
Abstract
Ample in vitro and in vivo experimental evidence supports the hypothesis that intercellular transmission of α-synuclein (αS) is a mechanism underlying the spread of αS pathology in Parkinson’s disease and related disorders. What remains unexplained is where and how initial transmissible αS aggregates form. In a previous study, we demonstrated that αS aggregates rapidly form in neurons with impaired nuclear membrane integrity due to the interaction between nuclear proaggregant factor(s) and αS and that such aggregates may serve as a source for αS seeding. In the present study, we identify histones as a potential nuclear proaggregant factor for αS aggregation in both apoptotic neurons and brains with αS pathology. We further demonstrate that histone-induced aggregates contain a range of αS oligomers, including protofibrils and mature fibrils, and that these αS aggregates can seed additional aggregation. Importantly, we demonstrate transmissibility in mouse brains from stereotaxic injection. This study provides new clues to the mechanism underlying initial pathological aggregation of αS in PD and related disorders, and could lead to novel diagnostic and therapeutic approaches.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Abnormal filamentous human α-synuclein (αS) aggregates are a pathological feature of Parkinson’s disease (PD) and related disorders [32, 33]. Increasing evidence suggests that cell-to-cell transmission of αS aggregates contributes to progression of PD [5, 6, 24, 27]. It remains an enigma, however, as to where and how the initial transmissible αS aggregates form. In a previous study, we demonstrated that filamentous αS aggregates rapidly form in neurons undergoing apoptosis, facilitated by a proaggregant factor or factors in nuclear material released upon disruption of nuclear envelope integrity [16]. Breakdown of apoptotic bodies results in release of αS aggregates, spread to neighboring healthy neurons, and seeding of additional aggregation. These findings suggest that a nuclear proaggregant factor exists that is responsible for facilitating rapid aggregation of cytoplasmic αS due to the loss of nuclear membrane integrity.
We hypothesize that a candidate for the proaggregant nuclear factor released upon the breakdown of the nuclear membrane during apoptosis is histone. Histones are a major class of nuclear proteins that have been shown to promote αS fibril formation in vitro, possibly due to their net positive charge [11]. Previous studies have also shown that histones are released into the cytoplasm during apoptosis [36]. To test this hypothesis, we used cellular models, animal models, and immunohistochemistry of brains with Lewy body pathology to examine the role of histones in αS aggregation. Our data show that histones associate with αS aggregates in apoptotic neurons and are a component of a subset of Lewy bodies in humans. More importantly, histone-induced αS aggregates are transmissible to neurons in both in vitro and in vivo conditions. Therefore, our study strongly suggests that histones may be a nuclear proaggregant factor that triggers αS aggregation.
Materials and methods
Cell culture and maintenance
Three neuronal cell models were used in the present study. The first two were 3D5 and αS-Flag derived from human dopaminergic neuroblastoma BE2-M17D cell line, which displays a neuronal phenotype under retinoic acid-induced differentiation. 3D5 cells express wild-type human αS upon TetOff induction and have been described previously [18]. αS-Flag is derived from BE2-M17D cell line with lentivirus overexpression of Flag-tagged αS. All neuronal cell models were maintained in DMEM/10% fetal bovine serum at 37 °C and 5% CO2, but 3D5 also required 2 μg⁄ml tetracycline. Neurons were seeded at a density of 3 × 106 cells/plate (100 × 20 mm, BD Biosciences) for biochemical analysis and 2 × 104 cells/well on coverslips in 24-well plates for immunocytochemistry. For differentiation, media were replaced with Neurobasal medium (Invitrogen), 2% B-27 supplement (Invitrogen), 2 mM l-glutamine (Sigma) and 10 μM RA (Sigma). To induce αS expression in 3D5 cells, tetracycline was deprived from the medium.
The third cell line used is the Lund human mesencephalic (LUHMES) cell line (LUHMES ATCC® CR-2927), which was differentiated into morphologically and biochemically mature dopamine-like neurons in advanced DMEM/F-12/Glutamax/N2 medium (Invitrogen) containing 2 ng/ml human recombinant glial cell-derived neurotrophic factor (R&D Systems), 1 mM cAMP (Sigma-Aldrich) and 1 μg/ml tetracycline as described previously [16, 23]. Cells with at least 4 days of differentiation were used for experiment.
Isolation of cytoplasmic proteins and subsequent western blot analysis
Neuronal cultures were harvested and centrifuged at 200×g for 15 min to obtain cell pellets. Cytoplasmic fractions were isolated using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific) following the manufacturer’s instructions, but with minor modifications. Briefly, cells were resuspended in cytoplasmic extraction reagent-I and incubated for 10 min, followed by the addition of cytoplasmic extraction reagent-II and centrifugation at 16,000×g for 5 min. Supernatants were considered the cytoplasmic fraction. The whole process was done on ice or at 4 °C. The bicinchoninic acid (BCA) assay was used for protein quantitation. The same amount of proteins (30 µg) from each sample was mixed with Tricine-SDS sample buffer (Invitrogen) and 2% β-mercaptoethanol, boiled for 5 min and resolved by SDS-PAGE using 10–20% Tris/Tricine gel (Bio-Rad). Precision-plus protein standards (Bio-Rad) were included as references. After gel electrophoresis, proteins were transferred onto polyvinylidene difluoride (PVDF) membranes for further probing with antibodies against αS (Syn1, 610787, BD Biosciences) or αS (NACP98, Mayo Clinic [7]), H1.2 (ab17677, Abcam), H3 (ab1791, Abcam). Western Lightning Plus ECL (PerkinElmer) or ECL™ Prime Western Blotting Detection Reagent (Fisher Scientific) was used for visualization of protein immunoreactivities. Data from at least 3 sets of independent experiments were analyzed by one-way ANOVA with Dunnett’s post hoc test for statistical significance.
Immunoprecipitation
Cytoplasmic fractions were isolated using NE-PER Nuclear and Cytoplasmic Extraction Reagents as described above, and then incubated overnight with Dynabeads® (Invitrogen) coated with rabbit IgG, antibodies to αS (NACP98, Mayo Clinic [7]), H1.2 (ab17677, Abcam), H3 (ab1791, Abcam), following the manufacturer’s instruction. Proteins bound to the beads were released by boiling in sample loading buffer and probed with antibodies of interest by western blotting.
Lentiviral plasmids and virus preparation
Lentiviral plasmids carrying αS-Flag [EX-G0543-Lv121] were purchased from Genecopoeia. The preparation of lentivirus carrying genes of interest was the same as described previously [15].
Immunocytochemistry and co-localization measurement for cell cultures
Cells grown on cover slips were rinsed with PBS, fixed in 4% paraformaldehyde, and permeabilized with 0.1 M Tris-buffered saline (TBS; pH 7.6) containing 0.5% Triton X-100 for 5 min. They were subsequently blocked with 3% skim milk in TBS, incubated with antibodies to proteins of interest in TBS containing 3% skim milk overnight at 4 °C and then incubated for 1 h with secondary antibodies. Immunolabeled cells were stained with nuclei stain DAPI (Invitrogen) for 10 min and observed by confocal fluorescence microscopy (Zeiss LSM 510, Carl Zeiss MicroImaging).
Confocal images (60 × objective magnification) of at least 20 cells randomly chosen from 5 fields (upper left, upper right, center, lower left and lower right) per group were captured. The same exposure time was used throughout the course of image acquisition. Zen 2009 Light Edition software (Zeiss) was used to evaluate co-localization between interesting proteins for each single cell. An Overlap Coefficient (Carl Zeiss Advanced Imaging Microscopy, Jena, Germany) was measured from each cell in each group. The data were analyzed for statistical difference between the two groups with Student t test.
Recombinant proteins
Recombinant human αS was freshly prepared as previously described [17]. Recombinant histone H1.0 (M2501S) was purchased from New England Biolabs (NEB).
Electron microscopy (EM)
Samples were processed for EM and immuno-EM using protocols described previously [17]. Briefly, samples were adsorbed onto carbon-coated copper grids, fixed with 2% formaldehyde in phosphate-buffered saline (PBS), negatively stained with 5% uranyl acetate, and examined with a Philips EM 2008 electron microscope (Phillips). For immunogold labeling, samples were adsorbed onto carbon-coated copper grids, fixed in 2% formaldehyde in PBS, blocked with PBS plus 5% normal goat serum, 0.1% bovine serum albumin, and 0.1% gelatin, and incubated for 1 h with a mixture of anti-H1.0 (ab125027, 1:10, Abcam) and anti-αS (610787, 1:10, BD Biosciences) antibodies. The grids were then incubated with a mixture of secondary antibodies containing anti-rabbit IgG (1:25) and anti-mouse IgG (1:25), respectively, conjugated with 5- and 10-nm colloidal gold. The immunogold-labeled samples were negatively stained with 2% uranyl acetate and examined by EM.
Stereotaxic surgery
All animal procedures were approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC) and were in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) revised 1996.
Recombinant protein solutions of αS and H1.0 were subjected to 100,000×g at 4 °C for 60 min to pellet any aggregated material as previously described [34]; the resultant supernatants were then measured by bicinchoninic acid assay (BCA) for determining protein concentration followed by 4-h incubation of αS/H1.0 mixtures, αS and H1.0 alone for preparation of injection materials. C57BL/6 mice (12 month of age) were anesthetized with 3% isoflurane and stereotaxically injected with 10 µg αS/H1.0 complexes (αS/H1.0 = 6/4), 6 µg αS or 4 µg H1.0 (NEB) per brain following methods of Luk et al. [25]. In addition, sham-operated mice with PBS injection were included as controls to exclude any effects due to surgery and injection. At least 7 mice were used for each group. A single needle insertion (coordinates: X = 2.0 mm; Y = 0.2 mm; Z = 0.8 and 2.6 mm, respectively) was used to deliver the inoculum to somatosensory cortex and dorsal neostriatum. Material was injected via a Hamilton syringe at a rate of 0.5 μl per min (5 μl total per site). After recovery from surgery, animals were monitored regularly. A week later, animals were anesthetized and subjected to transcardial perfusion with PBS and brain fixation with 10% formalin followed by paraffin embedding of brains. Coronal sections were cut at 5 microns thickness for immunohistochemical and immunofluorescent staining.
Immunohistochemical and immunofluorescent staining
Sections of paraffin-embedded tissue were sequentially subjected to deparaffinization, rehydration, pretreatment with 98% formic acid for 30 min (only for staining with NACP98 antibody), steaming in DAKO target retrieval solution (pH 6.1) for 30 min, digestion with Protease 24 for 8 min at room temperature (only for staining with LB509), and blocking with Protein Block (X0909, DAKO) for 1 h at room temperature. Following blocking of endogenous peroxidase with 3% hydrogen peroxide, the sections were incubated with 5% normal goat serum for 20 min to reduce non-specific labeling. Tissue sections were then incubated with antibodies to H1 (sc-8030, 1:50, Santa Cruz), H3 (ab1791, 1:1500, Abcam) and αS (LB509, 1:100, Invitrogen) for 45 min and Envision-Plus labeled polymer HRP, rabbit or mouse (DAKO) for 30 min. Peroxidase labeling was visualized with a solution containing 3, 3′-diaminobenzidine (DAB-Plus). The sections were subsequently counterstained with Lerner 1 hematoxylin (14-930-11, Fisher Scientific) and coverslipped with cytoseal mounting medium (8310-16, Richard-Allan Scientific).
For immunofluorescence, sections were incubated with Protein Block (X0909, DAKO) for 1 h at room temperature, followed by incubation with antibodies to MAP2 (Chicken, 1:3000, EnCor Biotechnology Inc.) plus H1 (sc-8030, 1:50, Ms, Santa Cruz) and αS (NACP98, 1:500, Rb), or H3 (ab1791, 1:500, Rb, Abcam) and αS (LB509, 1:100, Ms, Invitrogen) at 4 °C overnight, followed by incubation with three individual secondary antibodies (1:500, 1.5 h per antibody). For mouse brain sections, antibodies to H1.0 (ab125027, 1:100, Abcam) and αS (LB509, 1:100, Invitrogen) were used. Non-specific fluorescence signals were blocked by staining with Sudan Black stain for 2 min. Sections were coverslipped with Vectashield mounting media (H-1200, Vector Laboratories) and evaluated by confocal fluorescence microscopy.
Co-localization measurement for human brain sections
One section per human brain sample was used for immunofluorescence staining and subsequent confocal imaging. A total of 10 cases of diffuse Lewy body disease (DLBD) and 10 age- and sex-matched neurologically and pathologically normal controls with minimal or no Alzheimer or PD type pathology (Thal phase and Braak stage) were included. Images (20 × objective magnification) of multiple fields (at least 3 randomly chosen fields) per section from the basal forebrain of postmortem brain sections were captured. The same setting of exposure time was used throughout the course of image acquisition. We used Zen 2009 software to measure the Overlap Coefficient (Carl Zeiss Advanced Imaging Microscopy, Jena, Germany) between H1 and αS, or H3 and αS. The average of overlap coefficient measured from each section was collected. Differences between DLBD and control were assessed by Student’s t test.
Results
Histones H1 and H3 increase in cytoplasm in a time-dependent manner during neuronal apoptosis
We previously employed multiple neuronal cell models to show that during neuronal apoptosis, insoluble αS aggregates increase over time in cytoplasm and αS filaments can be found in apoptotic bodies. We also found that the formation of aggregates is associated with reciprocal diffusion and interaction between cytoplasmic αS and nuclear proaggregant factors due to the disruption of nuclear envelope integrity induced by apoptosis [16]. To determine if histones represent a nuclear proaggregant factor, we followed previous methods to induce apoptosis of differentiated 3D5 cells by exposing them to 25 nM staurosporine (STS) for 8, 16 and 24 h [16]. Cell lysates were subjected to SDS-PAGE and subsequent western blotting with antibodies to αS and two representative histone proteins—H1 and H3. As we expected, both H1 and H3 increase in cytoplasm in a time-dependent manner, which is concurrent with a gradual increase of cytoplasmic αS aggregates (Fig. 1 and Supplementary Fig. A1). Similar results were observed when apoptosis was induced by the PD-associated neurotoxins 1-methyl-4-phenylpyridinium (MPP) and 6-hydroxy-dopamine (6-OHDA) (Fig. 1 and Supplementary Fig. A1).

Increase of histones in cytoplasm is concurrent with αS aggregation during neuronal apoptosis. Differentiated 3D5 cells with 5 days of αS induction were exposed to staurosporine (STS), 1-methyl-4-phenylpyridinium (MPP) or 6-hydroxy-dopamine (6-OHDA) for 0, 8, 16, 24 h. Cytoplasmic proteins of each sample were extracted and subjected to SDS-PAGE and western blotting with antibodies to histone H1 and H3, as well as αS. Ponceau S staining was performed before immunostaining to demonstrate that all samples had comparable levels of total proteins. Statistical analysis of immunoreactivity of various proteins is shown in Fig. A1 in Supplemental Materials. Results demonstrate that H1 and H3 gradually increase in the cytoplasm during neuronal apoptosis, concurrent with gradual increase in αS aggregates. Number sign (#) denotes a non-specific Syn1-immunoreactive band on western blots of cell lysates
Histones H1 and H3 interact with αS in apoptotic neurons
To determine if histones interact with αS during neuronal apoptosis, differentiated 3D5 cells were exposed to STS for 24 h and then harvested. Cell lysates were prepared for immunoprecipitation with antibodies to αS, H1, H3 and rabbit IgG (rIgG), respectively. As expected, both H1 and H3 were detected in the immunoprecipitate of αS (IP/αS), and αS was detected in the reverse IP/H1 and IP/H3 (Fig. 2). In contrast, neither αS, H1 nor H3 was detected in the immunoprecipitate of IP/rIgG (Fig. 2). These results suggest that histone and αS interact in neurons undergoing apoptosis.

Co-immunoprecipitation experiments show that histone and αS interact in apoptotic neuronal cultures. Differentiated 3D5 cells with 5 days of αS induction were exposed to STS for 24 h. Cytoplasmic fractions were aliquoted and, respectively, incubated with antibodies to αS, histone H1, H3 or rabbit IgG to generate immunoprecipitates (IP), referred to as IP/αS, IP/H1, IP/H3 and IP/rIgG. Cell lysates lacking the aforementioned incubation were included as Input. A portion of each sample was probed with αS, histone H1, H3 to confirm enrichment of these proteins in IP products, and evaluate their interactions. IP/H1 and IP/H3 were shown to contain different sizes of αS-positive proteins corresponding to monomer (denoted by asterisk) and oligomers (denoted by number sign); and IP/αS was also shown to contain H1 (denoted by empty arrowhead) and H3 (denoted by empty arrow) proteins. Solid arrow and arrow head, respectively, denote the heavy chain (55 kDa) and light chain (25 kDa) of IgG in IP
Histones are associated with the formation of cytoplasmic αS aggregates in apoptotic neurons
To further confirm the association between histones and cytoplasmic αS aggregates in apoptotic neurons, differentiated 3D5 cells were subjected to treatment with STS or vehicle, followed by fixation and dual immunocytochemical staining with antibodies to H1 (sc-34464, Santa Cruz) and αS, or H3 (ab1791, Abcam) and αS. A well-known αS antibody (LB509 [19, 22, 31]) that is sensitive to aggregated forms of αS was used to demonstrate the distribution of αS aggregates. LB509 was generated against purified Lewy bodies as immunogen [13]. Using confocal microscopy, we observed that after STS treatment some αS translocate into the nucleus, which is consistent with our previous findings [16], leading to the co-localization between αS and H1 or H3 in nucleus. Simultaneously, H1 and H3 also diffuse into the cytoplasm, concurrent with the appearance of H1 and H3 immunopositive αS aggregates (arrow denoted structures in Fig. 3). In non-apoptotic cells treated with vehicle, αS and H1 or H3 remain in the cytoplasm and nucleus, respectively, and no αS aggregates can be observed (Fig. 3). Furthermore, the ratio of cells containing H1 or H3 co-immunopositive αS aggregates to total cells is, respectively, about 70 ± 10 and 17 ± 8%. Co-localization between histones and αS can also be detected in differentiated LUHMES dopamine cells after exposure to STS (Supplementary Fig. A2). These data suggest that H1 and H3 are associated with cytoplasmic αS aggregates in apoptotic neurons.

Immunocytochemical staining demonstrates that histones co-localize with αS aggregates in apoptotic neurons. Differentiated 3D5 cells with 5 days of αS induction were subjected to treatment with staurosporine (STS) or vehicle (Control), followed by dual immunocytochemical staining with antibodies to histone H1/αS or H3/αS. Co-localization of αS and H1 or αS and H3 can be observed in both nucleus and cytoplasm in cells with STS treatment, but not in control cells. Z-stack confocal imaging is used to confirm the intracellular location of αS aggregates. Arrows denote a cluster of αS aggregates immunopositive to H1 or H3. Scale bar 5 µm
Histones are associated with αS pathology in human brain
To determine if histones are associated with αS pathology in humans with Lewy-related pathology, tissue sections of basal forebrain from 10 patients with diffuse Lewy body disease (DLBD) and 10 age- and sex-matched normal controls were immunostained with antibodies to αS, histones H1 and H3. A subset of neurons in the basal nucleus of Meynert of DLBD had both nuclear and cytoplasmic immunoreactivity to H1 and H3 (Fig. 4a–c). Normal controls had only nuclear immunostaining. In addition, both H1 and H3 were associated with a subset of Lewy bodies and Lewy neurites (Fig. 4b, c). Co-localization analysis (Fig. 4d) demonstrated that overlap coefficients of both H1/αS and H3/αS in DLBD were significantly higher than those in controls. The overlap coefficient of H1/αS was much higher than that of H3/αS in DLBD cases, suggesting that of the two histone proteins, H1 may have a more significant role than H3 as a proaggregant factor for αS aggregation in DLBD. Therefore, we focused further investigations on histone H1.

Histone is associated with αS pathology in human brain tissues. Human specimens containing basal nucleus of Meynert from different cases with diffuse Lewy body disease (DLBD) and controls were probed with antibodies to αS and histone H1, or H3 by a immunohistochemistry and b, c immunofluorescence. i, ii in (a), respectively, show the representative pattern and intensity of H1 staining in brain tissue with αS pathology from DLBD patients and the same area from controls. Scale bar 100 µm. ii1 Enlarges the framed area in (ii) showing two H1 positive non-nuclear structures (arrow denoted). ii2 Shows two αS positive structures (arrows denoted) which are detected at the same location in the same area of an adjacent section of (ii1). Scale bar 60 µm. b, c Respectively, show the representative triple immunofluorescence staining results of αS/H1/MAP2 and αS/H3/MAP2 in brain tissue with αS pathology from DLBD patients (i1 to i5) and the same area from controls (ii1 to ii5). Both asterisk * and number sign “#” randomly denote some co-localized staining of αS/H1 and αS/H3, but asterisk also denotes a third co-localization with MAP2. Scale bar 20 µm. d Bar graph summarizes the co-localization analysis with Zen 2009 Light Edition software for H1/αS and H3/αS in aforementioned immunofluorescence staining studies of DLBD and controls
Histone H1 interacts with αS to form complexes with a filamentous ultrastructure
To determine if histone H1 can induce αS aggregation, recombinant αS, histone H1, or a mixture of both proteins were incubated at 37 °C for 10 min and immediately subjected to native gel electrophoresis and western blotting, as well as EM and immuno-EM. Duplicate samples were incubated at 37 °C for 4 h and then subjected to the same analysis. As a positive control, αS aggregates were prepared by constant shaking at 37 °C for one week. Native gel analysis demonstrates that αS and histone H1 can form complexes within 10 min of incubation, characterized as a single band near the cathode that is immunopositive for both H1 and αS (Fig. 5a). In contrast, H1 and αS each remained at the cathode or migrate close to the anode and are immunopositive only for H1 or αS (Fig. 5a). EM showed that after only 10 min incubation with H1, αS formed annular structures (Fig. 5b) similar to protofibrils as described previously [37], as well as short filamentous structures. After 4 h of incubation with H1, αS formed mature fibrils (Fig. 5b, panel i2) that were morphologically similar to fibrils formed from αS subjected to a week of constant shaking (Fig. 5b, panel i3). In contrast, samples of αS or H1 alone did not form distinctive structures after 4 h of incubation (Fig. 5b, panel i4 & i5). The filamentous structures observed in αS and H1 mixtures were decorated by both 5 nm gold particles for H1 and 10 nm gold particles for αS (Fig. 5b, panels ii1& ii2). Dual-labeled filaments are not detected in controls with only αS or H1 (Fig. 5b panel ii3, ii4 & ii5). The fibrils formed by incubation of αS and H1 for 4 h (αS/H1 complexes) were sonicated for subsequent in vitro and in vivo experiments.

αS rapidly forms aggregates upon interaction with histone H1. Recombinant αS with or without histone H1 was mixed and incubated at 37 °C for 10 min and 4 h, respectively. a Native gel electrophoresis and Western blotting analysis were performed to assess histone H1-induced αS aggregates. Asterisk * denotes two different bands staying closest to the cathode, one is the complexes formed by mixing αS with histone H1, which are immunostained by both H1 and αS (also see arrow head denoted band); the other is the histone H1, which is negative with αS immunostaining. Number sign # denotes the band of fresh recombinant αS, which migrates towards anode. b Negative (i1 to i5) and immunonegative (ii1 to ii5) stained transmission electron micrographs of histone H1 induced αS aggregates and different controls. The αS aggregates prepared by a week of constant shaking at 37 °C were included as a positive control and named as “αS–Ag.” Fresh H1 and αS protein solutions incubated at 37 °C for 4 h, referred to as αS/4 h and H1/4 h, were used as negative controls. Arrows denote annular structures, resembling protofibrils; arrowhead denotes short filamentous structure. All samples labeled with dual immunogold (5- and 10-nm gold particles for H1 and αS, respectively) are shown in ii1 to ii5. Scale bar 100 nm
Histone-induced αS aggregates can spread to neurons and seed αS aggregation
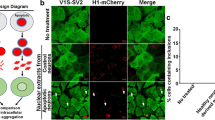
To determine if histone-induced αS aggregates can be spread in the brain, fresh recombinant H1 and αS were labeled with Lightning-Link® Rapid DyLight® 550 and DyLight® 650 from Innova Biosciences, respectively (referred to as “H1-550″ and “ αS–650″). The two samples were briefly mixed and incubated for 4 h at 37 °C to form complexes/aggregates of αS-650/H1-550. The samples of αS-650 and H1-550 alone, and αS/H1 mixtures (without DyLight fluorescent dye labeling) were incubated in parallel to serve as negative and positive controls, respectively. All samples were then subjected to negative staining followed by EM observation. Both αS-650/H1-550 and αS/H1 formed comparable filamentous structures, whereas no visible structures were found in the samples of αS-650 and H1-550 alone (Fig. 6a). Therefore, DyLight fluorescent dye-labeled αS and H1 were considered suitable to follow uptake, seeding and spreading.

Histone-induced αS aggregates can spread into neurons and seed αS aggregation in vitro. a Freshly prepared histone H1 and αS were, respectively, labeled with Lightning-Link® Rapid DyLight® 550 and DyLight® 650 (Innova Biosciences), referred to as “H1-550″ and “ αS-650″). The two samples were briefly mixed and incubated for 4 h at 37 °C to form complexes/aggregates of αS-650/H1-550; the latter were then subjected to negative staining followed by EM observation. The samples of αS-650 and H1-550 alone, and αS/H1 mixtures (without DyLight fluorescent dye) were prepared in parallel to serve as negative and positive controls. Scale bar 100 nm. b The filamentous αS-650/H1-550 complexes were added to differentiated M17D cells overexpressing αS-Flag. Cells fed with only αS-650 or H1-550 were used as controls. After 3 days, cells were fixed and immunostained with primary antibody against Flag (F3165, Sigma), followed by secondary antibody conjugated with AlexaFluor 488 and DAPI counterstaining. Results show that both αS-650 and H1-550 co-localize in cells fed with αS-650/H1-550 complexes, indicative of their internalization. Moreover, these cells have abundant intracellular αS-Flag aggregates, whose distribution is similar to that of intracellular αS/H1 complexes. Scale bar: 10 um. Z-stack confirms the intracellular location of seeded aggregates, which is further enlarged above the Z-stack picture. Arrows denote co-localization of αS-Flag aggregates and αS/H1 complexes. c Bar graph summarized the co-localization analysis with Zen 2009 software from at least 20 randomly chosen cells per group which shows that the overlap coefficient of αS-Flag aggregates and αS-650 is significantly higher in αS-650/H1-550 group compared with another two control groups
Differentiated M17D neuronal cells overexpressing αS fused with Flag tag (referred to as “αS-Flag”) were incubated with the αS-650/H1-550 complexes for 3 days and then subjected to immunocytochemistry with antibodies to Flag. As a control, neurons were treated only with αS-650 and H1-550 alone. Co-localization of αS-650 and H1–550 was observed in 92 ± 6% cells of the group treated with αS-650/H1-550 complexes/aggregates, indicative of highly efficient internalization (Fig. 6b). Moreover, these cells had abundant intracellular αS-Flag aggregates, whose distribution was similar to that of intracellular αS-650/H1-550 complexes. Quantitative analysis with Zen 2009 from at least 20 randomly chosen cells per group showed that the overlap coefficient of αS-Flag aggregates and αS-650 was significantly higher in αS-650/H1-550 compared to the two controls (Fig. 6c). These results suggest that H1-induced αS aggregates can be internalized in neurons and seed αS aggregation.
Histone-induced αS aggregates can be internalized by neurons in vivo
Wild-type C57BL/6 mouse brains were stereotactically injected into somatosensory cortex and dorsal neostriatum with sonicated αS/H1 complexes or with fresh recombinant αS and H1 alone. After one week, the animals were killed and brains harvested, fixed and embedded in paraffin for immunohistochemical studies. Coronal sections were processed for double labeling immunofluorescence microscopy. Human αS pathology was observed at the site of the injection of sonicated αS/H1 complexes using immunohistochemistry. Perinuclear αS deposits were detected in neurons in the neocortex (Fig. 7a, panel a1), while thread-like or dot-like αS deposits were detected in the white matter (Fig. 7a, panel a2). Dual immunofluorescence demonstrated that αS pathology was immunopositive for H1 (Fig. 7a, asterisk-marked cells), indicating that the αS/H1 complexes were stable to sonication. In contrast, sections from mice injected with recombinant αS or H1, alone, did not show neuronal or neuritic αS pathology.

Histone-induced αS aggregates can spread to neurons upon inoculation into mouse brain. Wild-type C57BL/6 mouse brains were inoculated with αS/H1 aggregates, fresh recombinant αS or histone H1 via stereotaxic brain injection and harvested a week later. a–c Paraffin sections of brains injected with (a) αS/H1 aggregates, b fresh recombinant αS or c fresh recombinant H1 were, respectively, subjected to immunohistologic staining with antibody to human αS (LB509) to demonstrate human αS pathology. Scale bar 2 mm. Panels a1, a2, b1, b2, c1 and c2 enlarge framed areas labeled 1 and 2 in a–c. Scale bar 60 µm. The framed areas in (a1) and (a2) are further enlarged to show intra-neuronal αS deposits and the thread- or dot-like αS deposits. Panels (a1-1 to a1-3), (a2-1 to a2-3), (b1-1 to b1-3), (b2-1 to b2-3), (c1-1 to c1-3) and (c2-1 to c2-3) are brain regions corresponding to a1, a2, b1, b2, c1 and c2 in adjacent sections, which were subjected to immunofluorescence staining with primary antibodies to human αS (mouse monoclonal) and H1 (rabbit polyclonal), and goat secondary antibodies to mouse (Alexa Fluor 568) and rabbit (Alexa Fluor 647). Scale bar 10 µm
Discussion
A decade ago, Goers et al. reported that histones could accelerate αS aggregation in vitro [11], but whether histones might play a role in inducing pathological αS in PD and related disorders has never been addressed. This is partly because histones and αS rarely interact in healthy neurons due to their distinct subcellular locations—histones in the nucleus and αS in the cytoplasm. Our previous study provided a theoretical basis for a role for nuclear proaggregant factor(s) in the formation of cytoplasmic αS aggregates upon loss of nuclear membrane integrity [16]. The present study identifies histones as a potential proaggregant nuclear factor involved in αS aggregation in neurons undergoing apoptosis and possibly in neurons with sub-lethal nuclear membrane abnormalities that lead to leakiness.
We found that in apoptotic neurons, histones H1 and H3 diffuse into the cytoplasm in a time-dependent manner and interact with αS to form cytoplasmic aggregates. More importantly, histones (especially H1) co-localized with a subset of Lewy bodies and Lewy neurites in DLBD. Because H1/αS had a comparatively higher overlap coefficient compared with H3/αS, we speculate that H1 may play a more important role in the formation of pathological αS. This might be due to different roles of H1 and H3 in nucleosomes [30]. It is known that H3 is a core protein of the nucleosome that is tightly wrapped around by DNA. It can be unmasked to interact with other proteins only after DNA is removed. In contrast, H1 serves as a linker histone that is less tightly associated with DNA. Therefore, it is possible that H1 is more labile and more likely to interact with αS in the nucleus and more likely to diffuse to cytoplasm upon disruption of nuclear membrane integrity, as occurs during apoptosis.
We showed that histone accelerated αS aggregation into filamentous structures as reported previously by Goers et al. [11], and further demonstrated that histone-induced aggregates contain a range of αS oligomers, including protofibrils and mature fibrils. More interestingly, we found that histone-induced αS aggregates are transmissible to neurons both in vitro and in vivo. In cultured neurons, histone-induced αS aggregates triggered aggregation of endogenous αS, demonstrating its seeding activity. We were unable to determine if histone-induced αS aggregates (αS/H1) have seeding activity after injection into mouse brain because no antibody is currently available that can distinguish endogenous mouse αS from injected human αS. To address this issue, an animal model needs to be developed in which mouse and human αS can be differentiated using tagged αS. Alternatively, considering that human αS is not likely to cause seeding in mouse brain, sonicated mouse αS/H1 fibrils might be used to promote in vivo seeding followed by immunostaining for S129-phosphorylated αS, a strategy successfully employed in previous studies [24, 26]. It will also be important to determine if mouse αS and H1 can form fibrils comparable to those observed after exposing human αS to human H1.
It is worth noting that histones might play roles beyond their proaggregant effect. It has been shown that the positive charge of histones renders them capable of crossing cellular membranes [12]. It has also been shown that the positively charged transfection reagent Lipofectamine can mediate uptake of αS aggregates into neuronal cells [29]. Thus, it is intriguing to speculate that endogenous histones, like the experimental agent Lipofectamine, might facilitate internalization of αS aggregates and, thus, favor cell-to-cell propagation.
Evidence suggests that histones can be secreted into extracellular fluids from a range of cell types, including neurons [1], glia [28] and some activated immune cells [2, 9, 35] in response to cell stress. Moreover, extracellular histones are significantly elevated in response to brain injury [4, 20], which is a risk factor for PD [3, 10]. There is increasing evidence that αS can be secreted into extracellular fluids by non-classical, endoplasmic reticulum- and Golgi-independent protein export pathways [21] and that the level of extracellular αS is increased with cellular stress and brain injury [8, 14]. Thus, it is reasonable to hypothesize that under conditions of cell stress or brain injury, histones may interact with αS in extracellular fluids to facilitate the formation of transmissible aggregates that could facilitate more widespread cell-to-cell propagation.
Although transmission of αS oligomers or aggregates has been previously reported, most studies used aggregates from pathologic brain samples or αS prepared in vitro by prolonged (several days to a week) shaking of recombinant αS [6, 24, 27]. In contrast, αS aggregates used in present study form quickly after incubating αS with histone. Therefore, a secondary outcome of our study is the identification of a faster method for preparing experimental αS aggregates to study transmission in animal models.
While previous work [11] and the present study provide compelling evidence that histones have proaggregant properties with respect to αS aggregation, there may be other nuclear factors that promote αS aggregation during neuronal apoptosis. In-depth studies are warranted to define the nature of other potential nuclear proaggregant factors.
Overall, our study suggests histones as prime candidates for one of the nuclear proaggregant factors involved in the formation of transmissible αS aggregates during neuron apoptosis. This study provides new insights into the pathogenesis of initial pathological αS seeds in PD and related disorders, and could lead to novel diagnostic and therapeutic approaches. Specifically, detection of excessive histone release from cells into body fluids (e.g., cerebrospinal fluid) might be a new strategy for early detection of disease-associated processes that favor αS pathology. It remains to be determined if limiting or preventing histone release could be an upstream target to decrease αS aggregation and subsequent propagation.
References
Bolton SJ, Perry VH (1997) Histone H1; a neuronal protein that binds bacterial lipopolysaccharide. J Neurocytol 26:823–831
Brix K, Summa W, Lottspeich F, Herzog V (1998) Extracellularly occurring histone H1 mediates the binding of thyroglobulin to the cell surface of mouse macrophages. J Clin Invest 102:283–293. doi:10.1172/JCI1614
Chase A (2015) Parkinson disease: traumatic brain injury increases the risk of Parkinson disease. Nat Rev Neurol 11:184. doi:10.1038/nrneurol.2015.39
Chen R, Kang R, Fan XG, Tang D (2014) Release and activity of histone in diseases. Cell Death Dis 5:e1370. doi:10.1038/cddis.2014.337
Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ (2012) Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener 7:42. doi:10.1186/1750-1326-7-42
Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA 106:13010–13015. doi:10.1073/pnas.0903691106
Dickson DW, Liu W, Hardy J, Farrer M, Mehta N, Uitti R, Mark M, Zimmerman T, Golbe L, Sage J, Sima A, D’Amato C, Albin R, Gilman S, Yen SH (1999) Widespread alterations of alpha-synuclein in multiple system atrophy. Am J Pathol 155:1241–1251
Emmanouilidou E, Elenis D, Papasilekas T, Stranjalis G, Gerozissis K, Ioannou PC, Vekrellis K (2011) Assessment of alpha-synuclein secretion in mouse and human brain parenchyma. PLoS ONE 6:e22225. doi:10.1371/journal.pone.0022225
Gabler C, Blank N, Hieronymus T, Schiller M, Berden JH, Kalden JR, Lorenz HM (2004) Extranuclear detection of histones and nucleosomes in activated human lymphoblasts as an early event in apoptosis. Ann Rheum Dis 63:1135–1144. doi:10.1136/ard.2003.011452
Gardner RC, Burke JF, Nettiksimmons J, Goldman S, Tanner CM, Yaffe K (2015) Traumatic brain injury in later life increases risk for Parkinson disease. Ann Neurol 77:987–995. doi:10.1002/ana.24396
Goers J, Manning-Bog AB, McCormack AL, Millett IS, Doniach S, Di Monte DA, Uversky VN, Fink AL (2003) Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 42:8465–8471. doi:10.1021/bi0341152
Hariton-Gazal E, Rosenbluh J, Graessmann A, Gilon C, Loyter A (2003) Direct translocation of histone molecules across cell membranes. J Cell Sci 116:4577–4586. doi:10.1242/jcs.00757
Iwatsubo T (2003) Aggregation of alpha-synuclein in the pathogenesis of Parkinson’s disease. J Neurol 250 Suppl 3:III11–14. doi:10.1007/s00415-003-1303-x
Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ (2010) Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem 113:1263–1274. doi:10.1111/j.1471-4159.2010.06695.x
Jiang P, Gan M, Ebrahim AS, Castanedes-Casey M, Dickson DW, Yen SH (2013) Adenosine monophosphate-activated protein kinase overactivation leads to accumulation of alpha-synuclein oligomers and decrease of neurites. Neurobiol Aging 34:1504–1515. doi:10.1016/j.neurobiolaging.2012.11.001
Jiang P, Gan M, Yen SH, Moussaud S, McLean PJ, Dickson DW (2016) Proaggregant nuclear factor(s) trigger rapid formation of alpha-synuclein aggregates in apoptotic neurons. Acta Neuropathol. doi:10.1007/s00401-016-1542-4
Jiang P, Ko LW, Jansen KR, Golde TE, Yen SH (2008) Using leucine zipper to facilitate alpha-synuclein assembly. Faseb J 22:3165–3174. doi:10.1096/fj.08-108365
Ko LW, Ko HH, Lin WL, Kulathingal JG, Yen SH (2008) Aggregates assembled from overexpression of wild-type alpha-synuclein are not toxic to human neuronal cells. J Neuropathol Exp Neurol 67:1084–1096. doi:10.1097/NEN.0b013e31818c3618
Kramer ML, Schulz-Schaeffer WJ (2007) Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci 27:1405–1410. doi:10.1523/JNEUROSCI.4564-06.2007
Kutcher ME, Xu J, Vilardi RF, Ho C, Esmon CT, Cohen MJ (2012) Extracellular histone release in response to traumatic injury: implications for a compensatory role of activated protein C. J Trauma Acute Care Surg 73:1389–1394. doi:10.1097/TA.0b013e318270d595
Lee HJ, Patel S, Lee SJ (2005) Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci 25:6016–6024. doi:10.1523/JNEUROSCI.0692-05.2005
Lee HJ, Shin SY, Choi C, Lee YH, Lee SJ (2002) Formation and removal of alpha-synuclein aggregates in cells exposed to mitochondrial inhibitors. J Biol Chem 277:5411–5417. doi:10.1074/jbc.M105326200
Lotharius J, Barg S, Wiekop P, Lundberg C, Raymon HK, Brundin P (2002) Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J Biol Chem 277:38884–38894. doi:10.1074/jbc.M205518200
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. doi:10.1126/science.1227157
Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012) Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med 209:975–986. doi:10.1084/jem.20112457
Mason DM, Nouraei N, Pant DB, Miner KM, Hutchison DF, Luk KC, Stolz JF, Leak RK (2016) Transmission of alpha-synucleinopathy from olfactory structures deep into the temporal lobe. Mol Neurodegener 11:49. doi:10.1186/s13024-016-0113-4
Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DM, Hasegawa M (2013) Prion-like spreading of pathological alpha-synuclein in brain. Brain 136:1128–1138. doi:10.1093/brain/awt037
Mishra B, von der Ohe M, Schulze C, Bian S, Makhina T, Loers G, Kleene R, Schachner M (2010) Functional role of the interaction between polysialic acid and extracellular histone H1. J Neurosci 30:12400–12413. doi:10.1523/JNEUROSCI.6407-09.2010
Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M (2010) Seeded aggregation and toxicity of {alpha}-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem 285:34885–34898. doi:10.1074/jbc.M110.148460
Ramakrishnan V (1997) Histone structure and the organization of the nucleosome. Annu Rev Biophys Biomol Struct 26:83–112. doi:10.1146/annurev.biophys.26.1.83
Rockenstein E, Nuber S, Overk CR, Ubhi K, Mante M, Patrick C, Adame A, Trejo-Morales M, Gerez J, Picotti P, Jensen PH, Campioni S, Riek R, Winkler J, Gage FH, Winner B, Masliah E (2014) Accumulation of oligomer-prone alpha-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 137:1496–1513. doi:10.1093/brain/awu057
Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M (1998) Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 251:205–208
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci USA 95:6469–6473
Volpicelli-Daley LA, Luk KC, Lee VM (2014) Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc 9:2135–2146. doi:10.1038/nprot.2014.143
Watson K, Edwards RJ, Shaunak S, Parmelee DC, Sarraf C, Gooderham NJ, Davies DS (1995) Extra-nuclear location of histones in activated human peripheral blood lymphocytes and cultured T-cells. Biochem Pharmacol 50:299–309
Wu D, Ingram A, Lahti JH, Mazza B, Grenet J, Kapoor A, Liu L, Kidd VJ, Tang D (2002) Apoptotic release of histones from nucleosomes. J Biol Chem 277:12001–12008. doi:10.1074/jbc.M109219200
Zhang H, Griggs A, Rochet JC, Stanciu LA (2013) In vitro study of alpha-synuclein protofibrils by cryo-EM suggests a Cu(2 +)-dependent aggregation pathway. Biophys J 104:2706–2713. doi:10.1016/j.bpj.2013.04.050
Acknowledgements
The authors thank Monica Castanedes-Casey, Virginia Phillips for their histologic supports. This study was supported by the National Institute of Health (P50-NS072187, R01-NS073740 and R21-NS099757), the Mangurian Foundation Lewy Body Dementia Program at Mayo Clinic (Dickson, Jiang & Yen). All authors have no actual or potential conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jiang, P., Gan, M., Yen, SH. et al. Histones facilitate α-synuclein aggregation during neuronal apoptosis. Acta Neuropathol 133, 547–558 (2017). https://doi.org/10.1007/s00401-016-1660-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-016-1660-z