
Abstract
Introduction
Influenza infects millions of people each year causing respiratory distress and death in severe cases. On average, 200,000 people annually are hospitalized in the United States for influenza related complications. Tissue inhibitor of metalloproteinase-1 (TIMP-1), a secreted protein that inhibits MMPs, has been found to be involved in lung inflammation. Here, we evaluated the role of TIMP-1 in the host response to influenza-induced lung injury.
Methods
Wild-type (WT) and Timp1-deficient (Timp1−/−) mice that were 8–12 weeks old were administered A/PR/8/34 (PR8), a murine adapted H1N1 influenza virus, and euthanized 6 days after influenza installation. Bronchoalveolar lavage fluid and lungs were harvested from each mouse for ELISA, protein assay, PCR, and histological analysis. Cytospins were executed on bronchoalveolar lavage fluid to identify immune cells based on morphology and cell count.
Results
WT mice experienced significantly more weight loss compared to Timp1−/− mice after influenza infection. WT mice demonstrated more immune cell infiltrate and airway inflammation. Interestingly, PR8 levels were identical between the WT and Timp1−/− mice 6 days post-influenza infection.
Conclusion
The data suggest that Timp1 promotes the immune response in the lungs after influenza infection facilitating an injurious phenotype as a result of influenza infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Influenza is a highly contagious respiratory pathogen that is characterized as a febrile illness causing cough, headache, a general sense of malaise, and inflammation of the upper respiratory tract [1]. Seasonal influenza infection results in over 200,000 hospitalizations and 30,000–50,000 deaths in the United States alone [2]. Globally, influenza infection is a public health concern that necessitates annual vaccinations for prevention [3]. Unfortunately, vaccinations can sometimes be ineffective as seen in both the 2014–2015 and 2016–2017 flu seasons [4]. Moreover, the influenza vaccine only provides temporary protection as the influenza virus undergoes antigenic drift allowing mutated strains to evade the memory conferred by influenza immunizations from prior years [1]. Furthermore, antigenic shift from re-assortment of the viral genome originating from human and swine viral genome along with avian viral genes has resulted in novel influenza strains leading to the last three pandemics [5].
Tissue inhibitor of metalloproteinase-1 (TIMP-1) is a glycoprotein belonging to the four-member family of TIMPs (TIMP1 through TIMP4) [6, 7]. TIMPs are composed of an N-terminal domain and C-terminal domain stabilized by three disulfide bonds [8, 9]. The N-terminal portion of TIMPs noncovalently binds to MMPs at the zinc-binding domain facilitating inhibition of MMP proteolytic activity [9, 10]. Although TIMP-1 is more known as an endogenous inhibitor of matrix metalloproteinases (MMPs), it also displays MMP-independent behaviors. TIMP-1 influences proliferation of fibroblasts and keratinocytes, anti-apoptotic activity in human Burkitt Lymphoma cell lines, anoikis in human breast epithelial cells and differentiation of oligodendrocyte progenitor cells independent of MMP activity [11,12,13,14]. Accordingly, CD63 has been identified as a receptor for TIMP-1 [11].
TIMP-1 expression increases following bleomycin injury and localizes to inflammatory foci of the injured lung suggesting a role in regulating inflammation [12]. Moreover, TIMP-1 deficiency confers protection to corneal infection and lung injury from Pseudomonas aeruginosa in mice [13]. TIMP-1 has also been implicated in myocarditis resulting from Coxsackievirus B3 infection and in exerting control over the migration of immune cells [14]. Herein, we investigated the role of TIMP-1 in regulating lung inflammation after influenza infection. Our results demonstrate that TIMP-1 promotes the immune response resultant from influenza-induced acute lung injury.
Methods
Mice Influenza Experiments
Wild-type (WT) and Timp1 deficient (TIMP1−/−) C57BL/6 mice were bred and housed in the Cedars-Sinai Medical Center vivarium. Mouse adapted H1N1 influenza, A/PR/8/34 (PR8), was propagated within incubated embryonated chicken eggs as previously described [15]. Stock PR8 was aliquoted and stored at − 80 °C. A working solution of PR8 was made by diluting in PBS and maintained on ice prior to infection. Adult male mice (8–12 weeks old) were anesthetized with isoflurane followed by intranasal administration of PR8 (500 PFU in 50 µl). All animal experiments were performed in accordance with the regulations of the Institutional Animal Care and Use Committee at Cedars-Sinai Medical Center.
Lung and Bronchial Alveolar Lavage (BAL) Fluid Collection
Post-infection mice were euthanized with a mixture of ketamine (300 mg/kg) and xylazine (30 mg/kg) through intraperitoneal injection followed by cervical dislocation. An incision in the trachea was made for placement of a 26-gauge angiocath followed by instillation of 1 ml of sterile PBS. The BAL was collected, and the process was repeated two additional times (total instillation of 3 ml of PBS). After centrifugation, the supernatant was collected and placed in − 80 °C for downstream assays. BAL cell counts were determined with an automated cell counter (Bio Rad TC 20™). Differential cell counts were determined with standard morphological criteria of cytospins stained using Differential Quik Stain (Modified Giemsa) Kit (Polysciences Inc; Warrington, PA).
After the BAL collection, left lungs were removed for protein and RNA analysis. Right lungs were perfused and fixed with neutral buffer formalin (1:10) followed by paraffin embedding and tissue sectioning. Sections (5 µm) were stained with H&E (Newcomer Supply, Middleton, WI).
Protein Measurement in BAL
BAL IgM levels were quantified by ELISA (Mouse IgM ELISA Quantitation Set; Bethyl Laboratories Montgomery, TX) according to the manufacture’s recommendations. Total protein from BAL fluid was measured using DC™ Protein Assay (Bio Rad; Hercules, CA). In brief, BAL obtained from WT and Timp1−/− mice was thawed and diluted 1:10 using PBS. Following a 15-min incubation, the absorbance was measured via spectrophotometer. Results were multiplied by 10 to account for the dilution factor.
PCR of Lung Tissue
RNA extraction was performed on whole WT and Timp1−/− lungs using TRIzol® reagent (Life Technologies). To quantify PR8 and TIMP-1 expression, cDNA was synthesized using the High-Capacity Reverse Transcription Kit (Fischer Scientific). SYBR™ Green PCR Master Mix (Fischer Scientific) was combined with cDNA and both forward and reverse primers for PR8 (CATCCTGTTGTATATGAGGCCCAT and GCACTGCAGCGTAGACGCTT) and TIMP-1 (GCAACTCGGACCTGGTCATAA and CGGCCCGTGATGAGAAACT). GAPDH expression served as an internal control. PR8 and TIMP-1 primers were generated and purchased through Integrated DNA Technologies (IDT). qPCR was performed using ViiA 7 (Applied Biosystems), and results were analyzed using Data Assist v3.01.
Lung Injury Scoring of Histology
Blinded histological scoring was implemented to assess of the severity of airway inflammation and hemorrhage in WT (n = 5) and Timp1−/− (n = 5) lungs as previously described [16]. Sections of each of the lobes from the right lung were randomly selected for pathological assessment. Scores were determined by the estimated percentage of the section of lung affected by inflammation determined by the collection of inflammatory cells, destruction to the parenchyma, such as the alveolar space, and loss of integrity of the epithelial cell barrier of the airway. Hemorrhage was scored based upon the presence and degree of red blood cell infiltration. Scores from each of the lobes of each lung were averaged resulting in the final histological score for each animal. The degree of inflammation and hemorrhage were determined based on the following numerical scale; 1 = minimal/mild, 2 = moderate, 3 = marked, 4 = severe.
Statistics
Statistical significance was determined using either Student’s t test and two-way analysis of variance. P values < 0.05 were noted as significant.
Results
Timp1 is Induced and Protective During Influenza Infection
TIMP-1 expression is induced by a wide array of stimuli [12, 17,18,19]. Therefore, we measured Timp1 expression in the lungs after influenza infection. In an unperturbed mouse, the lungs have very low levels of Timp1 (Fig. 1). However, influenza infection causes a large induction of Timp1. As expected, Timp1−/− mice had no expression.

TIMP-1 expression increases in mice after influenza infection. WT and Timp1−/− mice were infected with PR8, and lungs were harvested on day 4 after infection and processed for PCR. *P < 0.05; N = 3 for uninfected conditions; N = 4 for PR8 infected conditions
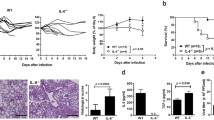
To determine the biological consequence of the Timp1 induction after influenza infection, we compared the WT and Timp1−/− mouse response to influenza infection. Body weight loss measurement is an excellent indicator of disease severity, and Timp1−/− mice had significantly less weight loss than WT mice after influenza infection (Fig. 2a). The differences were first identified at day 5 after infection and persisted through day 8.

TIMP-1 facilitates lung injury after influenza infection. a Mice were weighed at baseline and daily after infection with PR8. *P < 0.05; N = 22 for WT mice; N = 27 for Timp1−/− mice. b. WT and Timp1−/− mice were infected with PR8. On day 6 after infection, mice were sacrificed, and the lungs were processed for H&E staining. Each column is an independent mouse. Scale bar = 200 µm. c, d WT and Timp1−/− lungs were subjected to histological scoring for c inflammation and d hemorrhage, respectively. *P < 0.05; **P < 0.01
Timp1 Facilitates Airway Inflammation After Influenza Infection
After influenza infection, mice experienced extensive lung injury and tissue remodeling. Noticeable infiltration of immune cells dispersed in a peribronchiolar distribution was seen in WT and Timp1 −/− lungs after influenza infection (Fig. 2b). However, the WT lungs were shown to have considerably increased inflammatory infiltrates compared to Timp1 −/− mice. Histological scoring regarding the severity of airway inflammation in the lungs was significantly increased in WT mice in comparison to Timp1−/− mice (Fig. 2c; 3.4 ± 0.4 vs. 1.8 ± 0.2, respectively; P < 0.01). Histological scoring of hemorrhage was also higher in WT lungs versus Timp1−/− lungs (Fig. 2d; 2.6 ± 0.51 vs. 1.2 ± 0.2, respectively; P < 0.05).
As a measure of the immune response, total BAL cell counts and the cell differential were determined after influenza infection. Consistent with the histological findings on hemorrhage (Fig. 2d), the cytospins revealed a striking increase in the amount of red blood cells in WT comparison to Timp1−/− mice (Fig. 3a). WT mice also had double the total cell count compared to Timp1−/− mice (1.42 ± 0.07 × 106 vs. 0.70 ± 0.12 × 106 cells, respectively; P < 0.01) (Fig. 3b). In addition, Timp1−/− compared to WT mice had less neutrophils (0.48 ± 0.09 × 106 vs. 0.88 ± 0.09 × 106 cells, respectively; P < 0.05; Fig. 3c) and macrophages (2.10 ± 0.65 × 106 vs. 5.39 ± 0.58 × 106 cells, respectively; P < 0.01; Fig. 3d) in the BAL.

TIMP-1 promotes lung inflammation after influenza infection. WT (N = 4) and Timp1−/− (N = 4) mice were infected with PR8 virus. On day 6 after infection, mice were sacrificed, and cells in the bronchoalveolar lavage were processed for: a cytospins, arrowhead denotes RBCs; b total cell count; c neutrophil counts; and d macrophages counts. *P < 0.05; **P < 0.01; scale bar = 400 µm
Lung Leakage and Viral Load was Similar Between WT and Timp-1 −/− Mice After Influenza Infection
To assess the lung barrier integrity after injury, we measured the translocation of large proteins into the airspaces as previously described [20]. In brief, WT and Timp1−/− BAL was subjected to DC™ protein assay to detect protein content and ELISA for IgM levels. In contrast to the differences in lung inflammation, WT and Timp1−/− mice showed no differences in total protein or IgM concentrations after influenza infection (Fig. 4a, b, respectively). We also measured the viral load between WT and Timp1−/− mice by PCR for a viral specific gene, and there was no significant difference in PR8 expression between WT and Timp1−/− mice (Fig. 5).

Barrier function was no different between conditions after influenza infection. WT and Timp1−/− mice were infected with PR8. On day 6 after infection, mice were sacrificed and BAL was processed for: a total protein and b IgM levels

Viral load is identical between WT and Timp1−/− mice after influenza infection. WT and Timp1−/− mice were infected with PR8. On day 6 after infection, mice were sacrificed, and the lung homogenates from mice were processed for influenza expression by PCR
Discussion
TIMP-1 is known to be involved in a plethora of biological processes demonstrating its ability to exercise pleiotropic effects [21]. Here, we have established that TIMP-1 promotes the immune response during PR8 induced lung injury. We demonstrated the induction of TIMP-1 in PR8 infected mice. This increase in TIMP-1 expression facilitated the inflammatory response as Timp−/− mice had less weight loss and lung inflammation after PR8 infection.
Although our study focuses on influenza, these results phenocopy the effects seen after P. aeruginosa infection with less injury in Timp1−/− mice compared to WT conditions [22]. Interestingly, an opposite phenotype was displayed in bleomycin and LPS models of acute lung injury with worsened lung inflammation in Timp1−/− compared to WT mice [12]. The etiology of these differences is unclear and will require a better understanding of the molecular pathways that drive the respective phenotypes. However, one possibility is that TIMP-1 regulates differing pathways dependent on the nature of the inciting insult (e.g., infectious vs. sterile injury) [23].
TIMPs are best characterized for their ability to inhibit MMPs [24]. Indeed, TIMP-1 regulation of MMP activity has been implicated in conferring resistance against P. aeruginosa corneal infections [13]. However, TIMP-1 can also behave in a cytokine-like fashion, influencing cell growth, apoptosis, and regulating other cytokines to promote cell survival [21, 25]. It is possible TIMP-1 stimulates inflammation and cytokine-induced processes in influenza-induced lung injury in an MMP-independent manner. This presents an opportunity to advance additional studies necessary to elucidate the function of TIMP-1.
Increased levels of TIMP-1 in the blood correlates with poor disease-free survival in breast cancer patients [26] and worsened disease in cirrhotic patients [27]. Therefore, future studies could determine if TIMP-1 could be used to prognosticate the severity of illness after influenza infection. Furthermore, having a better understanding of the mechanism of action could lead to development of TIMP-1 as a therapeutic in the treatment of influenza.
In summary, our data demonstrate that TIMP-1 expression increased and had a pro-inflammatory role during influenza-induced lung injury. Although TIMP-1 is known primarily for inhibiting MMP function, it is plausible that TIMP-1 function during influenza-induced lung injury is MMP-independent. Further investigation is imperative in order to elucidate regulation of inflammation via TIMP-1 expression and determine its ability to risk stratify or be used therapeutically in patients infected with influenza.
References
Taubenberger JK, Morens DM (2008) The pathology of influenza virus infections. Annu Rev Pathol 3:499–522
Vemula SV, Zhao J, Liu J, Wang X, Biswas S, Hewlett I (2016) Current approaches for diagnosis of influenza virus infections in humans. Viruses 8:96
HIPC-CHI Signatures Project Team, HIPC-I Consortium (2017) Multicohort analysis reveals baseline transcriptional predictors of influenza vaccination responses. Sci Immunol 2
Paules CI, Sullivan SG, Subbarao K, Fauci AS (2018) Chasing seasonal influenza—the need for a universal influenza vaccine. N Engl J Med 378:7–9
Taubenberger JK, Morens DM (2010) Influenza: the once and future pandemic. Public Health Rep 125(Suppl 3):16–26
Brew K, Nagase H (2010) The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta 1803:55–71
Egea V, Zahler S, Rieth N, Neth P, Popp T, Kehe K, Jochum M, Ries C (2012) Tissue inhibitor of metalloproteinase-1 (TIMP-1) regulates mesenchymal stem cells through let-7f microRNA and Wnt/beta-catenin signaling. Proc Natl Acad Sci USA 109:E309–E316
Nagase H, Meng Q, Malinovskii V, Huang W, Chung L, Bode W, Maskos K, Brew K (1999) Engineering of selective TIMPs. Ann N Y Acad Sci 878:1–11
Gardner J, Ghorpade A (2003) Tissue inhibitor of metalloproteinase (TIMP)-1: the TIMPed balance of matrix metalloproteinases in the central nervous system. J Neurosci Res 74:801–806
Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP (1997) Tissue inhibitors of metalloproteinases: structure, regulation and biological functions. Eur J Cell Biol 74:111–122
Jung KK, Liu XW, Chirco R, Fridman R, Kim HR (2006) Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J 25:3934–3942
Madtes DK, Elston AL, Kaback LA, Clark JG (2001) Selective induction of tissue inhibitor of metalloproteinase-1 in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 24:599–607
Lee MM, Yoon BJ, Osiewicz K, Preston M, Bundy B, van Heeckeren AM, Werb Z, Soloway PD (2005) Tissue inhibitor of metalloproteinase 1 regulates resistance to infection. Infect Immun 73:661–665
Crocker SJ, Frausto RF, Whitmire JK, Benning N, Milner R, Whitton JL (2007) Amelioration of coxsackievirus B3-mediated myocarditis by inhibition of tissue inhibitors of matrix metalloproteinase-1. Am J Pathol 171:1762–1773
Brauer R, Chen P (2015) Influenza virus propagation in embryonated chicken eggs. J Vis Exp. https://doi.org/10.3791/52421
Gibson-Corley KN, Olivier AK, Meyerholz DK (2013) Principles for valid histopathologic scoring in research. Vet Pathol 50:1007–1015
Trim JE, Samra SK, Arthur MJ, Wright MC, McAulay M, Beri R, Mann DA (2000) Upstream tissue inhibitor of metalloproteinases-1 (TIMP-1) element-1, a novel and essential regulatory DNA motif in the human TIMP-1 gene promoter, directly interacts with a 30-kDa nuclear protein. J Biol Chem 275:6657–6663
Campbell CE, Flenniken AM, Skup D, Williams BR (1991) Identification of a serum- and phorbol ester-responsive element in the murine tissue inhibitor of metalloproteinase gene. J Biol Chem 266:7199–7206
Manoury B, Caulet-Maugendre S, Guenon I, Lagente V, Boichot E (2006) TIMP-1 is a key factor of fibrogenic response to bleomycin in mouse lung. Int J Immunopathol Pharmacol 19:471–487
Brauer R, Ge L, Schlesinger SY, Birkland TP, Huang Y, Parimon T, Lee V, McKinney BL, McGuire JK, Parks WC, Chen P (2016) Syndecan-1 attenuates lung injury during influenza infection by potentiating c-met signaling to suppress epithelial apoptosis. Am J Respir Crit Care Med 194:333–344
Ries C (2014) Cytokine functions of TIMP-1. Cell Mol Life Sci 71:659–672
Kim KH, Burkhart K, Chen P, Frevert CW, Randolph-Habecker J, Hackman RC, Soloway PD, Madtes DK (2005) Tissue inhibitor of metalloproteinase-1 deficiency amplifies acute lung injury in bleomycin-exposed mice. Am J Respir Cell Mol Biol 33:271–279
Shen H, Kreisel D, Goldstein DR (2013) Processes of sterile inflammation. J Immunol 191:2857–2863
Moore CS, Crocker SJ (2012) An alternate perspective on the roles of TIMPs and MMPs in pathology. Am J Pathol 180:12–16
Guedez L, Stetler-Stevenson WG, Wolff L, Wang J, Fukushima P, Mansoor A, Stetler-Stevenson M (1998) In vitro suppression of programmed cell death of B cells by tissue inhibitor of metalloproteinases-1. J Clin Invest 102:2002–2010
Wurtz SO, Moller S, Mouridsen H, Hertel PB, Friis E, Brunner N (2008) Plasma and serum levels of tissue inhibitor of metalloproteinases-1 are associated with prognosis in node-negative breast cancer: a prospective study. Mol Cell Proteomics 7:424–430
Busk TM, Bendtsen F, Nielsen HJ, Jensen V, Brunner N, Moller S (2014) TIMP-1 in patients with cirrhosis: relation to liver dysfunction, portal hypertension, and hemodynamic changes. Scand J Gastroenterol 49:1103–1110
Funding
This work was funded by the NIH Grants HL120947 (P.C.), HL103868 (P.C.), and HL137076 (P.C.), Samuel Oschin Comprehensive Cancer Institute Lung Cancer Research Award (P.C.), and the American Heart Association Grant-in-Aid (P.C.).
Author information
Authors and Affiliations
Contributions
JRA—Executed experiments, generated and collected data, and interpreted results. LG—Aided in interpretation of data and helped collect BAL samples and lung tissue from mice. YH—Executed experiments and maintained the mouse colony. RB—Aided in interpretation of data. TP—Executed experiments and aided in interpretation of data. SC—Helped with experimental design and interpretation of data. FS—Helped with experimental design and interpretation of data. PC—Supervised the project, designed experiments, interpreted data, and edited this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests.
Ethical Approval
All animal experiments were performed in accordance with the regulations of the Institutional Animal Care and Use Committee at Cedars-Sinai Medical Center.
Rights and permissions
About this article

Cite this article
Allen, J.R., Ge, L., Huang, Y. et al. TIMP-1 Promotes the Immune Response in Influenza-Induced Acute Lung Injury. Lung 196, 737–743 (2018). https://doi.org/10.1007/s00408-018-0154-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00408-018-0154-2