. Proc Natl Acad Sci USA 112(2):494–499
Shinohara ML, Kim JH, Garcia VA et al (2008) Engagement of the type I interferon receptor on dendritic cells inhibits T helper 17 cell development: role of intracellular osteopontin. Immunity 29(1):68–78
Zhao K, Zhang M, Zhang L et al (2016) Intracellular osteopontin stabilizes TRAF3 to positively regulate innate antiviral response. Sci Rep 6:23771
Rizzello C, Cancila V, Sangaletti S et al (2022) Intracellular osteopontin protects from autoimmunity-driven lymphoma development inhibiting TLR9-MYD88-STAT3 signaling. Mol Cancer 21(1):215
Christensen B, Petersen TE, Sørensen ES (2008) Post-translational modification and proteolytic processing of urinary osteopontin. Biochem J 411(1):53–61
Kaartinen MT, Pirhonen A, Linnala-Kankkunen A et al (1999) Cross-linking of osteopontin by tissue transglutaminase increases its collagen binding properties. J Biol Chem 274(3):1729–1735
Yokosaki Y, Tanaka K, Higashikawa F et al (2005) Distinct structural requirements for binding of the integrins alphavbeta6, alphavbeta3, alphavbeta5, alpha5beta1 and alpha9beta1 to osteopontin. Matrix Biol 24(6):418–427
Bayless KJ, Davis GE (2001) Identification of dual alpha 4beta1 integrin binding sites within a 38 amino acid domain in the N-terminal thrombin fragment of human osteopontin. J Biol Chem 276(16):13483–13489
Weber GF, Ashkar S, Glimcher MJ et al (1996) Receptor-ligand interaction between CD44 and osteopontin (Eta-1). Science 271(5248):509–512
Sun BS, Li Y, Zhang ZF et al (2013) Osteopontin combined with CD44v6, a novel prognostic biomarker in non-small cell lung cancer undergoing curative resection. Ann Thorac Surg 96(6):1943–1951
Kim JS, Bashir MM, Werth VP (2012) Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: a unique molecular signature. J Invest Dermatol 132(7):1825–1832
Raineri D, Dianzani C, Cappellano G et al (2020) Osteopontin binds ICOSL promoting tumor metastasis. Commun Biol 3(1):615
Mukherjee BB, Nemir M, Beninati S et al (1995) Interaction of osteopontin with fibronectin and other extracellular matrix molecules. Ann N Y Acad Sci 760:201–212
Christensen B, Nielsen MS, Haselmann KF et al (2005) Post-translationally modified residues of native human osteopontin are located in clusters: identification of 36 phosphorylation and five O-glycosylation sites and their biological implications. Biochem J 390(Pt 1):285–292
Schytte GN, Christensen B, Bregenov I et al (2020) FAM20C phosphorylation of the RGDSVVYGLR motif in osteopontin inhibits interaction with the αvβ3 integrin. J Cell Biochem. https://doi.org/10.1002/jcb.29708
Jono S, Peinado C, Giachelli CM (2000) Phosphorylation of osteopontin is required for inhibition of vascular smooth muscle cell calcification. J Biol Chem 275(26):20197–20203
Minai-Tehrani A, Chang SH, Park SB et al (2013) The O-glycosylation mutant osteopontin alters lung cancer cell growth and migration in vitro and in vivo. Int J Mol Med 32(5):1137–1149
Christensen B, Zachariae ED, Scavenius C et al (2016) Transglutaminase 2-catalyzed intramolecular cross-linking of osteopontin. Biochemistry 55(2):294–303
Higashikawa F, Eboshida A, Yokosaki Y (2007) Enhanced biological activity of polymeric osteopontin. FEBS Lett 581(14):2697–2701
Forsprecher J, Wang Z, Goldberg HA et al (2011) Transglutaminase-mediated oligomerization promotes osteoblast adhesive properties of osteopontin and bone sialoprotein. Cell Adh Migr 5(1):65–72
Bos LDJ, Ware LB (2022) Acute respiratory distress syndrome: causes, pathophysiology, and phenotypes. Lancet 400(20358):1145–1156
Wang J, Li X, Wang Y et al (2022) Osteopontin aggravates acute lung injury in influenza virus infection by promoting macrophages necroptosis. Cell Death Discov 8(1):97
Hayek SS, Roderburg C, Blakely P et al (2021) Circulating osteopontin levels and outcomes in patients hospitalized for COVID-19. J Clin Med 10(17):3907
Tonello S, D’Onghia D, Apostolo D et al (2023) Baseline plasma osteopontin protein elevation predicts adverse outcomes in hospitalized COVID-19 patients. Viruses 15(3):630
Kasetty G, Papareddy P, Bhongir RKV et al (2019) Osteopontin protects against lung injury caused by extracellular histones. Mucosal Immunol 12(1):39–50
Hirano Y, Aziz M, Yang WL et al (2015) Neutralization of osteopontin attenuates neutrophil migration in sepsis-induced acute lung injury. Criti Care. https://doi.org/10.1186/s13054-015-0782-3
Zhu Y, Wei Y, Chen J et al (2015) Osteopontin exacerbates pulmonary damage in influenza-induced lung injury. Jpn J Infect Dis 68(6):467–473
Zhao H, Chen Q, Huang H et al (2019) Osteopontin mediates necroptosis in lung injury after transplantation of ischaemic renal allografts in rats. Br J Anaesth 123(4):519–530
Kapur R, Kasetty G, Rebetz J et al (2019) Osteopontin mediates murine transfusion-related acute lung injury via stimulation of pulmonary neutrophil accumulation. Blood 134(1):74–84
Takahashi F, Takahashi K, Shimizu K et al (2004) Osteopontin is strongly expressed by alveolar macrophages in the lungs of acute respiratory distress syndrome. Lung 182(3):173–185
Yu ZX, Ji MS, Yan J et al (2015) The ratio of Th17/Treg cells as a risk indicator in early acute respiratory distress syndrome. Crit Care 19(1):82
Chen L, Yang J, Zhang M et al (2023) SPP1 exacerbates ARDS via elevating Th17/Treg and M1/M2 ratios through suppression of ubiquitination-dependent HIF-1α degradation. Cytokine 164:156107
Humbert M, Guignabert C, Bonnet S et al (2019) Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 53(1):1801887
Lorenzen JM, Nickel N, Krämer R et al (2011) Osteopontin in patients with idiopathic pulmonary hypertension. Chest 139(5):1010–1017
Bellan M, Piccinino C, Tonello S et al (2021) Role of osteopontin as a potential biomarker of pulmonary arterial hypertension in patients with systemic sclerosis and other connective tissue diseases (CTDs). Pharmaceuticals (Basel) 14(5):394
Kölmel S, Hobohm L, Käberich A et al (2019) Potential involvement of osteopontin in inflammatory and fibrotic processes in pulmonary embolism and chronic thromboembolic pulmonary hypertension. Thromb Haemost 119(8):1332–1346
Meng L, Liu X, Teng X et al (2019) Osteopontin plays important roles in pulmonary arterial hypertension induced by systemic-to-pulmonary shunt. FASEB J 33(6):7236–7251
Mura M, Cecchini MJ, Joseph M et al (2019) Osteopontin lung gene expression is a marker of disease severity in pulmonary arterial hypertension. Respirology 24(11):1104–1110
Anwar A, Li M, Frid MG et al (2012) Osteopontin is an endogenous modulator of the constitutively activated phenotype of pulmonary adventitial fibroblasts in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 303(1):L1–L11
Peng LY, Yu M, Yang MX et al (2020) Icotinib attenuates monocrotaline-induced pulmonary hypertension by preventing pulmonary arterial smooth muscle cell dysfunction. Am J Hypertens 33(8):775–783
Matsui Y, Rittling SR, Okamoto H et al (2003) Osteopontin deficiency attenuates atherosclerosis in female apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 23(6):1029–1034
Zhao H, Wang Y, Qiu T et al (2020) Autophagy, an important therapeutic target for pulmonary fibrosis diseases. Clin Chim Acta 502:139–147
Wu M, Schneider DJ, Mayes MD et al (2012) Osteopontin in systemic sclerosis and its role in dermal fibrosis. J Invest Dermatol 132(6):1605–1614
Wolak T, Kim H, Ren Y et al (2009) Osteopontin modulates angiotensin II-induced inflammation, oxidative stress, and fibrosis of the kidney. Kidney Int 76(1):32–43
Huang R, Hao C, Wang D et al (2021) SPP1 derived from silica-exposed macrophage exosomes triggers fibroblast transdifferentiation. Toxicol Appl Pharmacol 422:115559
Phan THG, Paliogiannis P, Nasrallah GK et al (2021) Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell Mol Life Sci 78(5):2031–2057
Kumar A, Elko E, Bruno SR et al (2022) Inhibition of PDIA3 in club cells attenuates osteopontin production and lung fibrosis. Thorax 77(7):669–678
Takahashi F, Takahashi K, Okazaki T et al (2001) Role of osteopontin in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 24(3):264–271
Hatipoglu OF, Uctepe E, Opoku G et al (2021) Osteopontin silencing attenuates bleomycin-induced murine pulmonary fibrosis by regulating epithelial-mesenchymal transition. Biomed Pharmacother 139:111633
Pardo A, Gibson K, Cisneros J et al (2005) Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PloS Med 2(9):e251
Hou J, Ji J, Chen X et al (2021) Alveolar epithelial cell-derived Sonic hedgehog promotes pulmonary fibrosis through OPN-dependent alternative macrophage activation. FEBS J 288(11):3530–3546
Tardelli M, Zeyda K, Moreno-Viedma V et al (2016) Osteopontin is a key player for local adipose tissue macrophage proliferation in obesity. Mol Metab 5(11):1131–1137
Nau GJ, Guilfoile P, Chupp GL et al (1997) A chemoattractant cytokine associated with granulomas in tuberculosis and silicosis. Proc Natl Acad Sci USA 94(12):6414–6419
Latoche JD, Ufelle AC, Fazzi F et al (2016) Secreted phosphoprotein 1 and sex-specific differences in silica-induced pulmonary fibrosis in mice. Environ Health Perspect 124(8):1199–1207
Dong J, Ma Q (2017) Osteopontin enhances multi-walled carbon nanotube-triggered lung fibrosis by promoting TGF-β1 activation and myofibroblast differentiation. Part Fiber Toxicol 14(1):18
Khaliullin TO, Kisin ER, Murray AR et al (2017) Mediation of the single-walled carbon nanotubes induced pulmonary fibrogenic response by osteopontin and TGF-β1. Exp Lung Res 43(8):311–326
Hirsch FR, Scagliotti GV, Mulshine JL et al (2017) Lung cancer: current therapies and new targeted treatments. Lancet 389(10066):299–311
Raja R, Kale S, Thorat D et al (2014) Hypoxia-driven osteopontin contributes to breast tumor growth through modulation of HIF1α-mediated VEGF-dependent angiogenesis. Oncogene 33(16):2053–2064
Qian J, LeSavage BL, Hubka KM et al (2021) Cancer-associated mesothelial cells promote ovarian cancer chemoresistance through paracrine osteopontin signaling. J Clin Invest 131(16):e146186
Hu Z, Lin D, Yuan J et al (2005) Overexpression of osteopontin is associated with more aggressive phenotypes in human non-small cell lung cancer. Clin Cancer Res 11(13):4646–4652
Gu T, Ohashi R, Cui R et al (2009) Osteopontin is involved in the development of acquired chemo-resistance of cisplatin in small cell lung cancer. Lung Cancer 66(2):176–183
Jiang YJ, Chao CC, Chang AC et al (2022) Cigarette smoke-promoted increases in osteopontin expression attract mesenchymal stem cell recruitment and facilitate lung cancer metastasis. J Adv Res 41:77–87
Boldrini L, Donati V, Dell’Omodarme M et al (2005) Prognostic significance of osteopontin expression in early-stage non-small-cell lung cancer. Br J Cancer 93(4):453–457
Shijubo N, Uede T, Kon S et al (1999) Vascular endothelial growth factor and osteopontin in stage I lung adenocarcinoma. Am J Respir Crit Care Med 160(4):1269–1273
Mack PC, Redman MW, Chansky K et al (2008) Lower osteopontin plasma levels are associated with superior outcomes in advanced non-small-cell lung cancer patients receiving platinum-based chemotherapy: SWOG Study S0003. J Clin Oncol 26(29):4771–4776
Isa S, Kawaguchi T, Teramukai S et al (2009) Serum osteopontin levels are highly prognostic for survival in advanced non-small cell lung cancer: results from JMTO LC 0004. J Thorac Oncol 4(9):1104–1110
Blasberg JD, Pass HI, Goparaju CM et al (2010) Reduction of elevated plasma osteopontin levels with resection of non-small-cell lung cancer. J Clin Oncol 28(6):936–941
Sun SJ, Wu CC, Sheu GT et al (2016) Integrin β3 and CD44 levels determine the effects of the OPN-a splicing variant on lung cancer cell growth. Oncotarget 7(34):55572–55584
Goparaju CM, Pass HI, Blasberg JD et al (2010) Functional heterogeneity of osteopontin isoforms in non-small cell lung cancer. J Thorac Oncol 5(10):1516–1523
Cui R, Takahashi F, Ohashi R et al (2007) Abrogation of the interaction between osteopontin and alphavbeta3 integrin reduces tumor growth of human lung cancer cells in mice. Lung Cancer 57(3):302–310
Senger DR, Ledbetter SR, Claffey KP et al (1996) Stimulation of endothelial cell migration by vascular permeability factor/vascular endothelial growth factor through cooperative mechanisms involving the alphavbeta3 integrin, osteopontin, and thrombin. Am J Pathol 149(1):293–305
Blasberg JD, Goparaju CM, Pass HI et al (2010) Lung cancer osteopontin isoforms exhibit angiogenic functional heterogeneity. J Thorac Cardiovasc Surg 139(6):1587–1593
Shi L, Hou J, Wang L et al (2021) Regulatory roles of osteopontin in human lung cancer cell epithelial-to-mesenchymal transitions and responses. Clin Transl Med 11(7):e486
Qi J, Sun H, Zhang Y et al (2022) Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat Commun 13(1):1742
Pazolli E, Luo X, Brehm S et al (2009) Senescent stromal-derived osteopontin promotes preneoplastic cell growth. Cancer Res 69(3):1230–1239
Nallasamy P, Nimmakayala RK, Karmakar S et al (2021) Pancreatic tumor microenvironment factor promotes cancer stemness via SPP1-CD44 Axis. Gastroenterology 161(6):1998-2013.e1997
Matsubara E, Komohara Y, Esumi S et al (2022) SPP1 derived from macrophages is associated with a worse clinical course and chemo-resistance in lung adenocarcinoma. Cancers (Basel) 14(18):4374
Matsubara E, Yano H, Pan C et al (2023) The significance of SPP1 in lung cancers and its impact as a marker for protumor tumor-associated macrophages. Cancers (Basel) 15(8):2250
Vincent JL, Levi M, Hunt BJ (2022) Prevention and management of thrombosis in hospitalised patients with COVID-19 pneumonia. Lancet Respir Med 10(2):214–220
Gibellini L, De Biasi S, Paolini A et al (2020) Altered bioenergetics and mitochondrial dysfunction of monocytes in patients with COVID-19 pneumonia. EMBO Mol Med 12(12):e13001
van der Windt GJW, Hoogerwerf JJ, de Vos AF et al (2010) Osteopontin promotes host defense during Klebsiella pneumoniae-induced pneumonia. Eur Respir J 36(6):1337–1345
van der Windt GJW, Hoogendijk AJ, Schouten M et al (2011) Osteopontin impairs host defense during pneumococcal pneumonia. J Infet Dis 203(12):1850–1858
Ueno T, Miyazaki E, Ando M (2010) Osteopontin levels are elevated in patients with eosinophilic pneumonia. Respirology 15(7):1111–1121
Chang JH, Hung WY, Bai KJ et al (2016) Utility of plasma osteopontin levels in management of community-acquired pneumonia. Int J Med Sci 13(9):673–679
MacDonald L, Alivernini S, Tolusso B et al (2021) COVID-19 and RA share an SPP1 myeloid pathway that drives PD-L1+ neutrophils and CD14+ monocytes. JCI Insight 6(13):e147413
Flynn JL, Chan J (2001) Immunology of tuberculosis. Annu Rev Immunol 19:93–129
O’Regan AW, Hayden JM, Body S et al (2001) Abnormal pulmonary granuloma formation in osteopontin-deficient mice. Am J Respir Crit Care Med 164(12):2243–2247
Hernández-Bazán S, Mata-Espinosa D, Lozano-Ordaz V et al (2022) Immune regulatory effect of osteopontin gene therapy in a murine model of multidrug resistant pulmonary tuberculosis. Hum Gene Ther 33(19–20):1037–1051
Koguchi Y, Kawakami K, Uezu K et al (2003) High plasma osteopontin level and its relationship with interleukin-12-mediated type 1 T helper cell response in tuberculosis. Am J Respir Crit Care Med 167(10):1355–1359
van der Windt GJ, Wieland CW, Wiersinga WJ et al (2009) Osteopontin is not crucial to protective immunity during murine tuberculosis. Immunology 128(1):e766-776
Inomata S, Shijubo N, Kon S et al (2005) Circulating interleukin-18 and osteopontin are useful to evaluate disease activity in patients with tuberculosis. Cytokine 30(4):203–211
Hasibuan FM, Shiratori B, Senoputra MA et al (2015) Evaluation of matricellular proteins in systemic and local immune response to Mycobacterium tuberculosis infection. Microbiol Immunol 59(10):623–632
Nau GJ, Liaw L, Chupp GL et al (1999) Attenuated host resistance against Mycobacterium bovis BCG infection in mice lacking osteopontin. Infect Immun 67(8):4223–4230
Christenson SA, Smith BM, Bafadhel M et al (2022) Chronic obstructive pulmonary disease. Lancet 399(10342):2227–2242
Schneider DJ, Lindsay JC, Zhou Y et al (2010) Adenosine and osteopontin contribute to the development of chronic obstructive pulmonary disease. FASEB J 24(1):70–80
Lee SJ, Kim SH, Kim W et al (2014) Increased plasma osteopontin in frequent exacerbator and acute exacerbation of COPD. Clin Respir J 8(3):305–311
Shan M, Yuan X, Song LZ et al (2012) Cigarette smoke induction of osteopontin (SPP1) mediates T(H)17 inflammation in human and experimental emphysema. Sci Transl Med 4(117):117ra119
Gela A, Bhongir RK, Mori M et al (2016) Osteopontin that is elevated in the airways during COPD impairs the antibacterial activity of common innate antibiotics. PloS one 11(1):e0146192
Kuruvilla ME, Lee FE, Lee GB (2019) Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol 56(2):219–233
Fahy JV (2015) Type 2 inflammation in asthma–present in most, absent in many. Nat Rev Immunol 15(1):57–65
Delimpoura V, Bakakos P, Tseliou E et al (2010) Increased levels of osteopontin in sputum supernatant in severe refractory asthma. Thorax 65(9):782–786
Samitas K, Zervas E, Vittorakis S et al (2011) Osteopontin expression and relation to disease severity in human asthma. Eur Respir J 37(2):331–341
Kohan M, Bader R, Puxeddu I et al (2007) Enhanced osteopontin expression in a murine model of allergen-induced airway remodelling. Clin Exp Allergy 37(10):1444–1454
Trinh HKT, Nguyen TVT, Kim SH et al (2020) Osteopontin contributes to late-onset asthma phenotypes in adult asthma patients. Exp Mol Med 52(2):253–265
Akelma AZ, Cizmeci MN, Kanburoglu MK et al (2014) Elevated level of serum osteopontin in school-age children with asthma. Allergol Immunopathol (Madr) 42(4):275–281
Hillas G, Loukides S, Kostikas K et al (2013) Increased levels of osteopontin in sputum supernatant of smoking asthmatics. Cytokine 61(1):251–255
Kanemitsu Y, Ito I, Niimi A et al (2014) Osteopontin and periostin are associated with a 20-year decline of pulmonary function in patients with asthma. Am J Respir Crit Care Med 190(4):472–474
Kohan M, Breuer R, Berkman N (2009) Osteopontin induces airway remodeling and lung fibroblast activation in a murine model of asthma. Am J Respir Cell Mol Biol 41(3):290–296
Simoes DC, Xanthou G, Petrochilou K (2009) Osteopontin deficiency protects against airway remodeling and hyperresponsiveness in chronic asthma. Am J Respir Crit Care Med 179(10):894–902
Yang HW, Park JH, Jo MS et al (2022) Eosinophil-derived osteopontin induces the expression of pro-inflammatory mediators and stimulates extracellular matrix production in nasal fibroblasts: the role of osteopontin in eosinophilic chronic rhinosinusitis. Front Immunol 13:777928
Puxeddu I, Berkman N, Ribatti D et al (2010) Osteopontin is expressed and functional in human eosinophils. Allergy 65(2):168–174
Xanthou G, Alissafi T, Semitekolou M et al (2007) Osteopontin has a crucial role in allergic airway disease through regulation of dendritic cell subsets. Nat Med 13(5):570–578
Gela A, Kasetty G, Mörgelin M et al (2016) Osteopontin binds and modulates functions of eosinophil-recruiting chemokines. Allergy 71(1):58–67
Kasetty G, Bhongir RKV, Papareddy P et al (2019) Osteopontin protects against pneumococcal infection in a murine model of allergic airway inflammation. Allergy 74(4):663–674
Kurokawa M, Konno S, Matsukura S et al (2009) Effects of corticosteroids on osteopontin expression in a murine model of allergic asthma. Int Arch Allergy Immunol 149(Supp 1):7–13
Uwadiae FI, Pyle CJ, Walker SA et al (2019) Targeting the ICOS/ICOS-L pathway in a mouse model of established allergic asthma disrupts T follicular helper cell responses and ameliorates disease. Allergy 74(4):650–662
Bellan M, Murano F, Ceruti F et al (2022) Increased levels of ICOS and ICOSL are associated to pulmonary arterial hypertension in patients affected by connective tissue diseases. Diagnostics (Basel) 12(3):704
Tanaka C, Fujimoto M, Hamaguchi Y et al (2010) Inducible costimulator ligand regulates bleomycin-induced lung and skin fibrosis in a mouse model independently of the inducible costimulator/inducible costimulator ligand pathway. Arthritis Rheum 62(6):1723–1732
Fan X, He C, Jing W et al (2015) Intracellular osteopontin inhibits toll-like receptor signaling and impedes liver carcinogenesis. Cancer Res 75(1):86–97
Shinohara ML, Lu L, Bu J et al (2006) Osteopontin expression is essential for interferon-alpha production by plasmacytoid dendritic cells. Nat Immunol 7(5):498–506
Acknowledgements
This work was supported by grant 82002100 (to Yiyi Yang) from the National Natural Science Foundation of China (Beijing, China).
Funding
This work was supported by Grant No 82002100 (to Yiyi Yang) from the National Natural Science Foundation of China (Beijing, China).
Author information
Authors and Affiliations
Contributions
QJ and ZH conceived, wrote, edited, and prepared the figures and tables in this manuscript. QJ, YO, and YY wrote the manuscript. XC, SY, and ZH edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jia, Q., Ouyang, Y., Yang, Y. et al. Osteopontin: A Novel Therapeutic Target for Respiratory Diseases. Lung 202, 25–39 (2024). https://doi.org/10.1007/s00408-023-00665-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00408-023-00665-z