
Abstract
Hexanucleotide repeat expansions in C9orf72 are the most common genetic cause of amyotrophic lateral sclerosis (C9 ALS). The main hypothesized pathogenic mechanisms are C9orf72 haploinsufficiency and/or toxicity from one or more of bi-directionally transcribed repeat RNAs and their dipeptide repeat proteins (DPRs) poly-GP, poly-GA, poly-GR, poly-PR and poly-PA. Recently, nuclear import and/or export defects especially caused by arginine-containing poly-GR or poly-PR have been proposed as significant contributors to pathogenesis based on disease models. We quantitatively studied and compared DPRs, nuclear pore proteins and C9orf72 protein in clinically related and clinically unrelated regions of the central nervous system, and compared them to phosphorylated TDP-43 (pTDP-43), the hallmark protein of ALS. Of the five DPRs, only poly-GR was significantly abundant in clinically related areas compared to unrelated areas (p < 0.001), and formed dendritic-like aggregates in the motor cortex that co-localized with pTDP-43 (p < 0.0001). While most poly-GR dendritic inclusions were pTDP-43 positive, only 4% of pTDP-43 dendritic inclusions were poly-GR positive. Staining for arginine-containing poly-GR and poly-PR in nuclei of neurons produced signals that were not specific to C9 ALS. We could not detect significant differences of nuclear markers RanGap, Lamin B1, and Importin β1 in C9 ALS, although we observed subtle nuclear changes in ALS, both C9 and non-C9, compared to control. The C9orf72 protein itself was diffusely expressed in cytoplasm of large neurons and glia, and nearly 50% reduced, in both clinically related frontal cortex and unrelated occipital cortex, but not in cerebellum. In summary, sense-encoded poly-GR DPR was unique, and localized to dendrites and pTDP43 in motor regions of C9 ALS CNS. This is consistent with new emerging ideas about TDP-43 functions in dendrites.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hexanucleotide repeat expansions of GGGGCC in chromosome 9 open reading frame 72 (C9orf72) are the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) [11, 41]. The repeat is bi-directionally transcribed and translated through a repeat-associated non-AUG-dependent (RAN) translation mechanism, giving rise to sense and antisense RNA foci and five different sense- and antisense-encoded dipeptide repeat proteins (DPRs): poly-Gly-Ala (GA), poly-Gly-Arg (GR), poly-Pro-Arg (PR), poly-Pro-Ala (PA), and poly-Gly-Pro (GP) (Fig. 1) [3, 14, 35, 36, 60]. The main pathogenic mechanisms are likely to relate to a gain of toxicity from one or more of the RNA or DPR products, or from C9orf72 protein haploinsufficiency. Of particular interest are the arginine-containing DPR poly-GR and poly-PR, which are positively charged, hydrophilic, and have been reported to be active inside nuclei and are able to disturb phase transition of proteins with low complexity domains [24] and impair nucleocytoplasmic trafficking [22, 24, 26, 29, 56]. Several nuclear pore-related proteins including RanGap, Lamin B1, and Nup205 can enhance or protect against such neurotoxicity caused by the repeat expansions in different disease models [13, 19, 57]. Models of loss of C9orf72 function in mice have mainly shown peripheral immune-related phenotypes, but no apparent neurodegeneration [4, 7, 18, 21, 23, 39, 47], while gain of function models using cells [12, 22, 25, 33, 42, 45, 52, 59, 60], yeast [19], flies [13, 34, 49, 54, 57], and mice [8, 18, 28, 38, 40, 44, 58] have indeed shown neurodegeneration. Currently, the most active debates in the field relate to the contributions of haploinsufficiency, relative contributions to toxicity of repeat-expanded RNAs and their DPR protein products, toxicity of arginine-containing DPRs, and contributions to toxicity from sense and antisense strands. The answers to these questions are key to future therapy development, since gain of function mechanisms can be targeted by antisense oligonucleotides (ASO), small molecules or therapeutic antibodies to reduce toxic products, but these therapies might worsen disease if loss of C9orf72 function is a significant contributor.

Proposed disease mechanisms of ALS/FTD caused by hexanucleotide repeat expansions in C9orf72. The GGGGCC hexanucleotide repeat in intron 1 of C9orf72 is bi-directionally transcribed and translated into seven potentially toxic products: sense (GGGGCC) RNA foci, antisense (GGCCCC) RNA foci, and 5 unique polydipeptide repeat proteins: poly-GA, poly-GP, poly-GR, poly-PA, and poly-PR
While cellular and animal models have enabled study of molecular mechanisms, it is critical to study actual patient nervous systems. Several studies of human tissues of C9 ALS have been performed. C9 ALS neuropathology is clearly in the spectrum of TDP-43 proteinopathies [1, 5, 20]. C9orf72 haploinsufficiency is supported by evidence showing decreased expression of C9orf72 mRNAs [15, 50] and abnormalities of C9orf72 protein [51, 53]. Aggregations of all five DPR proteins have been observed in C9 ALS nervous systems [3, 14, 36, 60]. However, several studies have shown that TDP-43 pathology rather than DPR pathology correlates to neurodegeneration [9, 30, 31, 45]. Neuropathological evidence of intra-nuclear accumulation such as dot-like aggregates of arginine-containing poly-GR or poly-PR DPRs that would support their nuclear toxicity has been controversial, with some reports supporting their presence [32, 45, 52], and others suggesting non-specificity [9, 31, 36]. Using RanGap and Nup205, one study reported abnormalities that consisted of discontinuous nuclear membranes and diffuse nuclear protein deposition consistent with defective nucleocytoplasmic transport [57]. Another group showed lack of nuclear membrane staining of Importin β1 and RanGTPase as markers for nucleocytoplasmic transport proteins in C9-ALS [53]. Neuropathological confirmation of these nuclear markers has not been established.
To study these critical neuropathological aspects of C9 ALS, we systematically evaluated all the main neuropathological proteins—C9orf72 protein, all five DPRs, and nuclear membrane markers RanGap, Lamin B1, Importin β1, Ran-GTPase and Nup205—in our short postmortem-interval ALS brain and spinal cord biorepository. We compared the accumulation of these proteins to one other, compared clinically related to unrelated neuroanatomical regions, and compared them to pTDP-43, the signature protein in both C9 ALS and sporadic ALS (SALS). We find that of all the features, sense-encoded arginine-containing poly-GR aggregates are uniquely correlated with disease and that this association was especially apparent in dendrites.
Materials and methods
CNS tissues
Human tissues were obtained using a short-postmortem interval acquisition protocol that followed HIPAA-compliant informed consent procedures and were approved by Institutional Review Board (Benaroya Research Institute, Seattle, WA IRB# 10058 and University of California San Diego, San Diego, CA IRB# 120056). The ALS tissues were from patients who presented with ALS as their clinical phenotype, with or without FTD, and all had progressed and died from their motor impairment. All C9-ALS cases showed mislocalization of pTDP-43 in frontal and motor cortex and in the spinal anterior horn (Supplemental Table 1). We evaluated five C9 ALS cases (confirmation by repeat-primed PCR and Southern blotting), three sporadic ALS (SALS) cases, and three non-neurological controls (Table 1).
CNS regions, cell types, and relationships to disease
In brain, we studied the following regions: frontal cortex, motor cortex, parietal cortex, occipital cortex, retrosplenial granular cortex (RSGC), hippocampus, cerebellum, and olfactory bulb. In spinal cord, we examined anterior and posterior horns and white matter tracks at cervical, thoracic, and lumbosacral levels. We examined all layers of brain with special attention to layer 5 of the motor cortex and its Betz cells, and all regions of the spinal cord with special attention to Rexed lamina IX in the anterior horn and its spinal motor neurons. Cell classification to neuron or glia was based on morphology. To classify a cell as neuronal, at least three of the following four criteria were met: cytoplasm was large, there was lipofuscin, nuclear diameter was greater than 10 µm, and there was a single prominent nucleolus. To classify a cell as glial, nuclei were small, cytoplasm was scant and compact, and there was no lipofuscin and no prominent nucleolus. In our analysis, we regarded the following regions as clinically related: motor cortex, frontal cortex and anterior horn of spinal cord; the following regions as clinically unrelated: parietal cortex, occipital cortex and posterior horn of spinal cord; and the following regions as of uncertain clinical relationship: hippocampus, cerebellum, RSGC, and olfactory bulb.
Chromogenic in situ hybridization (CISH)
Transcription of C9orf72 leads to three separate transcripts that share homology in the 5′ region. Two of the transcripts, NM_18325.4 and NM_1256054.2 are significantly longer [3261 base pairs (bp) and 3356 bp, respectively] compared to the shorter isoform NM_145005.5, which is 1957 bp and has a unique 3′ untranslated region. The two longer isoforms encode identical proteins (54.3 kDa) as they only differ in their 5′ non-coding region. The short isoform mRNA lacks multiple exons in the central and 3′ coding regions, and is predicted to encode a smaller protein (24.7 kDa). The 3′ terminal exon of the short isoform mRNA extends beyond a splice site resulting in a novel 3′ UTR, compared to both longer isoforms. Probes were designed choosing unique regions to specifically identify either the long or short isoforms. To specifically target the short isoform, the novel 3′ UTR region from bp 859–1932 (NM_145005.5) was used allowing for the generation of 13 zz-pairs for hybridization. For the longer isoforms, bp 774–2109 (NM_18325.4) was used as it is shared between both of the longer isoforms and yielded 20 zz-pairs. Due to the shorter sequence of the short isoform and thus the shorter number of zz-pairs, this probe was expected to have a lower overall signal compared to the longer isoform.
Immunoblotting
For antibodies against DPR proteins, peptide antigens (C-Ahx-(GA)8-amide, C-Ahx-(GR)8-amide, C-Ahx-(PR)8-amide, C-Ahx-(GP)8-amide and C-Ahx-(PA)8-amide) were used to immunize rabbits. Antibodies specific for each DPR protein are herein annotated for Rb4333 as poly(GA), Rb4335 as poly(GP), Rb4996 as poly(GR), Rb15989 as poly(PA) and Rb15986 as poly(PR). The specificity of these antibodies was tested by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis and immunoblotting. Briefly, 100 ng of recombinant GST-(GA)10, GST-(GR)10, GST-(GP)10, GST-(PR)9 and GST-(PA)10 proteins were separated by 12% SDS-PAGE and were transferred to nitrocellulose membrane. While it has been suggested that poly-GP translated by sense repeat-containing RNA may be different from the one by antisense RNA with unique C-terminal peptides [60], we could not distinguish between the two with our poly-GP antibody. For detecting C9orf72, human or mouse tissues were homogenized in standard RIPA lysis buffer and 30 μg of total protein lysate was resolved on a 10% SDS-PAGE. Proteins were transferred to nitrocellulose membranes and probed with antibodies against C9orf72 (Proteintech #22637-1-AP, 1:1000) and GADPH (Millipore #CB1001, 1:1000). The membranes were washed in PBST and blotted with primary antibodies. After the membrane was incubated with HRP-conjugated secondary antibody, bands were visualized by the ECL plus Western Blotting Detection System (Pierce).
Immunohistochemistry (IHC) and immunofluorescence (IF)
For IHC, we used formalin-fixed paraffin-embedded sections with 6 µm thickness. On day 1, sections were deparaffinized with Citrisolv (Fisher Scientific #04-355-121) and hydrated with different dilutions of alcohol. Endogenous peroxidase activity was quenched with 0.06% H2O2 for 15 min. Antigen retrieval was performed in a high pH solution (Vector #H-3301) in a pressure cooker for 20 min at a temperature of 120 °C. For each antibody, we optimized antigen retrieval at pH = 6 and pH = 9, we chose the pH with stronger signal and/or lower background. Following antigen retrieval, sections were blocked with 1% FBS (Atlanta Biologicals #511150) and 0.1% Triton X-100 (Sigma #65H2616) in PBS for 25 min. The sections were incubated with primary antibodies overnight as follows: In-house generated (please see description from immunoblotting) rabbit primary antibodies for DPRs (poly-GA: 1/1000, poly-GP, poly-GR, poly-PR and poly-AP: 1/5000), RanGap (Santa Cruz #sc-28322 1:500), Lamin B1 (Abcam #16048 1:500), Importin β1 (BD Biosciences #610559 1:50.), Ran-GTPase (BD Biosciences #610340 1:50), Nup205 (Novus #NBP1-91247 1:50), and C9orf72 (Proteintech #66140-1-Ig, 1:1000). On the second day, after 60-min incubation with secondary antibody (Immpress reagent kit, anti-Rabbit, Vector) in room temperature, signals were detected using NovaRed (Vector #sk-4800) for 1–5 min. Counterstaining was performed with hematoxylin (Fisher #HHS128).
For single IF of RanGap or co-IF of DPRs with pTDP-43 or RanGap, the protocol on first day was identical to IHC except that the DPR primary antibodies were mixed with mouse phospho-TDP-43 (Cosmo Bio #TIP-PTD-01, 1/5000) or mouse RanGap (Santa Cruz #sc-28322, 1/500) for overnight incubation. On the second day, mixed donkey anti-rabbit (Jackson Immuno Research, Alex Flow 488 #126601: 1/500) and donkey anti-mouse (Jackson Immuno Research, Cy3 #125797: 1/500) secondary antibodies were added for 60 min in room temperature. After 10 min staining with DAPI (Life tech, #D21490), we quenched CNS auto-fluorescence with 0.1% Sudan Black dissolved in 70% ethanol for 10–30 s. Slides were cover-slipped using prolong Gold anti-fade mounting reagent (Life tech, #1681269). For quenching of poly-GR antibody, a mixture of poly-GR antibody with 20 times more concentrated poly-GR10 recombinant protein was incubated for 2 h in room temperature before starting the same IHC protocol for DPRs.
Visualization and quantification
For IHC visualization, all slides were scanned with Hamamatsu Nanozoomer 2.0HT Slide scanner. With the use of NDP.view 2 viewing software, scanned slides were evaluated at 40X magnification. We selected multiple random fields to have at least 100 neurons and glial cells for each layer of selected areas. For Betz cells and neurons of anterior and posterior horn of spinal cord, we evaluated at least 20 neurons and determined the percentage of neurons and glial cells that contain at least one cytoplasmic aggregate. For visualization by IF, we used an inverted Olympus FV1000 laser scanning confocal microscope. Maximum projections of z-stacks were compiled using Fiji [43].
For DPRs, we studied five C9 ALS, three SALS and three control nervous systems (Table 1). For each case, we evaluated at least 100 neurons of different layers of cortex, hippocampus, cerebellum, and olfactory bulb, while for Betz cells of layer 5 of motor cortex and anterior and posterior horn of spinal cord at least 20 neurons were evaluated per case. We examined percent of neurons with aggregates and in addition devised a quasi-logarithmic scale from 0 to 6 to rate the abundance of DPR aggregations to capture the harmonic range of abnormalities: 0, no aggregation; 1, 0.1–1%; 2, 2–5%; 3, 6–10%; 4, 11–30%; 5, 31–60%; 6, more than 60%. For co-localization of pTDP-43 and DPRs, we examined five C9 ALS, three SALS, and three controls and examined at least ten dendritic and perinuclear aggregations for each region, and calculated percentage of DPR aggregations that co-localized with pTDP-43.
For RanGap, Lamin B1, Importin β1, and Nup205, we studied five C9 ALS, three SALS, and three controls using IF and IHC. We randomly selected 50 neurons in layer 5 of motor cortex of each case and all the remaining spinal motor neurons in lumbar spinal cord were evaluated of each case. For RanGap and Lamin B1, we divided the pattern of staining into four categories: ring (signals around the circumference), diffuse (uniform signal), folded (displaying creases), and punctate (spotted around the nucleus). For RanGap, we also evaluated nuclear shape as being normal (round or slightly oval-shaped nuclei, with or without diffuse RanGap), moderately abnormal (oval or elongated shape, with one or two visible deep folds in it), or severely abnormal (misshapen or with very folded membrane). For Importin β1, we divided the pattern of staining into three categories: smooth (diffuse nuclear signal with absence of nucleolar staining), intermediate (some folding of nuclear envelope visible, or with irregular borders), and irregular (no visible nuclear borders). Nup205 appeared diffuse throughout the cytoplasm in all neurons and Ran-GTPase (using the only commercially available antibody validated for human IHC) did not give signal, and therefore, these could not be evaluated for nuclear signal.
For co-localization of poly-GR and RanGap, we counted 20 neurons containing poly-GR and 20 neurons without poly-GR in layer 5 of motor cortex of each of five C9 ALS cases with double IF to determine whether the presence of poly-GR staining correlated with the expression pattern of RanGap.
Statistics
For analysis of datasets with two variables, we employed a two-tailed Wilcoxon matched-pairs signed-rank test, a test for comparing non-parametric data. For analyses of datasets with three or more variables, we employed the Kuskal–Wallis test (or one-way ANOVA on ranks), a non-parametric method for testing two or more independent samples. Error bars are presented as s.e.m.. All data analyses and graphing were performed using the GraphPad Prism version 6.00 for Windows (GraphPad Software, La Jolla, CA).
Results
DPRs were distinctly seen in cytoplasm of neurons, but not in axons, glia or white matter
Inclusions of unconventionally translated DPRs from both sense (poly-GP, poly-GA, and poly-GR) and antisense (poly-GP, poly-PA and poly-PR) strand RNA repeats are well-known neuropathological hallmarks of C9 ALS [3, 9, 14, 30, 31, 36, 45, 60]. We assessed the five unique DPRs in C9 ALS nervous system using DPR-specific antibodies (Fig. 2a and Table 2). We observed three different morphologies of inclusions: (1) perinuclear cytoplasmic aggregations that were star-shaped or amorphous (Fig. 2b, d–f), (2) dendritic aggregations (Fig. 2c), and (3) diffuse cytoplasmic staining (Fig. 2g). The aggregations ranged in size from 1 to 20 µm, and larger aggregates were generally seen in larger neurons. Aggregations were 1–10 µm in cortex, 1–6 µm in olfactory bulb, 6–8 µm in spinal cord, and up to 20 µm in large neurons of hippocampus (Table 2). Most DPR aggregations were perinuclear in location and star or amorphous in shape (Fig. 2b, d–f). Poly-GA, poly-GP and poly-GR also had occasional dendritic aggregations. Dendritic aggregates from poly-GR were only observed in motor cortex. Poly-GP and poly-GA had diffuse cytoplasmic staining but these were rare. Poly-PA and poly-PR were rare (Fig. 2e–f).

All five polydipeptide repeat proteins (DPRs) are seen in neurons but not glia or axons throughout the CNS, but only poly-GR correlates with disease anatomy and cellular pathology. a Antibodies specifically recognize poly-GA, poly-GR, poly-GP, poly-PR, and poly-PA using SDS polyacrylamide gel electrophoresis and immunoblotting. b–g Immunohistochemistry (IHC) staining identifies that the main morphologies of the DPR aggregates are star-shaped or amorphous cytoplasmic perinuclear aggregations (d, e, and f) but occasionally dendritic aggregations (c) and diffuse cytoplasmic staining are seen (g). DPRs rarely seen in anterior horn of spinal cord, including poly-GR (b, insert). h DPRs especially poly-GA, poly-GP and poly GR are widely distributed throughout the CNS. (i) Only poly-GR is significantly more prevalent in disease-related regions than in disease-unrelated areas (p < 0.001). j–l immunofluorescence (IF) staining shows dendritic poly-GR inclusions co-localize with pTDP-43. m–o IF staining shows perinuclear poly-GR inclusions co-localize with pTDP-43. p Most of the dendritic-type poly-GR (22/26) and none of the dendritic type poly-GA (0/20) and poly-GP (0/20) co-localize with pTDP-43 in motor cortex. q Occasionally star-shaped or amorphous poly-GR (17/100), poly-GA (1/100), and poly-GP (5/100) aggregates are co-localized with pTDP-43 in motor cortex and it is significantly more in poly-GR than in poly-GA (p = 0.005)
In the brain, we found aggregations of all the DPRs and these were more abundant in layers 2–6, the layers with prominent neurons, than in layer 1, which mainly has glial cells and neuronal fibers, or in subcortical white matter, which is comprised mainly of axons and glial cells, especially oligodendrocytes (Supplemental Fig. 1a–e, and Supplemental Data). In the olfactory bulb, we observed poly-GA and poly-GP, but no other DPRs in all layers except glomerular layer (Supplemental Fig. 1f). In the cerebellum, we observed all DPRs and we found these in molecular, granular and Purkinje cells (Supplemental Fig. 1g). In hippocampus, we observed all DPRs and we found them in layers with prominent neurons—hilus, dentate granule, and pyramidal cell layers—but not elsewhere, where there are mainly glial cells and neuron fibers (Supplemental Fig. 1h). In the spinal cord, by contrast, we saw only few poly-GA, poly-GP, and poly-GR, and no poly-PA or poly-PR (Supplemental Data). We observed these in neurons of both anterior and posterior horns, and did not observe them in the tracts, which similar to subcortical white matter, are comprised mainly of axons and glial cells, especially oligodendrocytes (Supplemental Fig. 1i).
Thus, DPRs were in neurons and grey matter, not in axons, glia, or white matter.
DPRs were widely distributed, but only poly-GR correlated to clinically related regions and had unique co-localization with pTDP-43 especially in dendrites
To better understand the significance of DPRs in C9 ALS pathogenesis, we determined whether there is correlation between DPR and regions of clinical involvement. As stated above, we regarded motor cortex, frontal cortex and anterior horn of spinal cord as clinically related regions, and parietal cortex, occipital cortex and posterior horn of spinal cord as clinically unrelated regions. When we compared neuroanatomical distributions in clinically related regions to unrelated regions, only poly-GR was significantly more abundant in areas related to disease (p value < 0.001) (Fig. 2h–i, Supplement Fig. 1k–l, and Supplemental Data). We verified specificity of poly-GR aggregation detection by quenching antibody with recombinant GST-(GR)10 proteins (Supplemental Fig. 1j). The effect of poly-GR was so robust in fact that when considering all DPRs together, there seemed to be an overall significant difference (p value = 0.003), although all of the effect was attributable to poly-GR alone. We verified such correlation between poly-GR and region of degeneration by replicating the experiment on three different sections from the same regions in three different IHC trials.
We further compared the correlation of DPRs and pTDP-43 by co-localizing poly-GR, poly-GA and poly-GP with pTDP-43. The low abundance of poly-PR and poly-PA did not permit similar analysis. We separately scored dendritic and perinuclear aggregations (Fig. 2j–o). We observed dendritic-type poly-GR inclusions only in motor cortex of C9 ALS nervous systems and, more strikingly, 22 of 26 dendritic-type poly-GR inclusions co-localized with pTDP-43 (Fig. 2p and Supplemental Table 2). By contrast, 0 of 20 dendritic type poly-GA inclusions and 0 of 20 poly-GP dendritic inclusions co-localized with pTDP-43 (Fig. 2p). We also found 17 of 100 perinuclear poly-GR inclusions co-localized with pTDP-43 in motor cortex of C9 ALS nervous systems, and this compared to 1 of 100 poly-GA inclusions and 5 of 60 inclusions poly-GP (Fig. 2q). Although most dendritic type poly-GR inclusions co-localized with pTDP-43, only 4 of 100 dendritic pTDP-43 inclusions co-localized with poly-GR (none of 100 involved poly-GA or poly-GP), showing the overall complexity of TDP-43 proteinopathy in C9 ALS.
Therefore, poly-GR was localized in dendrites and cytoplasm and was uniquely correlated with disease in C9 ALS in terms of both neuroanatomical and cellular pathology.
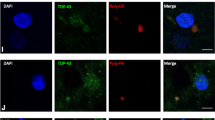
Arginine-containing DPR immunoreactivity in nuclei was not disease-specific
In model systems, short arginine-containing DPRs were shown to be able to accumulate in nucleoli and cause cell death by affecting RNA biogenesis [22] and protein translation [52]. Nuclear inclusions of the poly-GR and poly-PR in patient tissues have also been identified [32, 45, 52], but disputed [9, 31, 36]. We specifically evaluated nuclear DPR pathology in neurons in layer 5 of motor cortex and spinal motor neurons in Rexed lamina IX of spinal cord. Indeed, we detected signals from both poly-GR- and poly-PR-binding antibodies in nuclei of C9 ALS nervous systems, but we also observed these in sporadic ALS and controls, using both IHC and IF (Fig. 3a–g) and there were no quantitative differences among the groups (Fig. 3h). To test this further, we quenched poly-GR antibody with 20 times purified recombinant GST-GR10 antigen and still observed nuclear signals while cytoplasmic perinuclear signals were indeed masked, indicating non-specificity of the immunostaining (Fig. 3c insert).

Immunostaining for arginine-containing DPRs is non-specific. a–g Immunostaining for poly-GR and poly-PR are as strong in controls and SALS as C9 ALS in terms of intra-nuclear staining. c (insert) Quenching poly-GR primary antibody with poly-GR10 antigen does not mask nuclear signal. h There are no differences comparing controls, SALS, and C9 ALS in the percentage of cells with poly-GR nuclear staining in spinal motor neurons and Betz cells in layer 5 of motor cortex
Therefore, arginine-containing DPR signals in nuclei of neurons appeared to be non-specific.
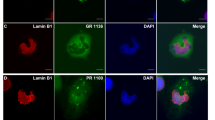
Nuclei and nuclear membrane morphology revealed by markers RanGap, Lamin B1, and Importin β1 were not abnormal in C9 ALS
Deficits in nucleocytoplasmic transport have recently been proposed to contribute to C9 ALS pathogenesis [13, 57]. However, only a few studies have determined whether nuclear membrane morphological abnormalities are seen in C9 ALS patients [13, 57]. We thus sought nuclear membrane proteins RanGap, Lamin B1, and Nup205 in neurons of layer 5 of motor and frontal cortex, spinal motor neurons, and Purkinje cells of cerebellum, and additionally Importin β1 and Ran-GTPase in spinal motor neurons. For RanGap and Lamin B1, we graded nuclear marker expression pattern as ring, diffuse nuclear, folded, and punctate around the nucleus (Fig. 4a–h). Using either RanGap or Lamin B1 as readouts, we could not find any differences among C9 ALS, SALS, and controls in either motor cortex or anterior horn of the spinal cord (Fig. 4i–j and Supplement Fig. 2a–h and m–n, respectively). Additionally, we evaluated the expression of the nuclear envelope protein Importin β1 in spinal motor neurons, graded as smooth, irregular, or intermediate (Supplemental Fig. 2o). While variation in staining pattern was demonstrated within individual spinal cords, we found no statistically significant differences among C9-ALS, SALS, and controls (Supplemental Fig. 2p). We were unable to assess potential nuclear changes using Nup205 since signals were without specific nuclear staining (Supplementary Fig. 2i–l) or using Ran-GTPase since there was no signal from the only commercially available antibody validated for human IHC. When we compared images taken by confocal microscopy using either a single section or z-stack, we found that diffuse nuclear staining of RanGap observed using IHC might in fact be a three-dimensional effect (Fig. 4k–l and Supplemental Fig. 3). For completeness, we also evaluated nuclear shape using RanGap—as stated, we graded as normal, moderately abnormal, or severely abnormal. We found nuclear morphologies were slightly aberrant (not statistically significant) in both C9 ALS and SALS motor neurons (Supplemental Fig. 4) and consistent with changes from neurodegeneration. Since we had identified poly-GR was relevant to disease, we also sought correlation between poly-GR and alterations in nucleocytoplasmic transport using RanGap as readout. The burden of ring, folded, diffuse nuclear, and punctate RanGap expression was similar in neurons with and without poly-GR (p = 0.7, 0.6, 0.8, and 0.9, respectively) (Fig. 4m–o).

Patterns of RanGap distribution are not altered in C9 ALS cases even in neurons containing poly-GR aggregates. a–h Similar patterns of RanGap staining are observed using IF staining (a–d) and IHC (e–h)—we categorized patterns as diffuse (a and e), ring (b and f), folded (c and g), and punctate (d and h). i, j There is no significant difference among controls, SALS, and C9 ALS cases in term of patterns of RanGap distribution in spinal motor neurons as well as motor neurons of motor cortex. k, l Confocal microscopy shows diffuse nuclear (k) and ring patterns are a three-dimensional effect. m–o Presence or absence of poly-GR inclusions do not alter the pattern of RanGap distribution
Thus, we could not detect nuclei abnormalities that were unique in C9 ALS.
C9orf72 protein was reduced in frontal and occipital cortex of C9-ALS, but not in cerebellum
Reduced C9orf72 expression in patients with GGGGCC repeat expansions have been reported at the mRNA [15, 50] and protein [51, 53] levels. The evidence for loss of C9orf72 protein has been difficult due to the quality of antibodies against C9orf72 [10]. We first characterized the specificity of our C9orf72 antibody in C9orf72 knockout mice (Fig. 5a–b) and using antibody quenching with purified recombinant C9orf72 proteins (Supplement Fig. 5a–b). Using immunohistochemistry staining, we found that C9orf72 protein was mainly cytoplasmic in neurons and glia in frontal cortex, motor cortex, cerebellum, and spinal cord (Fig. 5c–l). However, we did not see significant difference in intensity, distribution or morphology of C9orf72 staining between control, SALS and C9 ALS patients. To further quantitatively assess the level of C9orf72 protein, we extracted proteins from tissues acquired by a short-postmortem interval acquisition protocol and determined that the 54 kDa C9orf72 protein expression was significantly decreased not only in frontal cortex (disease related), but also in occipital cortex (disease unrelated) of C9orf72 nervous systems compared to healthy control or sporadic patients (Fig. 5m–p). Interestingly, we did not observe similar reduction of C9orf72 expression in cerebellum (Supplement Fig. 5c). The C9orf72 antibody used here is raised using amino acid 1–169 and should recognize both the full length and the predicted “short isoform” of C9orf72 protein. However, we were unable to detect the 25 kDa short isoform C9orf72 protein even though we detected strong signals of short and long mRNAs in human tonsillar epithelium by CISH (Supplement Fig. 5d–f).

C9orf72 protein is widely distributed in cytoplasm of neurons and glia throughout the CNS, but is reduced in both disease-related and disease-unrelated regions. a, b IF staining shows C9orf72 protein is present in neurons of granular, molecular and Purkinje layer of cerebellum of wildtype mice, which is absent in C9 knockout mice. c, d IHC stainings of C9or72 of Betz cells in layer 5 of motor cortex (c, insert) and motor neurons of anterior horn of spinal cord (d, insert) of C9 ALS case show diffuse cytoplasmic staining with C9orf72 antibody. e–l C9orf72 IHC staining shows no significant differences in diffuse cytoplasmic staining in Betz cells of motor cortex (layer 5), big neurons of frontal cortex (layer 5), Purkinje cells of cerebellum, and spinal motor neurons of anterior horn of spinal cord between C9 ALS and SALS. m–p Western blotting shows that C9orf72 protein expression is decreased in frontal cortex m, n and in occipital cortex (o, p) of C9 ALS compared to control and SALS cases
Thus, repeat expansions in C9orf72 lead to reduced C9orf72 protein expression in frontal and occipital cortex, but not in cerebellum.
Discussion
With the advent of exquisite fundamental therapeutic approaches such as offered by ASOs, therapeutic small molecules, and therapeutic antibodies, precise dissection of pathogenic mechanisms of neurodegeneration caused by GGGGCC repeat expansions in C9orf72 ALS is critical for identification of therapeutic targets. Of the two major hypotheses for pathogenic mechanisms—C9orf72 haploinsufficiency and gain of function from generation of one or more toxic product(s)—the gain of function mechanisms are amenable to such therapies while loss of function mechanisms could actually worsen disease. Two confounders make gain of function mechanisms especially difficult to unravel. One is that the repeat expansions are both transcriptionally and translationally active, generating both RNAs and DPRs. The other is that repeat expansions are bi-directionally active, generating essentially twice as many products. While experimental models have enabled dissection of these mechanisms, one critical measure is human neuropathology and in this study, we pursued this to characterize and compare various neuropathological hallmarks at the protein level.
The biological importance of DPRs is critically debated in C9 ALS pathogenesis. We found DPRs were neuron-specific, distributed widely in the CNS, more in brain than spinal cord, and sense-encoded much more than antisense-encoded, findings consistent with many other previously reported neuropathological studies [31, 32, 45, 60]. Most studies found DPR pathology did not correlate to disease-related regions, whereas critical studies have shown that TDP-43 aggregation does [31, 32, 45, 60]. However, importantly, in our analysis, we found that DPR pathology did correlate to disease-related regions, and when we sub-stratified by each individual DPR, we found this effect was entirely attributable to poly-GR. The different studies of poly-GR are compared and contrasted in Table 3. The most likely explanation for the different findings between our study and the others may have as much to do with definitions of clinically related and clinically unrelated regions of the nervous system as it does with different antibodies, short postmortem-intervals, and different quantification methods. We sought to define relationship to clinical disease in the clearest, strictest, and most direct manner so that we could identify a surest association (or lack of association). Since all of our nervous systems were from patients who presented clinically with an ALS-motor phenotype often with suspected FTD, we, therefore, strictly evaluated motor cortex, frontal cortex, and anterior horn of the spinal cord for disease-related regions; and occipital cortex, parietal cortex and posterior horn of the spinal cord for disease-unrelated regions. In the brain, we averaged two areas for analysis—frontal and motor, and occipital and parietal. In the spinal cord, we could only quantify neurons in the horns. Importantly, we defined regions such as hippocampus, cerebellum, RSGC, and olfactory bulb as having uncertain disease relation given their complexities, the heterogeneities of clinical phenotypes, and the changing views of their involvement in disease, and these were not included in our quantitative analysis.
Poly-GR aggregations also had significant co-localization with pTDP-43. Most DPR aggregations are in the perinuclear cytoplasm and either star-shaped or amorphous, and only 5–10% are localized in dendrites. But of the poly-GR dendritic deposits, 80% co-localized with pTDP-43—none of the other DPR dendritic deposits did this. Poly-GR aggregates in dendrites have been noted in different parts of cortex and spinal cord by some reports [45], but not others [30]. To a lesser extent, poly-GR perinuclear deposits also co-localized with pTDP-43. Of the poly-GR deposits that were perinuclear, 20% co-localized with pTDP-43, compared to 1–10% of the other DPR perinuclear aggregates. Co-localization of dendritic poly-GR with pTDP-43 raises the possibility that poly-GR and pTDP-43 might interact indirectly though dendritic proteins binding both. We do not believe this is the case, since only small number of dendrites have pTDP-43, and of these, only 4% also have poly-GR aggregations. Current classifications of ALS are based on TDP-43 neuropathology, and based either on morphology [6, 37, 48], or anatomy [6]. It has already been shown that occasionally TDP-43 surrounds perinuclear poly-GA aggregations [36] and there are perinuclear poly-GR and poly-PA aggregations in spinal cord that co-localized with pTDP-43 [16]. Interestingly, we could not find GR dendritic-type aggregation in other parts of cortex in CNS and the majority of perinuclear aggregations of poly-GR, poly-GA and poly-GP were negative for pTDP-43 in other parts of CNS. TDP-43 is increasingly known to be involved in many neuritic functions including an essential role of mRNA transport into distal neurites [2, 17], endosomal trafficking and signaling in dendrites [46], and RNA granules in dendritic arbors [27]. Additional research will be required to find why poly-GR DPRs uniquely co-localize with pTDP-43 in dendrites.
Many disease models have implicated the toxicity of arginine-containing DPRs [29, 34, 52, 55, 56]. Arginine bestows unique biophysical properties and is thought to have greater toxic potential than alanine-containing DPRs (poly-GA and poly-PA) or than poly-GP, and a nuclear site of action has been proposed [22, 52]. They have been proposed to cause neurodegeneration by one or more of several mechanisms including compromise of nucleocytoplasmic transport, DNA damage, oxidative stress, inhibition of splicing, disruption spliceosome assembly, nucleolar stress, and dysfunction of membrane-less organelles [24]. While our neuropathological studies uniquely identified poly-GR consistent with these studies, ironically, they suggested the site of action was in the cytoplasm, especially dendrites, and not the nucleus, where we could not identify significant pathological abnormality in C9 ALS cases. What we did observe in the nucleus was non-specific immunostaining that did not quench with antibody masking and changes were as prevalent in SALS and control as in C9 ALS, findings that have also been shown by others [9, 31, 36].
Specific defects of nucleocytoplasmic transport in motor neuron degeneration are implicated in disease models, including yeast [19], flies [13, 57], and cells [57]. Thus far, two neuropathological studies have identified nuclear abnormalities in C9 ALS motor cortex, specifically mislocalized, diffuse nuclear, discontinuous, and large punctate RanGAP1 and Nup205 signals compared to smooth perinuclear staining observed in controls [57] and abnormalities of nuclear pore proteins Importin β1 and Ran-GTPase [53]. We sought to validate these findings by evaluating RanGap, Lamin B1, Importin β1, RanGTPase, and Nup205 with IHC and IF in neurons in different regions of the nervous system including disease-related and disease-unrelated areas. We examined both nuclear morphology and nuclear shape and found no differences between C9 ALS, SALS and controls. We found that diffuse nuclear staining that has been reported in abnormally high numbers in C9 ALS may be due to three-dimensional effects by z-stacking confocal images. RanGap, Lamin B1, and Importin β1 are nuclear membrane proteins and expressed in relatively similar patterns. Nup205 was diffusely cytoplasmic and we could not detect any nuclear signal [57] and we cannot comment on Ran-GTPase since the antibody previously used is no longer available, and the one that is available does not give signal. We also created another criterion using shape of the nucleus with RanGap immunostaining and while there seemed to be more aberrantly shaped nuclei in C9 ALS cases compared to controls, it seemed even stronger in SALS. This is further demonstrated by the staining pattern observed with Importin β1. While previous studies have implicated fragmentation of the nuclear envelope as a function of C9-ALS [53], we found significant variation of staining pattern in all patients including controls. We found the greatest degree of disruption visualized was in SALS, which ultimately was not significant statistically, suggesting that what trends we observed might be related to general cell degeneration. Because we had identified uniqueness of poly-GR, we performed double-IF using RanGap and poly-GR antibodies in motor cortex and lumbar spinal cord, but we were unable to see differences in patterns of RanGap staining between neurons containing poly-GR and neurons without poly-GR, and thus the presence of poly-GR aggregation in neurons did not alter the pattern of RanGap. The reasons for the differences between our study and the previous reports remain unclear.
We found C9orf72 protein was prominent in the cytoplasm of both neurons and glia in different areas of CNS and we could not detect unique differences in C9 ALS compared to SALS and controls in terms of intensity, distribution or morphology of staining. IHC lacks sensitivity for quantitation of expression and decreased C9orf72 expression has been reported both at the mRNA [15, 22] and protein [51, 53] levels. Using immunoblotting, we too found evidence suggestive of decreased expression of C9orf72 protein in disease-related frontal lobe areas, but we also found this in disease-unrelated occipital lobe regions. However, no reduction in C9orf72 protein was observed in cerebellum. This finding is probably consistent with that of Waite et al. [51], who also found reduced C9orf72 in cortex, but not in cerebellum. C9orf72 RNA abundance is highest in cerebellum [41], thus, it is possible that the decreased expression level of cerebellar C9orf72 mRNA might not be sufficient to cause a change at the protein level [51]. A puzzle in the field is that although cerebellum does not produce clinical symptoms in C9 ALS patients, it does curiously display significant neuropathology in terms of RNA foci and DPR protein aggregation. Importantly, three different isoforms of C9orf72 mRNAs have been characterized in C9 ALS and they can encode either a full-length- (54 kDa) or a short-isoform (25 kDa) protein. Previously reported was the existence of the short-isoform C9orf72 protein that normally was in the nuclear membrane but in C9 ALS and surprisingly also in SALS mislocalized to the plasma membrane [53]. We were not able to detect the 25kD short-isoform C9orf72 protein using an antibody generated with amino acid 1–169 that should detect both the full-length and predicted short-isoform proteins. Overall, while there are a number of lines of evidence showing decreased C9orf72 expression, we think that the lack of anatomic correlation to disease-related regions and evidence emerging from disease models [4, 7, 18, 21, 23, 39, 47] support that haploinsufficiency is not the major contributor to neurodegeneration.
In summary, we found poly-GR is unique among the DPRs in that it is more clearly associated with disease anatomy and pTDP-43 deposition. Further, our findings suggest that dendritic subcellular localization and presumably dendritic functions rather than nuclear ones are important to its role in pathogenesis.
References
Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobagyi T, Shaw CE (2011) p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol 122:691–702. https://doi.org/10.1007/s00401-011-0911-2
Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A et al (2014) Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81:536–543. https://doi.org/10.1016/j.neuron.2013.12.018
Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW 3rd, Rademakers R et al (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77:639–646. https://doi.org/10.1016/j.neuron.2013.02.004
Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y et al (2016) C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep 6:23204. https://doi.org/10.1038/srep23204
Boxer AL, Mackenzie IR, Boeve BF, Baker M, Seeley WW, Crook R, Feldman H, Hsiung GY, Rutherford N, Laluz V et al (2011) Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry 82:196–203. https://doi.org/10.1136/jnnp.2009.204081
Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, Suh E, Van Deerlin VM, Wood EM, Baek Y et al (2013) Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74:20–38. https://doi.org/10.1002/ana.23937
Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH, Suzuki-Uematsu S, Ghosh S, Singh A, Merkle FT et al (2016) Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med 8:347ra93. https://doi.org/10.1126/scitranslmed.aaf6038
Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME et al (2015) Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science (New York, NY) 348:1151–1154. https://doi.org/10.1126/science.aaa9344
Davidson Y, Robinson AC, Liu X, Wu D, Troakes C, Rollinson S, Masuda-Suzukake M, Suzuki G, Nonaka T, Shi J et al (2016) Neurodegeneration in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9orf72 is linked to TDP-43 pathology and not associated with aggregated forms of dipeptide repeat proteins. Neuropathol Appl Neurobiol 42:242–254. https://doi.org/10.1111/nan.12292
Davidson YS, Robinson AC, Rollinson S, Pickering-Brown S, Xiao S, Robertson J, Mann DMA (2017) Immunohistochemical detection of C9orf72 protein in frontotemporal lobar degeneration and motor neurone disease: patterns of immunostaining and an evaluation of commercial antibodies. Amyotroph Lateral Scler Frontotemporal Degener 1–10. doi:https://doi.org/10.1080/21678421.2017.1359304
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256. https://doi.org/10.1016/j.neuron.2011.09.011
Devlin AC, Burr K, Borooah S, Foster JD, Cleary EM, Geti I, Vallier L, Shaw CE, Chandran S, Miles GB (2015) Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nature Commun 6:5999. https://doi.org/10.1038/ncomms6999
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC et al (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525:129–133. https://doi.org/10.1038/nature14974
Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T, Daughrity L, Dunmore JH, Castanedes-Casey M, Chew J et al (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol 126:829–844. https://doi.org/10.1007/s00401-013-1192-8
Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S et al (2012) A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 11:54–65. https://doi.org/10.1016/s1474-4422(11)70261-7
Gomez-Deza J, Lee YB, Troakes C, Nolan M, Al-Sarraj S, Gallo JM, Shaw CE (2015) Dipeptide repeat protein inclusions are rare in the spinal cord and almost absent from motor neurons in C9ORF72 mutant amyotrophic lateral sclerosis and are unlikely to cause their degeneration. Acta Neuropathol Commun 3:38. https://doi.org/10.1186/s40478-015-0218-y
Ishiguro A, Kimura N, Watanabe Y, Watanabe S, Ishihama A (2016) TDP-43 binds and transports G-quadruplex-containing mRNAs into neurites for local translation. Genes Cells 21:466–481. https://doi.org/10.1111/gtc.12352
Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, Stauffer JE, Jafar-Nejad P, Drenner K, Schulte D et al (2016) Gain of toxicity from ALS/FTD-Linked repeat expansions in C9ORF72 Is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90:535–550. https://doi.org/10.1016/j.neuron.2016.04.006
Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW 3rd, Sun S, Herdy JR, Bieri G et al (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18:1226–1229. https://doi.org/10.1038/nn.4085
King A, Maekawa S, Bodi I, Troakes C, Al-Sarraj S (2011) Ubiquitinated, p62 immunopositive cerebellar cortical neuronal inclusions are evident across the spectrum of TDP-43 proteinopathies but are only rarely additionally immunopositive for phosphorylation-dependent TDP-43. Neuropathology 31:239–249. https://doi.org/10.1111/j.1440-1789.2010.01171.x
Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sa R, Schellevis RD, Waite AJ, Blake DJ, Veldink JH et al (2015) C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol 78:426–438. https://doi.org/10.1002/ana.24453
Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y, McKnight SL (2014) Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science (New York, NY) 345:1139–1145. https://doi.org/10.1126/science.1254917
Lall D, Baloh RH (2017) Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J Clin Investig. https://doi.org/10.1172/jci90607
Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A et al (2016) C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167(774–788):e717. https://doi.org/10.1016/j.cell.2016.10.002
Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C et al (2013) Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5:1178–1186. https://doi.org/10.1016/j.celrep.2013.10.049
Lin Y, Mori E, Kato M, Xiang S, Wu L, Kwon I, McKnight SL (2016) Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 167(789–802):e712. https://doi.org/10.1016/j.cell.2016.10.003
Liu-Yesucevitz L, Lin AY, Ebata A, Boon JY, Reid W, Xu YF, Kobrin K, Murphy GJ, Petrucelli L, Wolozin B (2014) ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. J Neurosci 34:4167–4174. https://doi.org/10.1523/jneurosci.2350-13.2014
Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, Yachnis AT, Ranum LP (2016) C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 90:521–534. https://doi.org/10.1016/j.neuron.2016.04.005
Lopez-Gonzalez R, Lu Y, Gendron TF, Karydas A, Tran H, Yang D, Petrucelli L, Miller BL, Almeida S, Gao FB (2016) Poly(GR) in C9ORF72-Related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 92:383–391. https://doi.org/10.1016/j.neuron.2016.09.015
Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K, Weng SM, Haass C, Kretzschmar HA, Edbauer D et al (2013) Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol 126:859–879. https://doi.org/10.1007/s00401-013-1181-y
Mackenzie IR, Frick P, Grasser FA, Gendron TF, Petrucelli L, Cashman NR, Edbauer D, Kremmer E, Prudlo J, Troost D et al (2015) Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol 130:845–861. https://doi.org/10.1007/s00401-015-1476-2
Mann DM, Rollinson S, Robinson A, Bennion Callister J, Thompson JC, Snowden JS, Gendron T, Petrucelli L, Masuda-Suzukake M, Hasegawa M et al (2013) Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun 1:68. https://doi.org/10.1186/2051-5960-1-68
May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, Grasser FA, Mori K, Kremmer E, Banzhaf-Strathmann J et al (2014) C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol 128:485–503. https://doi.org/10.1007/s00401-014-1329-4
Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IO, Pietrzyk J et al (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science (New York, NY) 345:1192–1194. https://doi.org/10.1126/science.1256800
Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K, Weng SM, Schludi MH, van der Zee J, Cruts M et al (2013) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 126:881–893. https://doi.org/10.1007/s00401-013-1189-3
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C et al (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science (New York, NY) 339:1335–1338. https://doi.org/10.1126/science.1232927
Nishihira Y, Tan CF, Onodera O, Toyoshima Y, Yamada M, Morita T, Nishizawa M, Kakita A, Takahashi H (2008) Sporadic amyotrophic lateral sclerosis: two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol 116:169–182. https://doi.org/10.1007/s00401-008-0385-z
O’Rourke JG, Bogdanik L, Muhammad AK, Gendron TF, Kim KJ, Austin A, Cady J, Liu EY, Zarrow J, Grant S et al (2015) C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron 88:892–901. https://doi.org/10.1016/j.neuron.2015.10.027
O’Rourke JG, Bogdanik L, Yanez A, Lall D, Wolf AJ, Muhammad AK, Ho R, Carmona S, Vit JP, Zarrow J et al (2016) C9orf72 is required for proper macrophage and microglial function in mice. Science (New York, NY) 351:1324–1329. https://doi.org/10.1126/science.aaf1064
Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, Weiss A, Wightman N, Salameh J, Kim J et al (2015) Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron 88:902–909. https://doi.org/10.1016/j.neuron.2015.11.018
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268. https://doi.org/10.1016/j.neuron.2011.09.010
Rossi S, Serrano A, Gerbino V, Giorgi A, Di Francesco L, Nencini M, Bozzo F, Schinina ME, Bagni C, Cestra G et al (2015) Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. J Cell Sci 128:1787–1799. https://doi.org/10.1242/jcs.165332
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. https://doi.org/10.1038/nmeth.2019
Schludi MH, Becker L, Garrett L, Gendron TF, Zhou Q, Schreiber F, Popper B, Dimou L, Strom TM, Winkelmann J et al (2017) Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol. https://doi.org/10.1007/s00401-017-1711-0
Schludi MH, May S, Grasser FA, Rentzsch K, Kremmer E, Kupper C, Klopstock T, Arzberger T, German Consortium for Frontotemporal Lobar D, Bavarian Brain Banking A et al (2015) Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol 130:537–555. https://doi.org/10.1007/s00401-015-1450-z
Schwenk BM, Hartmann H, Serdaroglu A, Schludi MH, Hornburg D, Meissner F, Orozco D, Colombo A, Tahirovic S, Michaelsen M et al (2016) TDP-43 loss of function inhibits endosomal trafficking and alters trophic signaling in neurons. EMBO J 35:2350–2370. https://doi.org/10.15252/embj.201694221
Sudria-Lopez E, Koppers M, de Wit M, van der Meer C, Westeneng HJ, Zundel CA, Youssef SA, Harkema L, de Bruin A, Veldink JH et al (2016) Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol 132:145–147. https://doi.org/10.1007/s00401-016-1581-x
Takeuchi R, Tada M, Shiga A, Toyoshima Y, Konno T, Sato T, Nozaki H, Kato T, Horie M, Shimizu H et al (2016) Heterogeneity of cerebral TDP-43 pathology in sporadic amyotrophic lateral sclerosis: evidence for clinico-pathologic subtypes. Acta Neuropathol Commun 4:61. https://doi.org/10.1186/s40478-016-0335-2
Tran H, Almeida S, Moore J, Gendron TF, Chalasani U, Lu Y, Du X, Nickerson JA, Petrucelli L, Weng Z et al (2015) Differential toxicity of nuclear RNA foci versus dipeptide repeat proteins in a drosophila model of C9ORF72 FTD/ALS. Neuron 87:1207–1214. https://doi.org/10.1016/j.neuron.2015.09.015
van Blitterswijk M, Gendron TF, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, Daughrity LM, Murray ME, Heckman MG, Jiang J et al (2015) Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol 130:863–876. https://doi.org/10.1007/s00401-015-1480-6
Waite AJ, Baumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ (2014) Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging 35(7):1779.e5–1779.e13. https://doi.org/10.1016/j.neurobiolaging.2014.01.016
Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB et al (2014) Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84:1213–1225. https://doi.org/10.1016/j.neuron.2014.12.010
Xiao S, MacNair L, McGoldrick P, McKeever PM, McLean JR, Zhang M, Keith J, Zinman L, Rogaeva E, Robertson J (2015) Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol 78:568–583. https://doi.org/10.1002/ana.24469
Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS et al (2013) Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci USA 110:7778–7783. https://doi.org/10.1073/pnas.1219643110
Yang D, Abdallah A, Li Z, Lu Y, Almeida S, Gao FB (2015) FTD/ALS-associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathol 130:525–535. https://doi.org/10.1007/s00401-015-1448-6
Yin S, Lopez-Gonzalez R, Kunz RC, Gangopadhyay J, Borufka C, Gygi SP, Gao FB, Reed R (2017) Evidence that C9ORF72 Dipeptide Repeat Proteins Associate with U2 snRNP to Cause Mis-splicing in ALS/FTD Patients. Cell Rep 19:2244–2256. https://doi.org/10.1016/j.celrep.2017.05.056
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61. https://doi.org/10.1038/nature14973
Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu YF, Katzman RB, Gass J, Murray ME, Shinohara M et al (2016) C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci 19:668–677. https://doi.org/10.1038/nn.4272
Zhang YJ, Jansen-West K, Xu YF, Gendron TF, Bieniek KF, Lin WL, Sasaguri H, Caulfield T, Hubbard J, Daughrity L et al (2014) Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol 128:505–524. https://doi.org/10.1007/s00401-014-1336-5
Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH et al (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA 110:E4968–4977. https://doi.org/10.1073/pnas.1315438110
Acknowledgements
This research was supported by Grants from ALS Association (5356S3), Target ALS (20134792), National Institute of Neurological Diseases and Stroke (NIH R01NS088578 and NS047101), and Pam Golden. JJ is a recipient of career development Grant from Muscular Dystrophy Association (479769) and was supported by postdoctoral training Grant (T32 AG00216) and postdoctoral fellowship (F32 NS087842) from the NIH. MB is supported by NIH NIGMS Award T32GM008666.
Author information
Authors and Affiliations
Corresponding author
Additional information
Shahram Saberi, Jennifer E. Stauffer and Jie Jiang contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
401_2017_1793_MOESM1_ESM.pdf
Supplemental Figure 1 Dipeptide repeat protein distribution in different CNS regions. (a-h) Burden of all DPRs in different layers of cortex. Motor cortex (a) and frontal cortex (b) are disease-related parts of cortex, while occipital cortex (c) and parietal cortex (d) are disease-unrelated parts. Retrosplenial granular cortex, cerebellum, hippocampus and olfactory bulb are of uncertain relation to disease. All DPRs are more abundant in layers of brain with prominent neuronal population (layers 2-6). No DPRs could be seen in subcortical white matter with mostly glial and oligodendrocyte population. (i) Burden of DPRs in different parts of lumbar spinal cord. No DPRs could be seen in white matter (columns) of spinal cord with mostly glial and oligodendrocyte population. (j) Specificity of poly-GR immunoreactivity is demonstrated by quenching antibody with recombinant GST-(GR)10 proteins. (k-l) When analyzed by actual percentage as opposed to grading of neurons that contain DPRs, poly-GR is still uniquely significantly more abundant in disease-related areas of CNS (PDF 12142 kb)
401_2017_1793_MOESM2_ESM.pdf
Supplemental Figure 2 RanGap, Lamin B1, Nup205 and Importin β1 immunohistochemical (IHC) staining in C9 ALS and controls. (a-d) IHC identifying RanGap in Betz cells (a-b) and spinal motor neurons (c-d) of C9 ALS (a and c) and controls (b and d). (e-h) IHC identifying Lamin B1 in Betz cells (e-f) and spinal motor neurons (g-h) of C9 ALS (e and g) and controls (f and h). Patterns of expression of Lamin B1 is very similar as RanGap. (i-l) IHC identifying Nup205 in Betz cells (i-j) and spinal motor neurons (k-l) of C9 ALS (i and k) and controls (j and l). Despite the prominent nuclear signal seen with RanGap and Lamin B1, the signal with Nup205 is mostly diffuse in the cytoplasm. (m-n) There is no significant differences between controls, SALS and C9-ALS cases in patterns of expression of Lamin B1 in spinal motor neurons. (o-p) IHC identifying Importin β1 in spinal motor neurons shows smooth, intermediate or irregular patterns (o) and there are no significant differences between controls, SALS and C9-ALS in patterns of expression (p) (PDF 9690 kb)
401_2017_1793_MOESM3_ESM.pdf
Supplemental Figure 3 Diffuse nuclear pattern of RanGap is a three-dimensional effect. (a–d) Examples of diffuse nuclear RanGap signal in the motor neurons of spinal cord in C9 ALS cases when confocal images are stacked. On the left side and at the bottom of each panel are the transversal views of each sample. Below (red line) and to the left (yellow line) are the corresponding horizontal and transversal sections of each neuron. (a´–d´) show nuclear ring pattern seen by a three-dimensional plane in which image is cut through the cell (a´, b´, c´ and d´ are different co-focal planes through a, b, c and d, respectively) (PDF 9846 kb)
401_2017_1793_MOESM4_ESM.pdf
Supplemental Figure 4 Aberrant nuclear shapes in C9 ALS and SALS patients. (a–i) Nuclear morphologies were evaluated based on RanGap IF staining. We classified them as normal (a–c), moderately aberrant (d–f), and severely aberrant (g–i). (j) Evaluating the shape of the spinal motor neuron nucleus with RanGap shows slightly more moderately and severely aberrant nucleus in ALS cases (both SALS and C9 ALS) in comparison to controls (PDF 16387 kb)
401_2017_1793_MOESM5_ESM.pdf
Supplemental Figure 5 Short and long C9orf72 mRNA and proteins. (a–b) C9orf72 protein detection with immunohistochemical staining shows significant decrease after quenching with recombinant C9orf72 proteins in human tonsil epithelial lining. (c) Western blotting shows that C9orf72 protein expression is not decreased in cerebellum of C9 ALS compared to control and SALS cases. (d–e) Short (d) and long (e) mRNA foci (small black dot-like signals) could be seen in the epithelium of human tonsil, using CISH (chromogenic in situ hybridization), but not in the same tissue without applying probes (d and e-insert). (f) No predicted short isoform C9orf72 protein is detected using antibodies generated with amino acid 1-169, which should recognize both long and the predicted short isoform C9orf72 protein (PDF 24307 kb)
Rights and permissions
About this article

Cite this article
Saberi, S., Stauffer, J.E., Jiang, J. et al. Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol 135, 459–474 (2018). https://doi.org/10.1007/s00401-017-1793-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-017-1793-8