
Abstract
Myeloid-derived cells play important modulatory and effector roles in multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE). Myeloid-derived suppressor cells (MDSCs) are immature myeloid cells, composed of monocytic (MO) and polymorphonuclear (PMN) fractions, which can suppress T cell activities in EAE. Their role in MS remains poorly characterized. We found decreased numbers of circulating MDSCs, driven by lower frequencies of the MO-MDSCs, and higher MDSC expression of microRNA miR-223 in MS versus healthy subjects. To gain mechanistic insights, we interrogated the EAE model. MiR-223 knock out (miR-223−/−) mice developed less severe EAE with increased MDSC numbers in the spleen and spinal cord compared to littermate controls. MiR-223−/− MO-MDSCs suppressed T cell proliferation and cytokine production in vitro and EAE in vivo more than wild-type MO-MDSCs. They also displayed an increased expression of critical mediators of MDSC suppressive function, Arginase-1(Arg1), and the signal transducer and activator of transcription 3 (Stat3), which herein, we demonstrate being an miR-223 target gene. Consistently, MDSCs from MS patients displayed decreased STAT3 and ARG1 expression compared with healthy controls, suggesting that circulating MDSCs in MS are not only reduced in numbers but also less suppressive. These results support a critical role for miR-223 in modulating MDSC biology in EAE and in MS and suggest potential novel therapeutic applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is a complex disease of unknown etiology involving central nervous system (CNS) inflammation, demyelination, and axonal damage. Most MS patients (about 85 %) initially show a relapsing-remitting disease course (RRMS), but around half of these later develop secondary progressive MS. MS is thought to be an autoimmune disease targeting CNS myelin, in part based on the success in using its prime animal model experimental autoimmune encephalomyelitis (EAE) to develop several currently approved MS therapies [41]. In EAE, and very likely in MS, myelin-specific CD4+ T cells, T helper (Th) 1 and Th17, are generated in peripheral lymphoid organs, where they are activated and then circulate in the blood stream before gaining access to the CNS [10]. Once inside the CNS compartment, myelin-specific T cells are reactivated after encountering the specific antigen and trigger the recruitment of a large number of monocyte/macrophages and neutrophils from the circulating blood, which results in CNS inflammation [43]. In recent years, it has become clear that a heterogeneous population of myeloid cells, called myeloid-derived suppressor cells (MDSCs), including immature granulocytes, monocytes, and dendritic cells, plays a very important role in shaping T cell responses [11]. There are two different types of MDSCs, as identified in both human and mice: polymorphonuclear MDSCs (PMN-MDSCs) morphologically and phenotypically similar to neutrophils and monocytic (MO-MDSCs) which are similar to monocytes [4]. MDSCs can suppress T cell activities and are reported to modulate the clinical course and pathology of EAE [48, 49]. The mechanisms by which these cells are sustained during relapsing and chronic neuroinflammation are poorly understood. Likewise, the immunomodulatory role of MDSCs in MS has not been well addressed up to now. A single report has shown that PMN-MDSCs from MS patients potently suppress the activation and proliferation of autologous T cell in vitro [18]. Immunosuppression by MDSCs involves multiple mechanisms, including depletion of l-arginine (a key nutritional factor needed for T cell proliferation) via arginase-1 (ARG1) and increased production of nitric oxide (NO) and reactive oxygen species (ROS) by inducible NO synthase (iNOS) and the NADPH oxidase (NOX), respectively [36, 37]. The signal transducer and activator of transcription 3 (STAT3) is another very important factor that drives the expansion of MDSCs in cancer [11] and their suppressive function by the regulation of ARG1 activity [45]. The possible role of these pathways in MDSCs has not been investigated in the context of CNS autoimmunity yet.
MicroRNAs (miRNAs) are a class of small, non-coding RNAs that regulate gene expression post-transcriptionally, either by targeting mRNA degradation or by inhibiting protein translation. The importance of miRNAs in biological processes is evidenced by their high conservation across species and the fact that they target almost a third of human genes [1, 28]. MiRNAs are implicated in the pathogenesis of several diseases, including MS [14, 21], and they represent potential therapeutic targets that can readily be manipulated [2]. In prior studies, we discovered that cell-free miR-223 is decreased in serum of MS patients compared to healthy controls [9]. In contrast, miR-223 expression has been found to be increased in peripheral blood cells (whole blood and regulatory T cells) from MS patients compared to healthy subjects, [6, 23] as well as in active MS lesions compared to normal CNS areas in controls subjects [22]. MiR-223 is highly expressed in the myeloid compartment and acts as an important modulator of myeloid cell development and functions [20]. MiR-223 negatively regulates myeloid progenitor proliferation, and lack of miR-223 was associated with MDSC accumulation in the presence of an infection or a growing tumor through the inverse modulation of its target gene myocyte enhancer factor 2C (Mef2c) [20, 30, 35]. MiR-223 expression is not confined to the MDSCs. In humans, miR-223 is expressed in granulocyte subsets [8, 12], and its expression decreases during monocytes/macrophage differentiation [29].
In this study, we show that RRMS patients displayed lower numbers of suppressive MDSCs, more specifically MO-MDSCs, in the peripheral blood which was associated with significantly higher expression of miR-223 in MDSCs in MS patients compared to healthy controls. In the EAE model, miR-223 knockout (miR-223−/−) mice developed a less severe disease characterized by the accumulation of MDSCs and lower production of IL-17A and GM-CSF by CNS-isolated T cells compared to control mice. MO-MDSCs isolated from miR-223−/− mice were more suppressive toward T cell proliferation and cytokine production in vitro and in EAE in vivo compared to MO-MDSCs from WT cells. Mechanistically, greater suppressive function of miR-223−/− MO-MDSCs was associated with higher expression of Arg1 and Stat3, which herein, we demonstrate being a miR-223 target gene. Consistently, MDSCs from MS patients had lower expression of STAT3 and ARG1. Our results support the contention that miR-223 can regulate the number and function of MO-MDSCs in EAE and MS, which in turn modulate T cell responses that favor the amelioration of CNS inflammation.
Materials and methods
Subjects
This project was approved by the Institutional Review Board (IRB) at Washington University in St. Louis. All subjects provided informed consent. This population was recruited at the John L. Trotter MS Center, Washington University School of Medicine, Department of Neurology, St. Louis (Missouri, USA). The characteristics of patients and healthy controls are summarized in Supplementary Table 1a, b. All the MS patients met the 2010 McDonald‘s criteria for MS diagnosis [40]. Of the 34 MS patients [24 untreated and 10 treated with glatiramer acetate (GA)] included in flow cytometric analyses and of the 11 subjects with MS used for cell sorting, four in the first group and two in the second were experiencing a relapse at the time of analysis. For gene expression studies in circulating MDSCs, healthy control subjects were matched for age and gender to the MS group. Untreated RRMS subjects had not taken disease-modifying therapies or corticosteroids for at least 3 months before and at the time of blood collection.
Flow cytometry analyses
Staining of human MO- and PMN-MDSCs was performed using anti-human antibodies to CD33 (Biolegend, 303408), CD14 (eBioscience, 25-0149-42), CD15 (Biolegend, 301904), and HLA-DR (Beckman Coulter, 2017-03-21) starting from 100 μl of heparinized whole human blood which was incubated with the antibodies for 30 min at RT. Subsequently, samples were fixed and lysed with Step Fix/Lyse Solution (Affimetrix, eBioscience) at room temperature (RT) for 30 min. To calculate absolute cell numbers, 100 μl of 123count eBeads Solution (Affimetrix, eBioscience) were added to each sample before flow cytometric analysis according to manufacture instructions. Samples were analyzed on a Gallios Flow Cytometer (Beckman Coulter). Staining of murine tissues used anti-mouse antibodies to CD11b (BD Pharmigen 553311), Ly-6G (eBioscience, 15-5931-82), Ly-6C (eBioscience, 47-5932-82), CD4 (Biolegend, 100443), CD3 (Biolegend, 100216), CD45.1 (Biolegend, 110716), CD40 (BD Pharmigen, 553790), CD86 (Biolegend, 105012), and CD274 (PD-L1) (BD Pharmigen, 55809). After isolation, cells were labeled with the various fluorochrome-conjugated antibodies and the survival marker Zombie Dye for 25 min at 4 °C, washed, and analyzed on a the Gallios Flow Cytometer. Data analysis was performed by gating on Zombie Dye negative cells. Staining for intracellular cytokines encompassed anti-mouse antibodies to IL-17A (Biolegend, 506908), IL-10 (Biolegend, 505028), GM-CSF (Biolegend, 505406), IFN-γ (Biolegend, 505810), and TNF-α (eBioscience 11-7321-81). Isolated cells were incubated in 96 well plates for 5–6 h with 50 ng/ml of PMA and 750 ng/ml of Ionomicin (both from Sigma) and Monensin (eBioscience) at 37 °C. Surface staining was performed for 25 min at 4 °C. Subsequently, cells were fixed in 2 % PFA for 10 min at RT and then permeabilized in Saponin Buffer for another 10 min at RT. Cytokine staining was performed in permeabilization buffer for 25 min at 4 °C. Samples were run on the Gallios Flow Cytometer, and data were analyzed using FlowJo (Tree Star Inc.).
Human MDSC sorting
For human MDSC sorting, PBMCs were isolated by Ficoll-density centrifugation and then processed for cell sorting using anti-human CD33 and HLA-DR antibodies on an FACS ARIA (BD Bioscience). CD33b+HLA-DRneg MDSCs were collected and frozen for RNA extraction.
Animals
Animal protocols were approved by the Washington University Animal Studies Committee. MiR-223−/− and littermate-derived wild-type (WT) mice (C57Bl/6 background) were obtained from Dr. Todd Fehniger and bred at Washington University. The miR-223 gene is located on the X chromosome, and mice were bred as miR-223−/y or littermate miR-223+/y (termed WT throughout the manuscript) males with heterozygous females. MiR-223−/− and WT mice deriving from these breeding pairs were used in all experiments. C57BL/6 mice transgenic for a TCR with specificity for the peptide MOG35–55 (TCRMOG mice, also referred to as 2D2 mice; The Jackson Laboratory) were screened by flow cytometric analysis of peripheral blood cells using a specific antibody to Vβ11 (BD Biosciences) [3]. Mice were 6–9 weeks of age at the initiation of the studies.
EAE induction and histology
Mice were immunized with MOG35–55 (Sigma Genosys) to induce EAE as previously done [39]. In adoptive transfer experiments, MOG35–55-specific Th1 cells were generated as described [13]. Briefly, donor mice were immunized with MOG35–55 peptide. Spleens were removed 14 days post-immunization, and CD4+ T cells were purified and maintained in culture in the presence of irradiated splenocytes, IL-12 and IL-2 (eBioscience). Cells were restimulated weekly, and after the sixth stimulation MOG-specific T cells were harvested, washed, and transferred intravenously at 10 × 106 cells/mouse to induce EAE. In some active EAE experiments, miR-223−/− or WT MO-MDSCs obtained in vitro (see below for methodological details) were injected (2 × 106 cells) intravenously on day 5 post-immunization. The control group was injected with PBS. Timing of MDSCs transfer in EAE was decided based on prior reports in the literature [49]. For both active and adoptive transfer EAE, mice were evaluated clinically in a blinded fashion using a scale from 0 to 5 as previously described [39].
Histological and immunofluorescence microscopy analyses
CNS tissues were dissected from mice perfused with buffered 4 % paraformaldehyde, then embedded in paraffin. Slides were stained with hematoxylin & eosin (H&E) [39] and with solochrome cyanine to stain myelin as previously described [24]. For the latter, sections were stained with Eriochrome Cyanine R (Sigma), washed, differentiated in 10 % iron (III) chloride (Sigma), and counter-stained with Van Gieson’s stain. The quantification of inflammation and demyelination was performed in a blinded fashion as previously reported [38]. Distributions of injured axons and demyelinated axons were identified with double-staining experiments using antibodies to nonphosphorylated neurofilaments (SMI32 at 1:200; Sternberger Monoclonals) and myelin basic protein (MBP; at 1:200; Zymed Laboratories Inc.), respectively. MBP and SMI32 staining were analyzed as the percentage area of positive staining (number of positive pixels/1 mm2) within the area of interest. For immunofluorescence assays on in vitro derived MO-MDSCs, the following primary antibodies were used: anti-mouse Gr1 (1:200; Abcam) and ARG1 (1:200; Abcam). Sections were blocked with 5 % horse serum and 0.1 % triton-X in PBS for 1 h, and primary antibodies were applied overnight at 4 °C degree. Next, sections were incubated with fluorescently labeled secondary antibodies for 1 h at RT (Invitrogen) and mounted on a slide using a Fluoroshield Mounting Medium with DAPI (Abcam). The total number of ARG1+Gr1+ cells was divided by the total number of Gr1+ cells to calculate the percentage of double positive cells. Images were acquired using a Nikon Eclipse 90i fluorescent and bright field microscope and analyzed for quantification with the Metamorph 7.7 software.
Mouse MDSC isolation
Murine MDSCs were generated in vitro from the bone marrow (BM) of miR-223−/− and WT mice. BM was flushed, and red blood cells were lysed with red blood cells lysis buffer (Sigma). To obtain MDSCs, the remaining cells were plated with medium supplemented with GM-CSF (40 ng/ml) and IL-6 (40 ng/ml) and cultured at 37 °C in 5 % CO2-humidified atmosphere for 4 days. PMN- and MO-MDSCs were purified from the mixed population of BM-derived cells by immunomagnetic separation using biotinylated anti-Ly6G and anti-Gr1 antibodies and streptavidin-conjugated MicroBeads with MiniMACS columns according to manufacturer’s protocol (Miltenyi Biotec). In vitro functional assays were done with PMN-MDSCs and MO-MDSC subsets. Cell purity was confirmed by flow cytometric analysis using anti-CD11b and Gr-1 antibodies (purity >95 %), and viability was assessed by Trypan blue dye exclusion.
T cell suppression assays
For Ag-specific proliferation, MOG-specific CD4+ T cells were purified from 2D2 mice by CD4-positive selection (L34T, Milteny Kit) with a final purity ≥95 %. CD4+ T cells (105 cells/well) were cultured at different ratios with PMN- or MO-MDSCs (8:1, 4:1, and 2:1; CD4+ T cells: MDSCs) in the presence of 20 μg/ml MOG35–55 and irradiated splenocytes from C57BL/6 mice (1:1 for T cells: irradiated splenocytes). For CFSE-based proliferation assay, CD4+ T cells were incubated with 1 mM CFSE for 15 min at 37 °C, quenched in culture medium for 30 min, and washed before use. After 96 h in culture, cells were stained for CD4 and analyzed by flow cytometry to assess CD4+ T cell proliferation by CFSE dilution. In the human suppression assay of T cell proliferation, CD4+ T cells were isolated by positive selection (Human CD4 Microbeads, Miltenyi; purity >95 %) from PBMCs of a control subject, labeled with CFSE (1 mM for 15 min at 37 °C in labeling buffer PBS/0.1 %BSA), and co-cultured (50,000 cells/well) with CD33+HLA-DRneg MDSCs (Human CD33 MicroBeads, Miltenyi; purity >95 %) at a 2:1 ratio. T cell proliferation was induced by plate bound anti-CD3 (10 μg/ml) and anti-CD28 (10 μg/ml) antibodies. T cell proliferation in the absence or presence of MDSCs was examined by CFSE dilution by flow cytometry after 5 days of culture.
T cell differentiation in vitro
Spleens from 6- to 10-week-old miR-223−/− and WT mice were harvested and dissociated into a single cell suspension. After erythrocyte lysis, CD4+CD62L+ T cells were sorted using a FACS ARIA (BD Bioscience). Purified naive CD4+CD62L+ T cells from miR-223−/− and WT mice were cultured with antigen presenting cells (APC) and MOG35–55 (20 μg/ml) as previously shown [19]. For standard Th1 differentiation, cells were cultured in the presence of IL-2 (10 ng/ml), IL-12 (20 ng/ml), and anti-IL-4 (10 μg/ml) (eBioscience). For standard Th17 differentiation, cells were cultured in the presence of IL-6 (20 ng/ml), TGF-β (3 ng/ml), IL-23 (10 ng/ml), anti-IL-4, and anti-IFN-γ (10 μg/ml) (eBioscience). After 5 days in culture, cells were collected for intracellular cytokine staining by FACS analysis.
Isolation of CNS inflammatory cells and MDSCs
Brain and spinal cord tissues from mice perfused with PBS were digested with collagenase IV (Sigma-Aldrich) for 30 min at 37 °C, resuspended in 37 % Percoll, and loaded between a 30 and 70 % Percoll gradient. After centrifugation at 2000g for 20 min, CNS mononuclear cells were retrieved from the 37/70 % Percoll interface as previously done [38]. PMN- and MO-MDSC subsets were purified from CNS mononuclear cells by immunomagnetic separation using biotinylated anti-Ly6G and anti-Gr1 antibody and streptavidin-conjugated MicroBeads as mentioned above (Miltenyi Biotec). Cell purity was >95 % by flow cytometric analysis using anti-CD11b and Gr-1 antibodies.
Quantitative real-time PCR
RNA was purified from murine tissues (spinal cord, brain, and lymphoid tissues) or human/mouse sorted MDSCs (PMN and MO-MDSCs) using the RNeasy Micro Kit (Qiagen), converted into cDNA using the High-Capacity cDNA Reverse Transcription Kit or the MicroRNA Reverse Transcription Kit for microRNAs (Applied Biosystems), and used at 50 ng/μl in quantitative real-time PCR (qRT-PCR) analysis (QuantStudio 3 Real-Time PCR System, Applied Biosystems). The ΔΔCt method was applied to determine differences in gene expression levels after normalization to the arithmetic mean of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) for Arg1 and Stat3 or RNU48 and U6 as internal standards for miR-223 expression. The TaqMan probes (Life technologies) were the following: Arg1 (Mm00475988_m1; Hs00968979_m1); Stat3 (Mm01219775_m1; Hs1047580_m1); GAPDH (Mm99999915_g1; Hs99999905_m1); miR-223 (TM 002295_mir); U6 (001973); and RNU48 (001006).
Western blot
The whole cell lysates from MO- and PMN-MDSCs were prepared as previously described [38], and total protein contents were measured using the Nanodrop (Thermo Scientific). Equal protein amounts (30 μg/lane) were loaded per lane for the MO- and PMN-MDSC samples. Protein was separated by electrophoresis on precast gel (Bio-Rad Laboratories), and transferred onto PVDF membranes (Millipore Corp.). The membranes were incubated overnight at 4 °C with rabbit anti-mouse monoclonal antibody targeting STAT3, anti-rabbit monoclonal antibody targeting pSTAT3, or an anti-rabbit anti-GAPDH (Cell Signaling) and then with the secondary anti-rabbit or anti-mouse antibodies HRP for 1 h. Signal was developed with a chemiluminescent substrate (Thermo Fisher Scientific), and Kodak M35 X-OMAT Automatic Processors was used for the detection of signals. Bands were quantitated in densitometry units after the processing with the ImageJ software (NIH).
3′ UTR luciferase reporter assays
The murine Stat3 3′ UTR was cloned into the psiCheck2 vector (Promega, Madison, WI) using PCR, and the control vector contained only GFP (empty). MiR-223 was cloned into the pMND vector [44]. Luciferase assays were performed using the Dual-Glo luciferase Assay (Promega) as described [44]. Renilla luciferase (experimental) was normalized to Firefly luciferase (transfection control) followed by the comparison of the Renilla/Firefly ratios of the control and Stat3 psiCheck2 sensor plasmids. MiR-223 was overexpressed via pMND in all conditions. 293T cells were co-transfected with the pMND-miR-223 over-expression vector and a pSICheck2 control or Stat3 3′ UTR and assayed after 48 h.
Data analysis
Data were analyzed by t test or the equivalent non-parametric Mann–Whitney U test when comparing statistical differences between two groups. A one-way ANOVA or the equivalent non-parametric Kruskal–Wallis test with post hoc comparisons were used to analyze data with more than two groups. EAE incidence data were analyzed by the Fisher exact test. P < 0.05 was considered significant. Analyses were performed using GraphPad Prism 6.
Study approval
Human studies were approved by the Human Research Protection Office (HRPO) at Washington University in St. Louis (HRPO approval number: 201506145). For animal studies, all experimental procedures were approved by the animal studies committees of the Washington University School of Medicine (study approval number: 20150040).
Results
Peripheral blood MDSCs are reduced in RRMS patients
We examined the number of MDSCs in the peripheral blood of RRMS patients, including untreated (n = 24) and treated with GA (n = 10) subjects, as well as healthy control subjects (n = 16) by flow cytometry. MDSCs in humans are characterized by CD33+HLA-DRneg expression, with two different subsets defined based on surface marker expression. Human MO-MDSCs are defined as CD33+HLA-DR−CD14+CD15− and PMN-MDSCs as CD33+HLA-DR−CD14−CD15+ [26]. The gating strategy that was used to define MDSC subsets is illustrated in Fig. 1a on representative subjects from each group. We observed statistical significant lower numbers of total MDSCs in the whole blood of MS patients in comparison with healthy subjects (mean ± SD: 46 ± 41 vs. 172 ± 104 cells/μl, respectively; P < 0.0001 after post hoc comparison). Patients treated with GA had a modest increase in the numbers of total MDSCs (62 ± 37 cells/μl) compared to untreated patients, but this was not significantly different (Fig. 1b). Furthermore, the number of MO-MDSCs was significantly decreased in MS patients compared to healthy controls (mean ± SD: 19 ± 21 vs. 60 ± 47 cells/μl, respectively; P = 0.0005), while GA treatment was associated with a slight increase compared to untreated patients (28 ± 26 cells/μl; Fig. 1c). Similar results were obtained when the percentages of MDSCs and MO-MDSCs of total blood leukocytes were considered instead of absolute numbers (Supplementary Fig. 1). No differences were observed into the PMN-MDSC subset, even though the number of this cell subset based on our gate strategy was very low (Fig. 1d). Importantly, total MDSCs isolated from the blood of a control subject were able to inhibit the proliferation of autologous T cells in vitro (Fig. 1e). This functionally demonstrates the suppressive activity of MDSCs that we previously characterized by flow cytometry.

Reduced number of peripheral blood MDSCs with higher miR-223 expression in relapsing-remitting MS patients compared to healthy controls. a Representative flow cytometric plots for MDSCs (upper panel) and MO-MDSCs (lower panel) in the whole blood of one RRMS patient and one healthy control individual. In both upper and lower panels, cells were gated on live cells based on FSC/SSC characteristics and singlet cells. Next, in the upper panel MDSCs were gated as CD33+ HLA-DR−. In the lower panel, HLA-DR− cells were displayed for CD14 and CD33 expression to identify the MO-MDSC subset defined as CD14+ CD33+ (all these cells are also CD15−). Percentages refer to gated cells. b–d Total numbers (cells/μl) of MDSCs, MO- and PMN-MDSCs in the whole blood of untreated RRMS patients (n = 24), GA patients (n = 10), and in healthy individuals (n = 16). MDSCs are defined as HLA-DR−CD33+; MO-MDSCs as HLA-DR−CD33+CD15−CD14+, and PMN-MDSCs as HLA-DR−CD33+CD15+CD14−. e Suppression of T cell proliferation assay. MDSCs were isolated from the blood of a control subject and co-cultured for 5 days with autologous CFSE-labeled CD4+ T cells (1:2 ratio) in the presence of anti-CD3 and anti-CD28. T cell proliferation in the absence or presence of MDSCs was examined by CSFE dilution by flow cytometry. f Scatter plot of the distribution of miR-223 expression levels in MDSCs from RRMS patients (n = 11) and healthy controls (n = 10). Red squares in the scatter plots in b–d and in f represent RRMS patients having a relapse. Error bars are mean ± SD. P values were calculated by Mann–Whiteny U test or Kruskal–Wallis tests with post hoc comparisons if more than two groups
MiR-223 expression is higher in MDSCs isolated from MS patients compared to healthy controls
Next, we assessed miR-223 expression in MDSCs isolated from untreated RRMS patients (n = 11) and healthy controls (n = 10) by qPCR. We focused our attention on miR-223, because we have previously shown that its expression is altered in peripheral blood of MS compared to healthy subjects [42]. MDSCs were isolated from peripheral blood mononuclear cells (PBMCs) by sorting based on the expression of the CD33 and HLA-DR markers. MDSCs isolated from MS patients had statistically higher expression of miR-223 in comparison with MDSCs from control subjects (mean fold change ± SD over controls: 1.69 ± 0.35 vs. 0.78 ± 0.15, P = 0.02) (Fig. 1f). These results demonstrate that MS patients have lower numbers of circulating suppressive MDSCs, in particular of the MO-MDSC subset and a dysregulation of MDSC miR-223 expression compared to healthy control subjects.
MiR-223 expression is upregulated in the spinal cord during clinical EAE
To gain more mechanistic insight into the potential role of miR-223 in MS, we utilized the EAE model. First, miR-223 expression was studied in spinal cord tissue isolated from naive and MOG35–55-immunized C57BL/6 mice at 5, 10, 15, and 30 day post-immunization (DPI; n = 4/group). MiR-223 expression was markedly increased in the spinal cord of immunized mice at the peak of disease (15 DPI) compared to naive mice (Fig. 2a, P = 0.02). While we did not find significant differences in miR-223 expression levels in the spleen, brain, and lymph nodes of naive and immunized mice at each time point, bone-marrow expression of miR-223 was lower from 10 to 30 DPI compared to baseline (Supplementary Fig. 2). The spinal cord is the major site of inflammation during EAE, and the observed increase of miR-223 expression during acute EAE could be related to inflammatory cell infiltration. Therefore, we analyzed miR-223 expression in mononuclear cells isolated from the spinal cord of mice with clinical EAE after sorting cell populations based on the expression of the CD11b and CD45 markers. MiR-223 was highly expressed in CD45dimCD11b+ myeloid cells (macrophages and microglia), but was also detectable at lower levels in CD45highCD11b− cells which are mainly infiltrating lymphocytes (Fig. 2b).

MiR-223 is expressed in myeloid cells in the spinal cord at the peak of EAE. a MiR-223 expression in the spinal cord taken from naïve mice and on days 5, 10 15, and 30 post-EAE immunization (n = 4 mice/time point) analyzed by quantitative RT-PCR (qRT-PCR). U6 snRNA was used as endogenous controls. b MiR-223 expression in CD45+CD11b− (lymphocytes) and CD45+CD11b+ (myeloid cell) populations sorted from infiltrating mononuclear cells in the spinal cord on day 15 post-immunization (peak of disease) and analyzed by qRT-PCR. miR-223 expression is shown as relative to miR-223 expression in myeloid and T cells isolated from naïve splenocytes. Error bars represent mean ± SEM. P values were calculated by Kruskal–Wallis H test
MiR-223 deficient mice develop attenuated EAE compared to WT mice
To further clarify the role of miR-223 in CNS autoimmunity, EAE was induced in miR-223−/− and littermate wild-type (WT) controls by active immunization with MOG35–55. MiR-223−/− mice showed delayed disease onset, and a significantly less severe disease compared to control mice (Fig. 3a; Supplementary Table 2). Concordant with reduced clinical severity, histological analyses showed a decreased number of inflammatory infiltrates and demyelination (as revealed by solochrome cyanine and myelin basic protein staining) in the spinal cord of miR-223−/− mice compared to WT (Fig. 3b–e, h). Axonal damage detected by immunostaining for SMI32 revealed fewer SMI32-positive axons present in the miR-223−/− group compared to the control group (P = 0.04; Fig. 3f–h). In conclusion, active EAE is less severe clinically and pathologically in miR-223−/− compared to controls.

Lack of miR-223 ameliorates EAE. a Active EAE was induced in miR-223−/− (n = 8) and WT mice (n = 5) with MOG35–55. MiR-223−/− displayed delayed and significantly less clinical EAE than WT mice (P < 0.0001 by two-way ANOVA). One representative experiment out of four performed is shown. Data are shown as mean clinical score ± SEM. b–e Inflammation and demyelination were assessed using H&E and solochrome cyanine staining of spinal cord sections from immunized WT (n = 5) (b, d) and miR-223−/− (c, e) mice (n = 5) at disease peak. MiR-223−/− mice showed less inflammatory cells (black arrows) infiltrating the parenchyma compared to WT mice. f, g Spinal cord sections from EAE affected WT and miR-223−/− mice were stained for myelin basic protein (in green) and SMI32 (in red), to detect myelin and damaged axons, respectively. 10x magnification. Scale bars 100 μm (b–g). h Quantification of inflammation, demyelination, and axonal damage. Data are shown as mean ± SEM (5 mice/group) from one experiment representative of three performed. Statistical significance determined by Mann–Whitney U test
Lack of miR-223 interferes with priming of T cells against myelin peptides in periphery
Given the reduction in EAE severity in miR-223−/− mice compared to WT controls, we asked if a lack of miR-223 would interfere with the peripheral T cell priming phase and/or with the effector CNS phase during EAE. Therefore, adoptive transfer EAE was induced by transferring wild-type MOG-specific CD4+ T cells, polarized in vitro under Th1 conditions, into miR-223−/− and control mice. No differences in disease clinical course were noted in WT and miR-223−/− mice with passive transfer EAE (Fig. 4a), demonstrating no effects of miR223-deficiency on recipient mice injected with myelin-reactive WT T cells. This suggests that less severe active EAE in miR-223−/− mice is very likely related to the inhibition of T cell activation against the myelin antigen after immunization. Next, we explored the role of miR-223 in T cell priming during EAE. Control and miR-223−/− mice were immunized to induce EAE, and draining lymph nodes (DLNs) were isolated 6 DPI. Lymph nodes draining the site of immunization in miR-223−/− mice were larger in size and had higher cellularity compared to WT mice (Supplementary Fig. 3a). Flow cytometric analyses of DLNs showed increased frequency of myeloid cells (CD11b+Gr1+ cells) in miR-223−/− mice relative to WT, with no differences in the number of T cells (Supplementary Fig. 3b–c). Intracellular production of IFN-γ, IL-17A, and GM-CSF, cytokines known to be relevant in EAE pathogenesis [41], was tested in CD4+ T cells from DLNs of miR-223−/− and WT mice on day 6 DPI (Fig. 4b). We observed significantly lower percentages of IFN-γ and GM-CSF producing CD4+ T cells along with a higher frequency of IL-17A-producing T cells, in DLNs from miR-223−/− mice relative to WT mice. In conclusion, the lack of miR-223 interferes with the induction phase of EAE and leads to altered cytokine production profiles by antigen-specific T cells.

Defective T cell priming in miR-223−/− mice is not due to an intrinsic T cell defect. a Clinical course of adoptively transferred EAE induced by the injection of activated MOG35–55-specific CD4+ T cells obtained from C57BL/6 mice into miR-223−/− or WT recipients (n = 5 mice/group). Data are mean clinical score ± SEM of all animals in each group. Data are representative from three experiments performed with similar results. b Scatter plot of the percentages of IFN-γ, IL-17A, and GM-CSF in draining lymph nodes CD4+ T cells of WT and miR-223−/− mice at day 6 post-immunization (n = 5 mice/group). Numbers indicate the percentage of cytokine-producing CD4+ T cells. Data are representative of three independent experiments. Error bars are mean ± SEM. c, d WT and miR-223−/−-derived naive CD4+ T cells cultured in vitro under Th1 or Th17 conditions were restimulated with PMA/ionomycin on day 5 to analyze IL-17A and IFN-γ intracellular production by flow cytometry. In d, numbers represent frequencies of IFN-γ- and IL-17A-producing CD4+ T cells. Data are representative of three independent experiments. Error bars are mean ± SEM. P values were calculated by Mann–Whitney U test
Lack of mir-223 is not associated with an intrinsic T cell defect
Next, we studied the mechanisms leading to defective T cell priming in miR-223−/− mice after immunization with MOG35–55. MiR-223 is expressed at high levels in myeloid cells isolated from the CNS during EAE, but is also expressed by infiltrating lymphocytes (Fig. 2b). Therefore, the altered cytokine production profile of miR-223-deficient T cells during EAE could be related to an intrinsic T cell defect due to the lack of miR-223. To test this hypothesis, we studied the abilities miR-223-deficient lymphocytes to differentiate into Th1 and Th17 cells. Naive CD4+CD62+CD44− T cells were isolated from miR-223−/− and WT mice and cultured in vitro in Th1 and Th17 differentiating conditions using standard protocols [19]. Intracellular staining of cytokine production demonstrated that Th1 and Th17 differentiation occurred normally in miR-223−/− CD4+ T cells (Fig. 4c, d). Taken together these data indicate that miR-223 does not function to regulate T cell encephalitogenicity, as miR-223−/− T cells do not show any intrinsic defect that could explain a defective activation in vivo against the myelin antigen during EAE. Our results suggest that the reduced T cell activation is a secondary event.
Lack of miR-223 is associated with the expansion of MO-MDSCs in the CNS during EAE
It is known that different subsets of immature myeloid cells with the capacity for the suppression of T cell activities accumulate in the spleen and CNS at EAE onset and peak [31, 48]. Specifically, PMN-MDSC and MO-MDSC numbers have been shown to progressively increase until the peak of EAE [18, 43]. In our study, we observed an expansion of the myeloid compartment in DLNs from miR-223−/− mice during EAE which could potentially affect T cell responses (Supplementary Fig. 3). First, we analyzed by flow cytometry absolute numbers of PMN- and MO-MDSCs in naïve WT and miR-223−/− lymphoid organs. In mice, these two subsets of MDSCs are defined based on the expression of Ly6C and Ly6G. No baseline differences in MDSC absolute numbers were detected in the spleen, bone marrow, or lymph nodes of naïve mice (Supplementary Fig. 4). Next, splenocytes and CNS mononuclear cells isolated from miR-223−/− and WT mice were characterized by flow cytometry during EAE. At the peak of the disease (15 DPI), absolute numbers of miR-223−/− PMN and MO-MDSCs were significantly higher in the spleen compared to WT counterparts (Fig. 5a, b). No differences were noted in the number of CD4+ T cells (Fig. 5b). Phenotypic characterization of CNS-isolated mononuclear cells from miR-223−/− and WT mice at EAE peak showed a significant increase of MO-MDSCs in miR-223−/− compared to WT mice (Fig. 5c), but no differences in the number of (PMN)-MDSCs between the two groups. Numbers of CNS-isolated CD4+ T cells were also similar in miR-223−/− and WT mice (Fig. 5c). We examined the expression of activation (CD40, CD86, and MHC-II) and inhibitory (PD-L1) markers on MO- and PMN-MDSCs isolated from the spleen and CNS at EAE peak by flow cytometry. We found reduced expression of activation markers and PD-L1 on miR-223−/− compared to WT MDSCs (Supplementary Fig. 5a, b) [48]. The production of the pro-inflammatory cytokines TNF-α and GM-CSF was decreased, while IL-10 (anti-inflammatory cytokine) was increased in miR-223−/− PMN and MO-MDSCs compared to WT (Supplementary Fig. 5c). To characterize encephalitogenic T cell responses within the CNS during EAE, we analyzed by intracellular staining the cytokine production profile of CD4+ T cells isolated from the spinal cord of miR-223−/− and control mice with clinical disease. At the peak of the disease (15 DPI), CNS-infiltrating CD4+ T cells from miR-223−/− mice showed significant lower levels of IL-17A and GM-CSF compared to WT mice, and a trend toward less IFN-γ production (Fig. 5d, e).

Expansion of MDSCs in the spleen and in the CNS of miR-223−/− mice during EAE. a–c Splenocytes and CNS-infiltrating cells from immunized WT and miR-223−/− mice on day 15 post-immunization (EAE peak) analyzed by flow cytometry. a Representative flow cytometric analysis of frequencies of MO and PMN-MDSC populations from the spleen (top) and CNS (bottom) for the indicated groups. The gating strategy first included Zombie−CD45+CD11b+. Next, MO-MDSCs were defined as CD11b+Ly6Chigh cells and PMN-MDSCs as CD11b+Gr1+. Quantification of the absolute numbers of MO or PMN-MDSCs and CD4+ T cells was calculated from the percentages based on the total number of b splenocytes or c CNS-infiltrating cells isolated/mouse. d Representative plots of intracellular staining of CNS-infiltrating CD4+ T cells for the indicated cytokines. e Intracellular production of IFN-γ, IL-17A, and GM-CSF by CNS-derived CD4+ T cells from WT and miR-223−/− mice with EAE analyzed by flow cytometry. Numbers indicate percentage of CD4+ cells. These results are from one representative experiment of three performed. P values were calculated by Mann–Whitney U test
MiR-223 regulates MDSC suppressive functions
The observed expansion of MDSCs in miR-223−/− mice in peripheral lymphoid organs and in the CNS coinciding with lower production of the pathogenic cytokines IL-17A and GM-CSF by CD4+ T cells after immunization with MOG35–55 suggests that miR-223 regulates the suppressive function in MDSCs. To evaluate the role of miR-223 in MDSC immunosuppressive functions, miR-223−/− and WT PMN- and MO-MDSCs were cultured in vitro at different ratios with 2D2 CD4+ T cells in the presence of MOG35–55. We measured 2D2 CD4+ T cell proliferation by CFSE dilution as well as cytokine production. MiR-223−/− MO-MDSCs had greater immune-suppressive effects on CD4+ T cell proliferation in comparison with WT MO-MDSCs, while no differences were noted for miR-223−/− and WT PMN-MDSCs (Fig. 6a). Similarly, miR-223−/− MO-MDSCs suppressed T cell production of IFN-γ, GM-CSF, and IL-17A more profoundly than WT MO-MDSCs (differences between the two groups were statistically significant for GM-CSF in all conditions, at the 1:8 ratio for IFN-γ, and for all other conditions, there was a consistent trend toward more suppression in miR-223−/− vs. WT MO-MDSCs) (Fig. 6b). Next, MO-MDSCs were isolated from the CNS of miR-223−/− and WT mice at EAE peak and cultured in vitro with 2D2 CD4+ T cells in the presence of MOG35–55. Consistent with what we observed previously, miR-223−/− MO-MDSCs obtained ex vivo from the inflamed CNS-suppressed MOG-specific proliferation more potently than WT MO-MDSCs (Fig. 6c). To study the role of miR-223 in modulating MDSC suppressive activity in vivo, miR-223−/− or WT MO-MDSCs were injected into EAE-immunized recipient C57BL/6 mice on day 5 post-immunization. We observed the suppression of the clinical disease course in MDSC-injected groups compared to the PBS-treated control group (Fig. 6d; P < 0.0001). However, mice that received miR-223−/− MDSCs showed the suppression of EAE that was persistent over time compared to those injected with WT MDSCs which ultimately developed maximal disease (Fig. 6d). CFSE labeling of MO-MDSCs before injection demonstrated their localization into the CNS during EAE (Fig. 6e). Furthermore, the analysis of cytokine production by CNS-isolated T cells during establish EAE (day 34 post-immunization) demonstrated the reduced production of the pro-inflammatory cytokines IL-17A, IFN-γ, and GM-CSF in miR-223−/− MDSCs compared to PBS-injected mice. No significant differences were noted in T cell cytokine production between the WT MO-MDSC-treated mice and control PBS groups (consistent with similar clinical scores at time of analysis on >day 30 post-immunization; Fig. 6f). All together, these data suggest that miR-223−/− MO-MDSCs are more efficient in suppressing CD4+ T cell proliferation and cytokine production in vitro and in vivo.

MiR-223−/− MO-MDSCs are more potent immuno-suppressors compared to WT in vitro and in vivo. a Proliferation and b cytokine production of PMN or MO-MDSCs isolated from the bone marrow of WT and miR-223−/− mice and co-cultured with CFSE-labeled CD4+ T cells from 2D2 transgenic mice (1:2, 1:4, and 1:8 ratios) in the presence of MOG35–55. Error bars are mean ± SEM. c Proliferation of MO-MDSCs isolated from the CNS of EAE WT and miR-223−/− mice and co-cultured with CFSE-labeled CD4+ T cells from 2D2 transgenic mice (1:1, 1:2, 1:4, 1:8, and 1:16 ratios) in the presence of MOG35–55. A representative experiment from a total of four is showed. P values were calculated by Mann–Whitney U test. d Clinical course of active EAE in C57BL/6 mice immunized with MOG35-55 and injected (day 5 post-immunization) with WT or miR-223−/− MO-MDSCs. Control group was injected with PBS (P < 0.0001 by two-way ANOVA; the graph refers to one experiment of a total of two performed with similar results). e. WT and miR-223−/− MO-MDSCs were labeled with CFSE before injection into EAE-immunized mice and then detected by flow cytometric analysis in CNS-infiltrating mononuclear cells during ongoing EAE. The PBS control group was CFSE negative. f Cytokine production by CNS-isolated T cells during establish EAE (>day 30 post-immunization) in PBS-, WT-, or miR-223−/− MDSC-injected groups (P ≤ 0.05 by Kruskal–Wallis H test)
Regulation of MDSC immunosuppressive activity by miR-223 is associated with increased Arg1 and Stat3 expression
Immunosuppression by MDSCs involves multiple mechanisms, including depletion of l-arginine via ARG1 and increased NO production by iNOS [49]. To evaluate how miR-223 deficiency impacts on mechanisms of MDSC immunosuppression in our system, we examined Arg1 and iNOS transcript expressions by real-time PCR in bone-marrow-derived MO- and PMN- MDSCs from WT and miR-223−/− mice (Fig. 7a, b). Overall, Arg1 expression was higher in MO-MDSCs compared to PMN-MDSCs in both genotypes. More importantly, the expression of Arg1 mRNA was significantly higher in MO-MDSCs isolated from miR-223−/− compared to WT mice (Fig. 7a). No difference in Arg1 expression was noted between WT and miR-223−/− PMN-MDSCs, and no differences were found in iNOS expression by WT and miR-223−/− MO- and PMN-MDSCs (data not shown). Immunofluorescence analysis by microscopy for ARG1 protein in miR-223−/− and WT MO-MDSCs confirmed higher ARG1 expression at the protein level in MO-MDSCs lacking miR-223 compared to WT (Fig. 7b). Recent work using different mouse tumor models suggested that Arg1 expression is modulated by STAT3 [45]. Therefore, we examined the expression of Stat3 in bone-marrow-derived MO- and PMN-MDSCs from WT and miR-223−/− mice by real-time PCR. Stat3 expression was found to be significantly higher in miR-223−/− compared to WT MO-MDSCs (Fig. 7c). Western blot analysis of STAT3 and phosphorylated (p)STAT3 in MO- and PMN-MDSCs confirmed an increased expression of STAT3 in miR-223−/− vs. WT MO-MDSCs at the protein level, while no differences were noted in PMN-MDSCs (Fig. 7d). Furthermore, we found that the pSTAT3/STAT3 ratio was higher in miR-223−/− compared to WT MO-MDSCs, which supports the notion that the absence of miR-223 in MO-MDSCs is associated with increased levels of activated STAT3. All together, these results suggest that miR-223−/− MO-MDSCs have more potent suppressive activity compared to the WT counterpart, which was related to higher ARG1 expression associated with increased STAT3 expression.

MiR-223−/− regulates Arg1 and targets Stat3. a qRT-PCR analysis of arginase 1 (Arg1) in PMN and MO-MDSCs isolated from WT and miR-223−/− mice (n = 3 mice/group). GAPDH probe was used as endogenous control. Error bars are mean ± SEM. A representative experiment from a total of four is showed. P values were calculated by Mann–Whitney U test. b Immunofluorescence staining by microscopy for ARG1 (in red) and Gr1 (in green) in BM-derived MO-MDSCs from WT and miR-223−/− mice. 60x magnification. Scale bars 25 μm. Quantification of immunofluorescence analyses as percentages of ARG1+ Gr1+ MO-MDSCs of total Gr1+ MO-MDSCs from WT and miR-223−/− mice counted/field. c qRT-PCR analysis of STAT3 expression in PMN and MO-MDSCs isolated from WT and miR-223−/− mice (n = 3 mice/group). GAPDH probe was used as endogenous control. d Western blot analysis of STAT3 and pSTAT3 expression levels in MO- and PMN-MDSCs derived from the bone marrow of WT and miR-223−/− mice. STAT3 levels were normalized on GAPDH, while pSTAT3 was normalized on STAT3. e Luciferase assays showing that miR-223 downregulates the expression of Stat3 by targeting its 3′UTR. MiR-223 was overexpressed via pMND in all conditions. 293T cells were co-transfected with the pMND-miR-223 over-expression vector and a pSICheck2 control or Stat3 3′ UTR and assayed after 48 h (N = 3 independent experiments, with n = 9 biological replicates, P < 0.0001 by paired t test)
MiR-223 targets the Stat3 3′ UTR
Using the web-based target prediction software program “microRNA” (http://www.microrna.org/microrna/home.do) to identify potential miR-223 targets, the transcription factor Stat3 was identified as a gene that could be negatively modulated by miR-223. Next, to determine whether Stat3 is a target of miR-223, we performed a luciferase assays using Stat3 3′ UTR. We found that miR-223 inhibited luciferase activity of a reporter containing Stat3 3′ UTR, but not that of an empty control reporter containing only GFP (Fig. 7e). Taken together, our data strongly link miR-223 to the STAT3-ARG1 pathway.
Decreased ARG1 and STAT3 in peripheral blood MDSCs form MS subjects compared to healthy controls
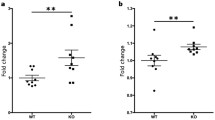
As reported above, RRMS patients display a significantly reduced number of total MDSCs and MO-MDSCs in peripheral blood compared to healthy controls and this was associated with an increased expression of miR-223 (Fig. 1a–f). To follow up on the increased expression of Arg1 and Stat3 in murine miR-223−/− MDCSs, we measured ARG1 and STAT3 expressions in human HLA-DR−CD33+ MDSCs sorted from peripheral blood of RRMS and age and gender-matched healthy control subjects (n = 8 and n = 10, respectively). Notably, the expression of ARG1 and STAT3 was significantly decreased in MDSCs from RRMS patients compared to healthy controls (mean fold change ± SD over controls for ARG1: 5.37 ± 5.9 vs. 1.22 ± 1.07, P = 0.03; STAT3: 0.77 ± 0.70 vs. 0.13 ± 0.13, P = 0.04) (Fig. 8a, b). These findings suggest that peripheral blood MDSCs in patients with RRMS are not only decreased in number, but might also be functionally impaired.

Lower ARG1 and STAT3 expressions in relapsing-remitting MS patients compared to healthy controls. a, b Scatter plot of the distribution of STAT3 and ARG1 levels in circulating MDSCs from RRMS patients (n = 8) and healthy controls (n = 10), respectively. Error bars represent the mean ± SD. P values were calculated by Mann–Whitney U test
Discussion
In the present study, we demonstrate that RRMS patients display reduced numbers of circulating suppressive MDSCs compared to healthy control subjects, with a more pronounced reduction in the MO-MDSC subset. Importantly, the expression of miR-223 was significantly increased in MDSCs from MS patients compared to healthy controls and this was associated with decreased expression of ARG1 and of the transcription factor STAT3. In parallel mouse studies, miR-223 deficiency led to reduced EAE severity and pathology which was associated with an increase in MDSC number in the spleen and CNS. Murine MO-MDSCs lacking miR-223 demonstrated increased suppressive function on T cell proliferation and cytokine production, with increased expression of Stat3 and Arg1 in mouse T cells. Furthermore, we provide evidence that Stat3 is a miR-223 target gene.
Although MS autoimmunity is thought to be primarily driven by T cells, myeloid-derived cells are the most abundant cells associated with CNS demyelinating lesions in MS [32]. Myeloid cells critically shape adaptive immune responses and the inflammatory environment during CNS autoimmunity [32]. MDSCs are a heterogeneous group of undifferentiated myeloid cells present in the bone marrow that during an inflammatory event can expand, circulate in the blood, and acquire ability to suppress T cell functions [32]. Immune-suppressive capability of MDSCs was described first in tumor-bearing mice [27], but MDSCs can also participate to the pathological processes of autoimmune diseases [32]. Published studies describe a role for MDSCs in the regulation of the immune responses during EAE [18, 31, 43, 48, 49]. Increased numbers of MO and PMN–MDSCs were reported in the spleen and blood in mice with EAE, and these cells were shown to gain access to the inflamed CNS [25, 31, 49]. Other investigators have shown that adoptive transfer of MO or PMN-MDSCs to mice primed to develop EAE ameliorated the clinical course and delayed disease onset. Improved clinical EAE was associated with decreased demyelination and inhibition of T cell responses with increased T cell apoptosis [18, 31].
Up to now, the role of MDSCs in MS has been poorly characterized. Here, we report that RRMS patients display significantly reduced numbers of circulating total MDSCs, with the main reduction in MO-MDSCs, compared to healthy controls. Treatment with GA moderately increased the number of MDSCs in peripheral blood, compared to untreated MS patients. Also noted was a trend toward lower numbers of PMN-MDSCs in MS. To our knowledge, ours is the first report that studied total MDSCs and MO-MDSCs in MS patients. Previous work by another group reported an enrichment of circulating PMN-MDSCs in MS patients with active disease, while no changes in their numbers were noted in RRMS patients during remission compared to healthy controls [18]. Different reasons could explain this discrepancy in regard of circulating PMN-MDSC frequencies in RRMS and healthy controls between this previous work and ours. These would include genetic differences in the cohorts studied, exposure to disease-modifying therapies, and/or possible differences in the isolation procedure or gating strategies for MDSCs. Herein, we have also demonstrated human MDSC suppressive activity on T cell proliferation in vitro. The analysis of the suppressive potential of peripheral blood MDSCs isolated from MS vs. healthy control subjects is an important future study to be done.
Decreased numbers of MDSCs and MO-MDSCs were associated with increased MDSC expression of miR-223 in MS patients compared to heathy controls. This is an important and novel finding suggesting the possible role of miR-223 in modulating MDSC biology in the context of autoimmunity. Dysregulation of several miRNAs has been observed in the blood and CNS tissues of MS compared to control subjects by microarray profiling [9, 42]. MiR-223 was found to be upregulated by microarray analysis in the whole blood [22] and in active brain lesions in MS patients compared to control subjects. A recent study showed the upregulation of miR-223 also in normal appearing white matter in MS patients [15]. MiR-223 modulates myeloid cell differentiation and number [8]. Specifically, it was shown to act as a negative regulator of granulocytic differentiation and activation in neutrophils [20] which leads to increased numbers of granulocyte/myeloid progenitors in the bone marrow of miR-223−/− mice [7, 20].
Our study shows that miR-223 expression is upregulated in the spinal cord of C57BL/6 mice with active EAE and it is highly expressed in myeloid cells isolated from the inflamed CNS. Furthermore, active EAE was attenuated clinically, histopathologically and with reduced frequencies of IFN-γ+, IL-17A+, and GM-CSF+ CNS-infiltrating CD4+ T cells in miR-223-deficient mice compared to littermate control mice. In contrast, when adoptive transfer EAE was induced by injecting encephalitogenic Th1 cells into WT and miR-223−/− recipient mice, we did not observe differences in the EAE clinical course between the two genotypes. In the setting of passive EAE, we analyzed MDSC accumulation in the spleen or CNS (day 15 post-transfer) and we did not observe differences between WT and miR-223−/− recipient mice in MDSC numbers as well as their suppressive function on T cell cytokine production (data not shown). This suggests that once WT T cells are primed against MOG35–55, lack of miR-223 does not interfere with subsequent effector events, including lymphocyte migration though the blood brain barrier, and re-activation in the CNS and subsequent CNS tissue damage. Therefore, we surmise that the effect of miR-223 deficiency on active EAE is very likely dependent on events leading to the generation of MDSCs during the peripheral phase of antigen-specific T cell activation. These suppressive cells could in turn express their modulatory potential on T cell functions either in the periphery and/or within the CNS, where they also accumulate during clinical disease. At 6 DPI, we observed increased accumulation of MDSCs in lymph nodes draining the site of immunization with significantly higher numbers of CD4+ T cells producing IL-17A in miR-223−/− compared to WT mice. This latter result was surprising given the well-known pathogenic role of IL-17A in EAE and difficult to reconcile with the fact that active EAE is attenuated in miR-223−/− mice. However, at EAE peak in the CNS, we observed a reduced number of Th17 cells in miR-223−/− compared to WT mice, which is in agreement with the observed reduced disease severity. Therefore, the higher numbers of IL-17A cells found in DLN in miR-223−/− did not result in increased CNS pathology, which could be explained by several different scenarios. The expansion of MDSCs in DLN at time of T cell priming in miR-223−/− mice could promote Th17 polarization as previously suggested in EAE [46] and be responsible for the higher numbers IL-17A producing T cells detected in DLNs before EAE onset in miR-223-deficient mice compared to WT. On the other end, MDSCs and T cells can reciprocally influence each other [33, 49]. In fact, IL-17A+CD4+ T cells can increase the suppressive activity of MDSCs through Arg1 upregulation [34]. MDSCs with enhanced suppressive activity might in turn lead to lower numbers of Th17 cells at later stages of EAE. Another possible scenario to explain the reduced clinical course despite increased numbers of Th17 cells generated in DLNs in miR-223−/− mice could be that the increased MDSC numbers led to the conversion of the Th17 cells into regulatory T cells (Tregs) [16], a subset of T cells capable of suppressing EAE [38]. Recently, it has been shown that Th17 cells can convert into Tregs [47]. Perhaps, this process is part of the mechanism of action of MDSCs in suppressing T cell functions [33]. In support of this hypothesis, we have preliminary results showing an increased number of CNS-infiltrating CD4+CD25+Foxp3+ T regs in the CNS during clinical EAE in miR-223−/− compared to WT mice (data not shown). The attenuation of active EAE in miR-223−/− mice was recently reported by another group [17]. Consistent with our results, they also found defective T cell priming when miR-223 is lacking and attributed this observation to a deficiency in T cell activation by myeloid dendritic cells (DCs). In contrast, our results suggest that miR-223 deficiency leads to defective T cell functions due to an expansion of MDSCs with enhanced suppressive activities.
Herein, we provide mechanistic information on the molecular pathways regulated by miR-223 in MDSCs. Furthermore, we provide evidence that Stat3 is a miR-223 target gene and lack of miR-223 in murine MDSCs is indeed associated with increased expression of Stat3 compared to WT cells. STAT3 plays important roles in the expansion and activities of MDSCs, as shown in several tumor models [5, 27]. Here, we speculate that miR-223 could also modulate MDSC accumulation in STAT3-mediated mechanisms in the setting of EAE and Arg1, a downstream STAT3 target gene and a potent T cell inhibitor [45]. In agreement with this notion, higher Stat3 expression in miR-223−/− murine MDSCs was associated with increased expression of Arg1 compared to the WT counterpart. This suggests that the hyper-activation of the STAT3-ARG1 pathway might be responsible for more potent immunosuppression by miR-223−/− compared to WT MDSCs. Taken together, these findings support the notion that miR-223 is a critical regulator of Arg1 and Stat3 genes which would impact on MDSC suppressive functions. Importantly, in this study, MDSCs isolated from RRMS had higher expression of miR-223 compared to those in healthy control subjects. This was associated with a decreased expression of STAT3 and ARG1. This inversely recapitulates what we have observed in the murine system and leads to the hypothesis that circulating MDSCs in RRMS are not only defective in number, but also in immunosuppressive capacity.
In conclusion, our study reports for the first time that the numbers of total circulating MDSCs and MO-MDSCs are reduced in RRMS patients compared to healthy controls and this was associated with higher MDSC expression of miR-223. Through validation in the murine MS model, we underscore the critical role of mir-223 in modulating MDSC numbers and suppressive activities, possibly through effects on STAT3 and Arg1 pathways. Understanding the specific molecular events that regulate MDSC number and function could lead to the design of new strategies of immune intervention in MS.
Abbreviations
- MS:
-
Multiple sclerosis
- miRNAs:
-
MicroRNAs
- MDSCs:
-
Myeloid-derived suppressor cells
- EAE:
-
Experimental autoimmune encephalomyelitis
- STAT3:
-
Signal transducer and activator of transcription 3
- ARG1:
-
Arginase-1
References
Ambros V (2004) The functions of animal microRNAs. Nature 431:350–355. doi:10.1038/nature02871
Bader AG, Brown D, Winkler M (2010) The promise of microRNA replacement therapy. Cancer Res 70:7027–7030. doi:10.1158/0008-5472.CAN-10-2010
Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK (2003) Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 197:1073–1081. doi:10.1084/jem.20021603
Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S et al (2016) Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 7:12150. doi:10.1038/ncomms12150
Condamine T, Ramachandran I, Youn JI, Gabrilovich DI (2015) Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med 66:97–110. doi:10.1146/annurev-med-051013-052304
De Santis G, Ferracin M, Biondani A, Caniatti L, Rosaria Tola M, Castellazzi M, Zagatti B, Battistini L, Borsellino G, Fainardi E et al (2010) Altered miRNA expression in T regulatory cells in course of multiple sclerosis. J Neuroimmunol 226:165–171. doi:10.1016/j.jneuroim.2010.06.009
Fazi F, Racanicchi S, Zardo G, Starnes LM, Mancini M, Travaglini L, Diverio D, Ammatuna E, Cimino G, Lo-Coco F et al (2007) Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell 12:457–466. doi:10.1016/j.ccr.2007.09.020
Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, Bozzoni I (2005) A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 123:819–831. doi:10.1016/j.cell.2005.09.023
Fenoglio C, Ridolfi E, Cantoni C, De Riz M, Bonsi R, Serpente M, Villa C, Pietroboni AM, Naismith RT, Alvarez E et al (2013) Decreased circulating miRNA levels in patients with primary progressive multiple sclerosis. Mult Scler 19:1938–1942. doi:10.1177/1352458513485654
Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH (2010) T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol 162:1–11. doi:10.1111/j.1365-2249.2010.04143.x
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12:253–268. doi:10.1038/nri3175
Gilicze AB, Wiener Z, Toth S, Buzas E, Pallinger E, Falcone FH, Falus A (2014) Myeloid-derived microRNAs, miR-223, miR27a, and miR-652, are dominant players in myeloid regulation. Biomed Res Int 2014:870267. doi:10.1155/2014/870267
Gimenez MA, Sim J, Archambault AS, Klein RS, Russell JH (2006) A tumor necrosis factor receptor 1-dependent conversation between central nervous system-specific T cells and the central nervous system is required for inflammatory infiltration of the spinal cord. Am J Pathol 168:1200–1209. doi:10.2353/ajpath.2006.050332
Guerau-de-Arellano M, Alder H, Ozer HG, Lovett-Racke A, Racke MK (2012) miRNA profiling for biomarker discovery in multiple sclerosis: from microarray to deep sequencing. J Neuroimmunol 248:32–39. doi:10.1016/j.jneuroim.2011.10.006
Guerau-de-Arellano M, Liu Y, Meisen WH, Pitt D, Racke MK, Lovett-Racke AE (2015) Analysis of miRNA in normal appearing white matter to identify altered CNS pathways in multiple sclerosis. J Autoimmune Disord 1:1–8
Hoechst B, Gamrekelashvili J, Manns MP, Greten TF, Korangy F (2011) Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood 117:6532–6541. doi:10.1182/blood-2010-11-317321
Ifergan I, Chen S, Zhang B, Miller SD (2016) Cutting edge: microRNA-223 regulates myeloid dendritic cell-driven Th17 responses in experimental autoimmune encephalomyelitis. J Immunol 196:1455–1459. doi:10.4049/jimmunol.1501965
Ioannou M, Alissafi T, Lazaridis I, Deraos G, Matsoukas J, Gravanis A, Mastorodemos V, Plaitakis A, Sharpe A, Boumpas D et al (2012) Crucial role of granulocytic myeloid-derived suppressor cells in the regulation of central nervous system autoimmune disease. J Immunol 188:1136–1146. doi:10.4049/jimmunol.1101816
Jager A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK (2009) Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol 183:7169–7177. doi:10.4049/jimmunol.0901906
Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, Brummelkamp TR, Fleming MD, Camargo FD (2008) Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 451:1125–1129. doi:10.1038/nature06607
Junker A, Hohlfeld R, Meinl E (2011) The emerging role of microRNAs in multiple sclerosis. Nat Rev Neurol 7:56–59. doi:10.1038/nrneurol.2010.179
Junker A, Krumbholz M, Eisele S, Mohan H, Augstein F, Bittner R, Lassmann H, Wekerle H, Hohlfeld R, Meinl E (2009) MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain J Neurol 132:3342–3352. doi:10.1093/brain/awp300
Keller A, Leidinger P, Lange J, Borries A, Schroers H, Scheffler M, Lenhof HP, Ruprecht K, Meese E (2009) Multiple sclerosis: microRNA expression profiles accurately differentiate patients with relapsing-remitting disease from healthy controls. PLoS One 4:e7440. doi:10.1371/journal.pone.0007440
Kiernan JA (1984) Chromoxane cyanine R. II. Staining of animal tissues by the dye and its iron complexes. J Microsc 134:25–39
King IL, Dickendesher TL, Segal BM (2009) Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood 113:3190–3197. doi:10.1182/blood-2008-07-168575
Kotsakis A, Harasymczuk M, Schilling B, Georgoulias V, Argiris A, Whiteside TL (2012) Myeloid-derived suppressor cell measurements in fresh and cryopreserved blood samples. J Immunol Methods 381:14–22. doi:10.1016/j.jim.2012.04.004
Kumar V, Patel S, Tcyganov E, Gabrilovich DI (2016) The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol 37:208–220. doi:10.1016/j.it.2016.01.004
Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20. doi:10.1016/j.cell.2004.12.035
Li T, Morgan MJ, Choksi S, Zhang Y, Kim YS, Liu ZG (2010) MicroRNAs modulate the noncanonical transcription factor NF-kappaB pathway by regulating expression of the kinase IKKalpha during macrophage differentiation. Nat Immunol 11:799–805. doi:10.1038/ni.1918
Liu Q, Zhang M, Jiang X, Zhang Z, Dai L, Min S, Wu X, He Q, Liu J, Zhang Y et al (2011) miR-223 suppresses differentiation of tumor-induced CD11b(+) Gr1(+) myeloid-derived suppressor cells from bone marrow cells. Int J Cancer 129:2662–2673. doi:10.1002/ijc.25921
Moline-Velazquez V, Cuervo H, Vila-Del Sol V, Ortega MC, Clemente D, de Castro F (2011) Myeloid-derived suppressor cells limit the inflammation by promoting T lymphocyte apoptosis in the spinal cord of a murine model of multiple sclerosis. Brain Pathol 21:678–691. doi:10.1111/j.1750-3639.2011.00495.x
Moline-Velazquez V, Vila-Del Sol V, de Castro F, Clemente D (2016) Myeloid cell distribution and activity in multiple sclerosis. Histol Histopathol 31:357–370. doi:10.14670/HH-11-699
Nagaraj S, Youn JI, Gabrilovich DI (2013) Reciprocal relationship between myeloid-derived suppressor cells and T cells. J Immunol 191:17–23. doi:10.4049/jimmunol.1300654
Novitskiy SV, Pickup MW, Gorska AE, Owens P, Chytil A, Aakre M, Wu H, Shyr Y, Moses HL (2011) TGF-beta receptor II loss promotes mammary carcinoma progression by Th17 dependent mechanisms. Cancer Discov 1:430–441. doi:10.1158/2159-8290.CD-11-0100
O’Connell RM, Zhao JL, Rao DS (2011) MicroRNA function in myeloid biology. Blood 118:2960–2969. doi:10.1182/blood-2011-03-291971
Parekh VV, Wu L, Olivares-Villagomez D, Wilson KT, Van Kaer L (2013) Activated invariant NKT cells control central nervous system autoimmunity in a mechanism that involves myeloid-derived suppressor cells. J Immunol 190:1948–1960. doi:10.4049/jimmunol.1201718
Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V (2010) Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol 22:238–244. doi:10.1016/j.coi.2010.01.021
Piccio L, Cantoni C, Henderson JG, Hawiger D, Ramsbottom M, Mikesell R, Ryu J, Hsieh CS, Cremasco V, Haynes W et al (2013) Lack of adiponectin leads to increased lymphocyte activation and increased disease severity in a mouse model of multiple sclerosis. Eur J Immunol 43:2089–2100. doi:10.1002/eji.201242836
Piccio L, Stark JL, Cross AH (2008) Chronic calorie restriction attenuates experimental autoimmune encephalomyelitis. J Leukoc Biol 84:940–948. doi:10.1189/jlb.0208133
Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, Fujihara K, Havrdova E, Hutchinson M, Kappos L et al (2011) Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 69:292–302. doi:10.1002/ana.22366
Procaccini C, De Rosa V, Pucino V, Formisano L, Matarese G (2015) Animal models of multiple sclerosis. Eur J Pharmacol 759:182–191. doi:10.1016/j.ejphar.2015.03.042
Ridolfi E, Fenoglio C, Cantoni C, Calvi A, De Riz M, Pietroboni A, Villa C, Serpente M, Bonsi R, Vercellino M et al (2013) Expression and genetic analysis of microRNAs involved in multiple sclerosis. Int J Mol Sci 14:4375–4384. doi:10.3390/ijms14034375
Rumble JM, Huber AK, Krishnamoorthy G, Srinivasan A, Giles DA, Zhang X, Wang L, Segal BM (2015) Neutrophil-related factors as biomarkers in EAE and MS. J Exp Med 212:23–35. doi:10.1084/jem.20141015
Sullivan RP, Leong JW, Schneider SE, Ireland AR, Berrien-Elliott MM, Singh A, Schappe T, Jewell BA, Sexl V, Fehniger TA (2015) MicroRNA-15/16 antagonizes Myb to control NK cell maturation. J Immunol 195:2806–2817. doi:10.4049/jimmunol.1500949
Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, Blosser RL, Tam AJ, Bruno T, Zhang H et al (2013) STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Investig 123:1580–1589. doi:10.1172/JCI60083
Yi H, Guo C, Yu X, Zuo D, Wang XY (2012) Mouse CD11b+ Gr-1+ myeloid cells can promote Th17 cell differentiation and experimental autoimmune encephalomyelitis. J Immunol 189:4295–4304. doi:10.4049/jimmunol.1200086
Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY et al (2008) TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 453:236–240. doi:10.1038/nature06878
Zhu B, Bando Y, Xiao S, Yang K, Anderson AC, Kuchroo VK, Khoury SJ (2007) CD11b+ Ly-6C(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J Immunol 179:5228–5237
Zhu B, Kennedy JK, Wang Y, Sandoval-Garcia C, Cao L, Xiao S, Wu C, Elyaman W, Khoury SJ (2011) Plasticity of Ly-6C(hi) myeloid cells in T cell regulation. J Immunol 187:2418–2432. doi:10.4049/jimmunol.1100403
Acknowledgments
We thank Anne H. Cross, MD for careful reading of the manuscript; Julia Sim and Angela Archambault, PhD for technical assistance and advices; Erin Longbrake, MD PhD for helping with patient enrollment in the study; all MS patients and healthy controls that donated blood for this project as well as the study coordinators that drew the blood: Samantha Lancia, Susan Fox, and Bridgette Clay. LP is a Harry Weaver Neuroscience Scholar of the National Multiple Sclerosis Society (NMSS, JF 2144A2/1) and supported by Fondazione Italiana Sclerosi Multipla (FISM; 2014/R/15). GFW was supported by R01NS083678. LP and GFW were funded by the Dana Foundation “Program in the Neuroimmunology and Brain Infections and Cancer”. CC was supported during the course of this study by a FISM fellowship (2012/B/1) and subsequently by a NMSS fellowship (FG 2010-A1/2). TAF was supported by R01AI102924. Patients were seen for this study in the Neuroclinical Research Unit (NCRU) supported by the National Institute of Health (CO6 RR020092) and Washington University Insitute of Clinical and Translational Sciences-Brain Behavioral and Performance Unit (TR000448).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
401_2016_1621_MOESM1_ESM.tif
Supplementary Fig. 1. Percentages of MDSCs and MO-MDSCs in the peripheral blood of healthy control subjects (n = 16), untreated and GA-treated RRMS patients (n = 10 and n = 24). a, b Percentages of MDSCs and MO-MDSCs were significantly lower in untreated RRMS patients compared to healthy controls. GA-treated patient showed a modest increase in MDSC and MO-MDSC percentages, but this was not statistically significant. Percentages were calculated relative to a gate which included whole leukocytes. Error bars represent the mean ± SD. P values were calculated by Kruskal–Wallis H test and post hoc analysis (TIFF 9217 kb)
401_2016_1621_MOESM2_ESM.tif
Supplementary Fig. 2. Mir-223 expression in the CNS and lymphoid tissues in C57BL/6 mice during EAE. MiR-223 expression in spleen, brain, lymph nodes, and bone marrow taken from C57BL/6 naïve mice and on days 5, 10 15, and 30 post-EAE immunization (n = 4 mice/time point) analyzed by quantitative RT-PCR. U6 snRNA was used as endogenous controls (TIFF 44468 kb)
401_2016_1621_MOESM3_ESM.tif
Supplementary Fig. 3. Size and cellularity of WT and miR-223−/− lymph node draining the immunization. a At day 6, post-immunization WT and miR-223−/− lymph node draining the site of immunization were isolated and measured in size. b Scatter plot of the percentage of myeloid cells (defined as CD11b+Gr1+ cells) from WT and miR-223−/− lymph nodes. Error bars represent mean ± SEM. A representative experiment from a total of three performed is showed. P values were calculated by Mann–Whitney U test (TIFF 18680 kb)
401_2016_1621_MOESM4_ESM.tif
Supplementary Fig. 4. Absolute number of MO and PMN-MDSCs in naive WT and miR-223−/− mice. Cells were isolated from bone marrow, spleen, blood, and lymph nodes from naive WT and miR-223−/− mice and analyzed by flow cytometry. Gates were set on Zombie−CD45+CD11b+ cells; MO-MDSCs were defined as CD11b+Ly6Chigh cells, PMN-MDSCs as CD11b+Gr1+. Absolute numbers of MO and PMN-MDSCs were calculated from the percentages based on the total number of cells (bone marrow, spleen, and lymph nodes) or on the total number of cells per 100 μl of blood. Error bars represent the mean ± SEM. A representative experiment from a total of two is showed. P values were calculated by Mann–Whitney U test (TIFF 24193 kb)
401_2016_1621_MOESM5_ESM.tif
Supplementary Fig. 5. Characterization of surface markers expression and cytokine profile of MDSCs from immunized WT and miR-223−/− mice. a, b MO and PMN-MDSCs in the spleen and CNS-infiltrating cells from MOG35–55 immunized mice (15 DPI) were examined for the expression of MHC II, CD40, CD86, and PD-L1 by flow cytometry. Expression of individual markers is shown based on gating for MO- (CD11b+Ly6G−Ly6Chigh) and PMN-MDSCs (CD11b+Ly6G+Ly6Clow). Histograms for the different surface markers on MO and PMN-MDSCs in the spleen (a) and in the CNS (b) are referring to representative mice from each group. Red and blue lines indicate mean fluorescence intensity (MFI) in WT and miR-223−/−, respectively. Scatter plots are compiling the data from one experiment (four mice/group) shown as percentages of MO and PMN-MDSCs expressing the specific marker. c Analysis of cytokine (TNF-α, IL-10, and GM-CSF) production in splenic MO and PMN-MDSCs from WT and miR-223−/− mice at 15 DPI. Numbers indicate the percentage of cytokine-producing MO and PMN-MDSCs. Error bars are mean ± SEM. P values were calculated by Mann–Whitney U test (TIFF 153485 kb)
Rights and permissions
About this article

Cite this article
Cantoni, C., Cignarella, F., Ghezzi, L. et al. Mir-223 regulates the number and function of myeloid-derived suppressor cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol 133, 61–77 (2017). https://doi.org/10.1007/s00401-016-1621-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-016-1621-6