
Abstract
Purpose
To scrutinize the characteristics of all cases with antenatally established diagnosis of cephalocele in two tertiary referral centers.
Methods
We retrospectively reviewed 65 cases diagnosed with cephaloceles and tabulated sonographic findings and autopsy recordings as well as medical charts of all survivors in terms of clinical outcome.
Results
The case notes of 65 fetuses were available for final analysis. Gestational age (GA) at diagnosis ranged from 10.4 to 38.1 weeks. Of our cohort, 53/65 cases (80%) had occipital protrusions, 10 (15%) were found to have frontal lesions, and another two had parietal cephaloceles. A total of 52 pregnancies were terminated or resulted in intrauterine fetal demise (78%). In 18 cases (11%), the cephalocele was part of underlying syndromic disorders (e.g., Meckel–Gruber syndrome). Thirteen pregnancies were continued until term, out of which all affected individuals were live-born. Neurosurgical intervention was prompted within the first 7 months postnatally.
Conclusions
In general, the outcome of fetuses with cephaloceles is rather poor as four out of five pregnancies were terminated. Postnatal outcome of all survivors in our cohort was rather determined by localization of the cele and more important by the presence and severity of concomitant malformations than the extent of the lesion.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cephaloceles—cranial meningoceles and encephaloceles—are characterized by a skull defect enabling herniation of intracranial tissue [1]. Their incidence is estimated 1:2000 to 1:5000 live births with marked geographic and ethnic differences. Whereas the prevalence of neural tube defects has substantially decreased since folic acid fortification has been recommended in the early 1990s, this issue has been controversially debated in terms of the development of cephaloceles [2, 3]. The underlying etiology of cranial herniations is not fully understood, but includes ethnic, genetic, and environmental factors. They merely constitute a failure of separation of the neural and surface ectoderm after the closure of the rostral neuropore has been completed during the 4th gestational week [4]. As previous studies suggested, the prognosis strongly depends on the anatomical site of the lesion, the presence and extent of neural contents as well as coexisting malformations [3]. It ranges from unremarkable physical and cognitive development to severe neuromotoric disabilities. It has been further shown that anatomic disruption and cognitive and physical impairment do not necessarily correlate [5]. Therefore, precise prenatal diagnosis as early as possible is indispensable in order to provide the expectant parents with adequate advice. The detection rate of cephaloceles during first trimester has been estimated 80%, they constitute 5% of all detected severe structural defects of the central nervous system (CNS) [1, 6]. Associated malformations or conditions comprise hydrocephalus, which is associated with the worst prognosis, Meckel–Gruber syndrome, trisomy 18, single-gene disorders, Walker–Warburg syndrome, cerebro-ocular-muscular syndrome (COMS), and toxic embryopathies [3].
The aim of our work was to evaluate the incidence, pre- and postnatal course and additional anomalies of prenatally detected cephaloceles in a mixed low- and high-risk population. The present retrospective study illustrates the issue of prenatal diagnosis of cephalocele and aims at correlating prenatal diagnostic findings with postnatal development or autopsy outcome of affected fetuses registered in two tertiary referral centers for prenatal medicine.
Methods
This retrospective study included all pregnancies with an abnormal appearance of the fetal CNS diagnosed antenatally by ultrasound examination between the years 1994 and 2012. Screening for fetal CNS abnormalities as an integral part of fetal anomaly survey is routinely offered to all pregnant women attending our prenatal referral centers at the University Hospital of Schleswig-Holstein, Campus Lübeck, and at the University Hospital Bonn. In our mixed-risk collective, the indications for ultrasound examination varied from first and second trimester anomaly screening to referral due to family history and due to suspected CNS anomalies for further investigation. All ultrasound examinations were performed with an Acuson Sequoia 128 XP (Siemens, Erlangen, Germany), an ATL HDI 5000 (Philips, Solingen, Germany), a Voluson 730 Expert (GE Healthcare, Milwaukee, USA), or IU22 (Philips, Solingen, Germany) ultrasound machines using a 2–5, a 4–7, or 5–9 MHz curved array abdominal transducer or a 4–8 MHz vaginal sector probe.
During the study period, all pregnancies with cephaloceles (encephaloceles and cranial meningoceles) were tabulated for further analysis. 65 pregnancies diagnosed between the years 1994–2012 were reviewed including prenatal imaging, postnatal studies, operative findings, pediatric charts of the survivors, and autopsy reports for terminated pregnancies. The average postnatal follow-up was 12 months. Necropsies after termination of the pregnancy (TOP), stillborn, or neonatal death were performed by experienced pathologists trained in fetal autopsy.
Maternal characteristics are summarized in Table 1. Emphasis must be placed on the history of precedent miscarriages, which was slightly increased (37%) in comparison to the German population (approximately 30%).
Results
During the study period, a total of 65 pregnancies were diagnosed with fetal cranial tissue herniation. Mean gestational age at diagnosis was 18 + 0 weeks of gestation (ranging from 10.4 to 38.1 weeks). There was a slight female preponderance (34 fetuses vs. 30 males). In one fetus, the gender could not be determined. Karyotyping was conducted in 51 of 65 cases. Chromosomal anomalies were found in three cases with, trisomy 18 (47, XX, +18), triple X syndrome (47, XXX), and a chromosomal deletion (46, XX, del(14)(q24-qter)), respectively. The remainder (45 of 51 cases) had normal chromosomes. Single-gene disorders were present or suspected in 18 fetuses: Meckel–Gruber syndrome (MKS) was diagnosed in 11 cases (affected siblings in three cases). In three fetuses of the same family, occipital encephalocele was diagnosed as a part of oro-facial-digital syndrome (OFDS) type IV (out of which two cases were already published by Rösing and co-workers [7]). In one family, two fetuses showed small occipital meningocele as a part of Joubert-related disorder, in one family with a previous affected sibling the fetus showed a small occipital encephalocele as a part of Walker–Warburg syndrome. Furthermore, one fetus affected by an occipital encephalocele showed signs of oculo-auriculo-vertebral dysplasia (Goldenhar-Gorlin syndrome). A single umbilical artery was found in 4 of the 65 study cases.
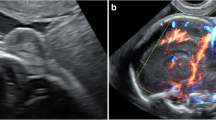
Fifty-three (80%) of the 65 fetuses had cephaloceles located occipitally, from those three fetuses had craniospinal dysraphism as part of disruptive anomalies (amniotic band sequence). Ten (15%) showed frontal lesions (eight with nasofrontal protrusion, one with infraorbital protrusion, and one with a nasoethmoidal protrusion), and two (3%) had a parietal lesion (Figs. 1, 2). Termination of pregnancy on parental request was undertaken. In 47 of 65 cases (72%) herniated brain tissue was visualized. Termination of the pregnancy on parental request was undertaken in 49 of 65 cases (75%) (among those, selective feticide was performed in two dichorionic twin pregnancies in which the unaffected co-twins were born by Cesarean section with favorable outcome), and intrauterine fetal death occurred in 3 of 65 (5%) cases. All couples were extensively counseled (together with neuropediatricians and neurosurgeons). The presence of accompanied cerebral and associated extracerebral anomalies was the main cause for parents to opt for termination of the pregnancy.

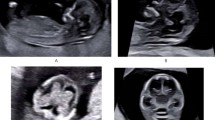
Different appearance of fetal cephaloceles. Occipital encephalocele at 30 weeks with marked posteriorly located cranial protrusion (a), corresponding transversal sonographic view (d) and MR image (g) both depicting a bony lesion enabling herniation of intracranial (cerebellar) tissue. The fetus was stillborn at 33 gestational weeks, the parents declined autopsy because of religious objections. Images b, e, h showing an occipital meningocele at 28 weeks clearly lacking any neural content. Parietal encephalocele at 33 weeks (c) communicating with meningeal sac through a skull defect as seen both on US (f) and MR images (i). The pregnancy in the latter two cases was terminated on parental request. Pathological examination during autopsy confirmed the prenatal diagnosis

Frontoethmoidal encephalocele at 34 gestational weeks. Rendered 3D image showing frontal protrusion (a) and corresponding MRI with herniated intracranial tissue (b). Gross appearance on autopsy, note the marked microcephalus (c)
In 13 of 65 (20%) pregnancies with prenatally detected cephalocele, in whom the parents opted for continuation of the pregnancy, prenatal short-term follow-up examinations were scheduled, and in 11 cases Cesarean section was planned at 38 completed weeks (Table 2). In another two cases, the mode of delivery was vaginal birth. All 13 children were live-born without gross impairment during neonatal period (all newborns presenting with 10-min Apgar scores of 9 or 10, respectively). One of the survivors had an additional X chromosome as stated above. In all cases, MRI was performed postnatally (prenatal MR imaging was available in four of these fetuses) and subsequent surgical interventions were conducted. Postnatal neurosonography and MRI confirmed prenatal diagnosis in all live-born fetuses. Neurosurgery was performed within the first 7 months postnatally. Outcome was strongly depending on the severity of concomitant cerebral anomalies.
Long-term pediatric follow-up was available in two of the survivors. In one case, postnatal and long-term outcome was rather favorable in spite of severe cerebro-cranial malformations that had been diagnosed antenatally. The child suffered from cranio-cervical meningocele, hypoplasia of the cerebellum, cervicothoracic syrinx, and complex cervico-spinal malformation. At the age of two weeks, neurosurgery was performed. At the age of 3 9/12 years, the child showed diminished cognitive and motor skills (particularly speech development). However, skills improved significantly with ergotherapy and logopedic support. Neurologic status was uneventful apart from discrete dysdiadochokinesis. The second case with frontoethmoidal encephalocele and additional arachnoidal cyst had neurosurgical resection of the cele at the age of 7 weeks. The concomitant arachnoidal cyst was resected at the age of 7 months. The girl showed impaired motor and cognitive skills and presented with seizures and macrocephaly and plagiocephalus. At the age of 18 months, the child demonstrated slightly retarded motor development and normal neurologic state.
In the remaining 11 cases, postnatal follow-up was available up to the age of 6–12 months. In all, but one case (tiny parietal meningocele) surgical resection of the herniated tissue and plastic covering of the defect was necessary. Four children needed additional ventriculoperitoneal shunting due to concomitant hydrocephalus. So far, all cases solely affected by meningeal protrusions showed a normal or only slightly impaired development (Table 2). Those infants who had resection of an encephalocele (containing cerebral tissue) were more likely to have neuromotoric sequelae. However, based on our data, there were no severely disabled individuals to report.
Discussion
This is a large cohort study of antenatal cases diagnosed with cephaloceles. The majority of fetuses (72%) enrolled in this study suffered from herniated brain tissue. Out of which, only six pregnancies continued until term, whereas nearly half of fetuses who had solely meningeal protrusions were live-born. Short-term (and in two cases long-term) follow-up revealed that sixty percent of the survivors showed normal development or only had a mild neuromotoric delay.
Prenatal sonographic diagnosis in most cases is based on the direct visualization of the cephalocele and the osseous defect, which is typically localized in the midline and rarely lateral in association with the cranial sutures. Overall, occipital cephaloceles constitute 75% of all craniocerebral bulgings [3]. In our patient population, we noticed a similar distribution (80% occipital defects vs. 6% parietal lesions). Another 14% were frontally located, which is in line with previous reports (Table 3). Interestingly, the latter are discussed to constitute not a genuine form of neural tube defect (NTD), but rather originate from environmental influences (e.g., reduced folic acid intake) and are more common in Southeast Asia [2]. In this regard, maternal passive smoking has recently been attributed to be an independent risk factor for three subtypes of NTD (anencephaly, spina bifida and most strongly encephalocele) [17, 18]. An occipitocervical lesion accompanied by features of Chiari II malformation (herniation of posterior fossa contents) has also been described as Chiari type III malformation (present in two live-born fetuses in our study) [19, 20].
Although the onset of cephaloceles reportedly occurs between 25 and 50 days of gestation (for anteriorly located lesions) and up to 60 days (for posterior defects), respectively, the median GA in our study at initial diagnosis was 18 + 0 gestational weeks (Table 2) which is in accordance to current literature [14]. In 43% the diagnosis was established before 20 completed weeks. The earliest GA at diagnosis was 10 + 2 weeks (together with 14 additional cases identified during first trimester). Recently, Sepulveda and co-workers stated that with detailed evaluation of skull contour all cases of cephaloceles are potentially amenable to early first trimester diagnosis [15]. An early diagnostic hint might be an enlargement of the rhombencephalic cavity, a structure that is readily seen between 8 and 12 gestational weeks [21].
Although cephaloceles as small as 3 mm in diameter could be detected during first trimester scan, the issue of correct antenatal diagnosis of any neural involvement remains challenging. Moreover, one has to bear in mind that progression from meningocele to meningoencephalocele with advancing pregnancy has been described [6, 22]. On the other hand, regression of an encephalocele has also been found [23]. Bronshtein and Zimmer reported on a case with a changing appearance of the cele (sliding of herniated brain tissue in a to and fro manner) [24]. Nevertheless, this has to be taken into account during counseling of the parents after early diagnosis of a cephalocele. It is of note that in our series, similar to Sepulveda’s findings (two live-borns), none of the affected pregnancies continued until term after early diagnosis of a cephalocele. Although there was a certain increase in relative size throughout pregnancy, we did not notice any change in gross appearance during repeated scans.
It has recently been shown that in the case of open spina bifida, as a more frequent neural tube defect, a biparietal diameter <5th percentile will tenfold increase the likelihood of a spinal dysraphism [25]. Applying these results on our study population, microcephaly was not a consistent finding, but if present it did worsen the prognosis as stated beneath. The main differential diagnoses of cephaloceles include cystic hygroma, teratoma, hemangioma, and scalp edema [26,27,28,29]. In this regard, detection rates of cephaloceles are frequently higher in case of high-risk family constellation, (e.g., affected relatives with Meckel–Gruber, Walker–Warburg, oro-facial-digital syndrome, Joubert syndrome and related disorders (JSRD), or Knobloch syndrome, respectively, as in the present study) [30]. In fact, in 13 of 65 cases enrolled in our study, a genetic predisposition was registered (consanguinity, affected siblings or family). However, familial recurrence of isolated non-syndromic cephalocele is an uncommon phenomenon [31] and was present in a single family in our series. Definitive prenatal diagnosis might be achieved by applying molecular diagnostic techniques (e.g., targeted DNA sequencing chromosomal microarray, and whole genome sequencing) [32, 33].
In isolated cephaloceles, the volume of protruding brain parenchyma and consecutive microcephaly constitutes the main impact on prognosis postnatally. There were several attempts for categorizing craniocerebral protrusions in either prenatal and postnatal cohorts, all of which tabulated affected individuals with respect to their gross appearance (cystic vs. solid nature or classification based on the origin and/or extent of herniated tissue) [5, 34,35,36]. Bannister et al. pointed out that large herniation sacs were not necessarily associated with a large mass of herniated tissue, and thus the size of the entire cele does not correlate with the prognosis for the fetus [14]. These findings are closely related to our observations, as we were able to show that the size of the cele (ranging from 5 × 6 to 66 × 56 mm) did not allow to draw reliable conclusions regarding both the course of pregnancy and the postnatal outcome in our cohort. Although 12/13 live-born fetuses needed surgical treatment, even those two children with larger herniations had no signs of severe developmental delay. On the other hand, it has been proposed that if >50% of intracranial tissue is herniated, postnatal survival is rather unlikely [13]. A large neurosurgical study revealed that after surgical intervention 50/85 patients (59%) showed normal development or only had mild delay, 14 (16%) had moderate, and 21 (25%) had severe delay, respectively [35]. Focusing on the impact of location of the skull defect, occipital encephaloceles are supposed to be associated with a higher frequency of hydrocephalus and seizures during postnatal life [3].
Although fetal neurosonography remains the primary imaging modality, magnetic resonance imaging (MRI) as an adjunctive tool to detailed CNS examination might be of help particularly in those cases with limited acoustic window. According to Kasprian et al., MR imaging is of value in assessment of gyration anomalies and brainstem morphology of externalized brain tissue as this has been reported to be subject of degenerative changes throughout pregnancy [16]. In our patient population, fetal MRI was performed beyond 20 completed weeks of gestation in eight fetuses out of which four were live-born (Fig. 1). There was no additional information added by MRI in these fetuses compared to the results of detailed neurosonography. Nevertheless, in case of continuation of the pregnancy or when additional cerebral malformations are suspected by ultrasound, fetal MRI is an adjunct for establishing a precise diagnosis.
Our study has some limitations which have to be pointed out. First the retrospective design of the study; second there is a certain bias because of the referral population focusing on more severe cases; third there is only a small number of pregnancies continued until term. Furthermore, the follow-up is limited and no systematic approach for assessment of the surviving infants was available.
Conclusions
In case of prenatally suspected cephalic anomalies (increased nuchal translucency, small skull (reduced biparietal diameter), enlarged rhombencephalic cavity), affected fetuses in prior pregnancies or a positive family history, early diagnosis of cranial herniations as early as the first trimester is feasible. After establishing the diagnosis, a thorough evaluation of accompanying anomalies of the fetus is mandatory and invasive testing should be offered including advanced moleculargenetic analysis. Although most of the affected pregnancies were terminated (Table 3), 20% the parents opted for continuation of the pregnancy. In our cohort, there were no severe neuromotoric disabilities (after neurosurgical intervention) reported. Parental counseling should be individually tailored in an interdisciplinary approach together with neuropediatricians, geneticists, and neurosurgeons. In those cases, repeated sonograms and MRI are strongly recommended.
References
Cameron M, Moran P (2009) Prenatal screening and diagnosis of neural tube defects. Prenat Diagn 29:402–411
Rowland CA, Correa A, Cragan JD, Alverson CJ (2006) Are encephaloceles neural tube defects? Pediatrics 118:916–923
Thompson DN (2009) Postnatal management and outcome for neural tube defects including spina bifida and encephalocoeles. Prenat Diagn 29:412–419
ten Donkelaar HJ, Bekker M, Renier WO, Hori A, Shiota K (2014) Neurulation and neural tube defects. In: ten Donkelaar HJ (ed) Clinical neuroembryology, 2nd edn. Springer, Heidelberg, pp 165–217
Goldstein RB, LaPidus AS, Filly RA (1991) Fetal cephaloceles: diagnosis with US. Radiology 180:803–808
Blaas HG, Eik-Nes SH (2009) Sonoembryology and early prenatal diagnosis of neural anomalies. Prenat Diagn 29:312–325
Rösing B, Kempe A, Berg C, Kahl P, Knöpfle G, Gembruch U, Geipel A (2008) Orofaciodigital syndrome Type IV (Mohr-Majewski): early prenatal diagnosis in siblings. Ultrasound Obstet Gynecol 31:457–460
Graham D, Johnson TR Jr, Winn K, Sanders RC (1982) The role of sonography in the prenatal diagnosis and management of encephalocele. J Ultrasound Med 1:111–115
Simpson DA, David DJ, White J (1984) Cephaloceles: treatment, outcome, and antenatal diagnosis. Neurosurgery 15:14–21
Chervenak FA, Isaacson G, Mahoney MJ, Berkowitz RL, Tortora M, Hobbins JC (1984) Diagnosis and management of fetal cephalocele. Obstet Gynecol 64:86–91
Jeanty P, Shah D, Zaleski W, Ulm J, Fleischer A (1991) Prenatal diagnosis of fetal cephalocele: a sonographic spectrum. Am J Perinatol 8:144–149
Wininger SJ, Donnenfeld AE (1994) Syndromes identified in fetuses with prenatally diagnosed cephaloceles. Prenat Diagn 14:839–843
Budorick NE, Pretorius DH, McGahan JP, Grafe MR, James HE, Slivka J (1995) Cephalocele detection in utero: sonographic and clinical features. Ultrasound Obstet Gynecol 5:77–85
Bannister CM, Russell SA, Rimmer S, Thorne JA, Hellings S (2000) Can prognostic indicators be identified in a fetus with an encephalocele? Eur J Pediatr Surg 10(Suppl 1):20–23
Sepulveda W, Wong AE, Andreeva E, Odegova N, Martinez-Ten P, Meagher S (2015) Sonographic spectrum of first-trimester fetal cephalocele: review of 35 cases. Ultrasound Obstet Gynecol 46:29–33
Kasprian GJ, Paldino MJ, Mehollin-Ray AR, Shetty A, Williams JL, Lee W, Cassady CI (2015) Prenatal Imaging of Occipital Encephaloceles. Fetal Diagn Ther 37:241–248
Wang M, Wang ZP, Zhang M, Zhao ZT (2014) Maternal passive smoking during pregnancy and neural tube defects in offspring: a meta-analysis. Arch Gynecol Obstet 289:513–521
Suarez L, Ramadhani T, Felkner M, Canfield M, Hendricks K (2011) Maternal smoking, passive tobacco smoke, and neural tube defects. Birth Defects Res A Clin Mol Teratol 91:29–33
Cama A, Tortori-Donati P, Piatelli GL, Fondelli MP, Andreussi L (1995) Chiari complex in children—neuroradiological diagnosis, neurosurgical treatment and proposal of a new classification (312 cases). Eur J Pediatr Surg 5(Suppl 1):35–38
Cakirer S (2003) Chiari III malformation: varieties of MRI appearances in two patients. Clin Imaging 27:1–4
van Zalen-Sprock RM, van Vugt JM, van Geijn HP (1996) First-trimester sonographic detection of neurodevelopmental abnormalities in some single-gene disorders. Prenat Diagn 16:199–202
Basaran A (2011) Diagnosis of occipital meningocele at 10 weeks of gestation and its natural course—imaging of meningoencephalocele in early postembryonic period. Ultraschall Med 32:622–623
Hanley ML, Guzman ER, Vintzileos AM, Leiman S, Doyle A, Shen-Schwarz S (1996) Prenatal ultrasonographic detection of regression of an encephalocele. J Ultrasound Med 15:71–74
Bronshtein M, Zimmer EZ (1991) Transvaginal sonographic follow-up on the formation of fetal cephalocele at 13–19 weeks’ gestation. Obstet Gynecol 78:528–530
Bernard JP, Cuckle HS, Stirnemann JJ, Salomon LJ, Ville Y (2012) Screening for fetal spina bifida by ultrasound examination in the first trimester of pregnancy using fetal biparietal diameter. Am J Obstet Gynecol 207(306):e301–e305
Shahabi S, Busine A (1998) Prenatal diagnosis of an epidermal scalp cyst simulating an encephalocoele. Prenat Diagn 18:373–377
Sherer DM, Perillo AM, Abramowicz JS (1993) Fetal hemangioma overlying the temporal occipital suture, initially diagnosed by ultrasonography as an encephalocele. J Ultrasound Med 12:691–693
Noriega CA, Fleming AD, Bonebrake RG (2001) A false-positive diagnosis of a prenatal encephalocele on transvaginal ultrasonography. J Ultrasound Med 20:925–927
Sepulveda W, Wong AE, Sepulveda S, Corral E (2011) Fetal scalp cyst or small meningocele: differential diagnosis with three-dimensional ultrasound. Fetal Diagn Ther 30:77–80
Mittermayer C, Lee A, Brugger PC (2004) Prenatal diagnosis of the Meckel–Gruber syndrome from 11th to 20th gestational week. Ultraschall Med 25:275–279
Ghonge NP, Kanika SS, Poonam B (2011) Familial occipital cephalocele in a fetus at 21 weeks’ gestation: imaging demonstration across 3 generations. J Ultrasound Med 30:1747–1751
Tallila J, Salonen R, Kohlschmidt N, Peltonen L, Kestila M (2009) Mutation spectrum of Meckel syndrome genes: one group of syndromes or several distinct groups? Hum Mutat 30:E813–E830
Yatsenko S, Davis S, Hendrix N et al (2013) Application of chromosomal microarray in the evaluation of abnormal prenatal findings. Clin Genet 84:47–54
Fleming AD, Vintzileos AM, Scorza WE (1991) Prenatal diagnosis of occipital encephalocele with transvaginal sonography. J Ultrasound Med 10:285–286
Lo BW, Kulkarni AV, Rutka JT, Jea A, Drake JM, Lamberti-Pasculli M, Dirks PB, Thabane L (2008) Clinical predictors of developmental outcome in patients with cephaloceles. J Neurosurg Pediatr 2:254–257
Docherty JG, Daly JC, Carachi R (1991) Encephaloceles: a review 1971–1990. Eur J Pediatr Surg 1(Suppl 1):11–13
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was not funded.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors. It is a retrospective archive study. The institutional review boards of both the universities of Bonn and Lübeck do not require formal approval for retrospective archive studies; therefore, an ethical approval was not sought.
Informed consent
Informed consent was obtained from all individual participants enrolled in this study.
Author contributions
UG, JW: Protocol/project development. UG, MK, UG, RAF, AK, AG, ChB, JW: Data collection or management. JW, FH, UG: Data analysis. JW, FH, UG: Manuscript writing/editing.
Rights and permissions
About this article

Cite this article
Weichert, J., Hoellen, F., Krapp, M. et al. Fetal cephaloceles: prenatal diagnosis and course of pregnancy in 65 consecutive cases. Arch Gynecol Obstet 296, 455–463 (2017). https://doi.org/10.1007/s00404-017-4424-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00404-017-4424-7