
Abstract
Several diverse proteins are linked genetically/pathologically to neurodegeneration in amyotrophic lateral sclerosis (ALS) including SOD1, TDP-43 and FUS. Using a variety of cellular and biochemical techniques, we demonstrate that ALS-associated mutant TDP-43, FUS and SOD1 inhibit protein transport between the endoplasmic reticulum (ER) and Golgi apparatus in neuronal cells. ER–Golgi transport was also inhibited in embryonic cortical and motor neurons obtained from a widely used animal model (SOD1G93A mice), validating this mechanism as an early event in disease. Each protein inhibited transport by distinct mechanisms, but each process was dependent on Rab1. Mutant TDP-43 and mutant FUS both inhibited the incorporation of secretory protein cargo into COPII vesicles as they bud from the ER, and inhibited transport from ER to the ER–Golgi intermediate (ERGIC) compartment. TDP-43 was detected on the cytoplasmic face of the ER membrane, whereas FUS was present within the ER, suggesting that transport is inhibited from the cytoplasm by mutant TDP-43, and from the ER by mutant FUS. In contrast, mutant SOD1 destabilised microtubules and inhibited transport from the ERGIC compartment to Golgi, but not from ER to ERGIC. Rab1 performs multiple roles in ER–Golgi transport, and over-expression of Rab1 restored ER–Golgi transport, and prevented ER stress, mSOD1 inclusion formation and induction of apoptosis, in cells expressing mutant TDP-43, FUS or SOD1. Rab1 also co-localised extensively with mutant TDP-43, FUS and SOD1 in neuronal cells, and Rab1 formed inclusions in motor neurons of spinal cords from sporadic ALS patients, which were positive for ubiquitinated TDP-43, implying that Rab1 is misfolded and dysfunctional in sporadic disease. These results demonstrate that ALS-mutant forms of TDP-43, FUS, and SOD1 all perturb protein transport in the early secretory pathway, between ER and Golgi compartments. These data also imply that restoring Rab1-mediated ER–Golgi transport is a novel therapeutic target in ALS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterised by degeneration and death of motor neurons. Multiple proteins are linked genetically to ALS, including superoxide dismutase 1 (SOD1) [55], TAR DNA binding protein (TDP-43) [68], and Fused in Sarcoma (FUS) [78]. TDP-43 and FUS are also associated with frontotemporal dementia and misfolded wildtype (WT) SOD1 is present in small granular aggregates in glia and motor neuron nuclei, although it has not been detected in the typical ubiquitinated TDP-43 pathological inclusions [13, 19, 20, 35]. Transgenic mice overexpressing mutant SOD1G93A develop features characteristic of ALS and are a widely used disease model. Many cellular defects have been implicated in the aetiology of ALS, including protein aggregation, endoplasmic reticulum (ER) stress, autophagy defects, RNA dysfunction and inhibition of axonal transport [54].
Efficient intracellular vesicle trafficking is essential for cellular survival. Proteins newly synthesised in the ER are packed into vesicles and transported to the Golgi apparatus via the ER–Golgi intermediate compartment (ERGIC) [37], and finally redistributed to their final destinations [15]. Hence ER–Golgi transport is a vital gateway to the endomembrane system. The ERGIC is a distinct organelle from the ER and cis-Golgi that concentrates and sorts protein cargo [1]. Functional ER–Golgi transport relies on coat protein complexes (COPs), that recruit cargo proteins [4], and deform the lipid bilayer of donor membranes into vesicles [43, 67]. COPII is essential for export from ER exit sites, and is composed of the GTPase Sar1 and two hetero-dimeric complexes, Sec23/Sec24 and Sec13/Sec31 [4, 38]. COPII vesicles move from the ER to ERGIC, and subsequently from ERGIC to Golgi. The latter, but not the former, step requires microtubules, comprised of tubulin [33]. Finally, COPII vesicles are docked via tethering factor p115 to receptor GM130, on the Golgi membrane.
Rab GTPases are master regulators of all intracellular vesicle trafficking events, and each Rab isoform has distinct target membranes [69]. Rab1 regulates ER–Golgi transport, including COPII vesicle budding, delivery, tethering, fusion to the Golgi [47, 56] and COPII function [63]. In yeast, the Rab1 homologue Ypt1 also mediates microtubule organisation and function, and loss of Ypt1 function results in microtubule defects [61]. Furthermore, Rab1/Ypt1 plays a central role in regulating the unfolded protein response (UPR), suggesting a regulatory mechanism linking vesicle trafficking to the UPR and ER homeostasis [75]. Inhibition of ER–Golgi transport also induces ER stress [48], providing a further link to the UPR.
Previously, we demonstrated that mutant TDP-43 (mTDP-43) [79] and mutant FUS (mFUS) [16] induce ER stress by an undefined mechanism. We also showed that mutant SOD1 (mSOD1) and aggregated WTSOD1 inhibit ER–Golgi transport, and consequently trigger ER stress from the cytoplasm, in cellular models of ALS [3, 72], although the molecular mechanisms involved remains unclear. Here, we demonstrate that mTDP-43, and mFUS also inhibit ER–Golgi transport. Furthermore, we also show that mTDP-43 and mFUS inhibit transport from the ER to ERGIC compartment, by preventing the incorporation of protein cargo into COPII vesicles. However, whilst mFUS was present within the ER, mTDP-43 was attached to the cytoplasmic face of the ER membrane, implying that mTDP-43 and mFUS inhibit transport by discreet mechanisms. In contrast, mSOD1 destabilised microtubules and inhibited transport from the ERGIC compartment to Golgi. Hence, these proteins each inhibit transport by different mechanisms. However, these processes are all Rab1-dependent, demonstrating that antagonism of Rab1 function is a common target shared by ALS-associated forms of SOD1, TDP-43 and FUS. Furthermore, overexpression of Rab1 restored ER–Golgi transport and reduced ER stress, mSOD1 inclusion formation and apoptosis in cells expressing mSOD1, mTDP-43 or mFUS, thus linking ER–Golgi transport inhibition to neurodegeneration. ER–Golgi transport was also inhibited in embryonic cortical and motor neurons obtained from SOD1G93A mice, thus validating this pathological mechanism in primary neurons and as a very early event in disease pathology. Moreover, Rab1 was also recruited to inclusions in spinal motor neurons displaying typical, ubiquitinated TDP-43 pathology in sporadic ALS (sALS) patients, thus implying that Rab1 misfolding and dysfunction is present in sporadic disease.
Materials and methods
Additional materials and methods can be found in Supplementary Materials.
VSVG assay to quantify ER–Golgi transport
Neuro2a cells were plated on 24-well plates with 13 mm coverslips. The following day, cells were co-transfected with TDP-43, FUS, or SOD1 and VSVG-tagged with fluorescent mCherry for indicated time points. Cells were incubated at 40 °C directly after transfection except in the case of the 72 h transfection experiments, where cells were first incubated at 37 °C for 48 h. The temperature was then shifted to 40 °C for a further 24 h after transfection to accumulate VSVG in the ER. Cycloheximide (Sigma, 01810, 20 µg/ml) was added and cells were shifted to the permissive temperature, 32 °C for 30 min. At each time interval, cells were washed with ice-cold PBS and fixed for immunocytochemistry as described. Twenty cells were scored in each experiment and all experiments were performed in triplicate. Image analysis was performed using Image J (http://rsbweb.nih.gov/ij/index.html): only single cells expressing both SOD1-EGFP/EGFP-TDP-43/HA-FUS and VSVG-mCherry were selected for analysis. Plugins were used and the measuring areas were selected above a threshold against background staining. After analysis, the Mander’s coefficient [36] in the range from 0 to 1.0 (representing 0–100 % overlapping pixels) was calculated to determine the degree of overlap between images. For Fig. 1e, the x axis values; 80, 40–80 and 40; refer to arbitrary units that represent the total pixel intensity quantified in each cells by using Image J. In each population, cells were separated into 3 categories, so a third of cells with the lowest pixel intensity were categorised as ‘low’, the next third was categorised as ‘medium’ and the remaining third were categorised as ‘high’ representing those cells with the greatest pixel intensities.

ER–Golgi transport is inhibited in cells expressing mTDP-43, mFUS and mSOD1. a Representative fluorescence images and z-stack orthogonal views of cells expressing EGFP-tagged TDP-43 and VSVG-mCherry stained with markers of ER (calnexin) or cis-Golgi (GM130). The degrees of co-localisation of VSVG with calnexin or GM130 in cells expressing b TDP-43, c FUS, and d SOD1 were quantified using Mander’s coefficient. e Cells expressing mSOD1 A4V or G85R (top), mTDP-43 Q343R (bottom left) or mFUS R522G (bottom right) were separated into intensity bins as shown on the x axis, and their Mander’s coefficients (VSVG and calnexin or GM130) were plotted. Mean ± SEM, n = 3. ***p < 0.001, *p < 0.05 difference with TDP-43 WT, EGFP vector alone or control untransfected cells (Untr). One-way ANOVA with Tukey’s post hoc test
In vitro ER-budding assay
A modified in vitro assay [81] was used to analyse ER vesicle budding. Briefly, perforated Neuro2a cells co-transfected with VSVG-mCherry and SOD1, TDP-43 or FUS vectors were incubated with rat liver cytosol and an energy regenerating system (40 mM creatine phosphate, 0.2 mg/ml creatine phosphokinase and 1 mM ATP) at 32 °C for 30 min. Identical samples were incubated at 4 °C as a measure of non-specific ER fragmentation. The cells were removed by low speed centrifugation at 4000g for 1 min, followed by 15,000g for 1 min, and budded vesicles in the resulting supernatant were recovered by centrifugation at 100,000g for 1 h. The levels of VSVG cargo in the budded vesicle fractions were quantified using western blotting. The resulting quantities of budded vesicles were normalised to the levels of ERGIC53 from each sample.
Fluorescence protease protection assay
The fluorescence protease protection assay was performed as described previously [34]. Briefly, Neuro2a cells were transfected with the indicated plasmids 18 h before analysis. Cells were washed three times with KHM buffer (110 mM potassium acetate, 20 mM Hepes, 2 mM MgCl2 in H2O) for 1 min each wash. Digitonin (Sigma), at 55 % purity, was dissolved in H2O by heating to 95–98° for 10 mg/ml stock. KHM buffer was removed and 1 ml of KHM buffer with 60 µM digitonin was added to cells. Fluorescence images were captured at regular intervals with ¼ s exposure; fluorescence exposure outside of this capture period was kept to a minimum to prevent photobleaching. Buffer was removed at 110 s after digitonin addition, and cells were washed briefly with KHM buffer. Proteinase-K (Qiagen, Victoria, Australia, stock 20 mg/ml) at 50 µg/ml in KHM buffer was added to cells. At 330 s, 1 % Triton-x-100 with proteinase-K in KHM buffer was added to the cells, and fluorescence images were captured at regular intervals using identical settings between samples.
Statistical analysis
All data are expressed as the mean ± standard error (SEM) and analysed for statistical significance by ANOVA followed by Tukey’s post hoc test or 2-tailed student t test (GraphPad Prism, La Jolla, CA). The differences were considered significant at p < 0.05.
Results
ER–Golgi transport is inhibited in cells expressing proteins associated with ALS
Vesicular stomatitis virus glycoprotein-ts045 (VSVG) is a widely used marker for ER–Golgi trafficking. At 40 °C, VSVG reversibly misfolds and accumulates in the ER; traffic to the Golgi is restored at 32 °C [25]. To examine whether mTDP-43 and mFUS inhibit ER–Golgi transport, Neuro2a cells were co-transfected with mCherry-tagged VSVG and either EGFP-tagged WTTDP-43 or mTDP-43 (A315T/Q343R/Q331K) or HA-tagged WTFUS or mFUS (P525L/R524S/R522G/R521G) constructs at 40 °C. Localisation of VSVG with the ER (calnexin) or Golgi (GM130) was detected immunocytochemically and quantified using Mander’s coefficient as previous [3]. In cells expressing WTTDP-43, EGFP, or untransfected cells, VSVG was efficiently transported to the Golgi, in contrast to mTDP-43 cells, where most VSVG remained in the ER (66, 61 and 47 %, respectively, p < 0.0001, Fig. 1a, b) and less transported to the Golgi (35, 42 and 48 %, respectively, p < 0.0001, Fig. 1b). Thus, three mTDP-43 proteins antagonise anterograde transport of VSVG from ER–Golgi. Immunoblotting revealed similar transfection efficiencies between EGFP, WTTDP-43 and mTDP-43 cells (Suppl. Fig. 1a), demonstrating that transport inhibition is independent of protein expression. Similarly, in cells expressing WTFUS or untransfected cells, VSVG was located predominantly in the Golgi (Suppl. Fig. 2a). In contrast, in cells expressing all four mFUS proteins, significantly less VSVG was transported from ER to Golgi (60 %, p < 0.0001 and p < 0.05 in R521G; Fig. 1c and Suppl. Fig. 2a). Again, similar transfection efficiencies were evident between WTFUS and mFUS (Suppl. Fig. 1b). Hence four ALS-associated mFUS proteins also inhibit ER–Golgi transport. Similarly, consistent with our previous studies in NSC-34 and SYSH5Y cells [3, 72], mSOD1 also inhibited transport of VSVG from ER to Golgi in Neuro2a cells (Fig. 1d), with similar transfection efficiencies (Suppl. Fig. 1c). The inhibition of ER–Golgi transport was not caused by non-specific over-expression of recombinant, mutant protein, because expression of mutant R311K Nck2 (Nck adaptor protein 2), a cytoplasmic adaptor protein [70] not previously linked to neurodegeneration, did not inhibit ER–Golgi transport (Suppl Fig. 2b).
To further confirm that mSOD1, mTDP-43 and mFUS specifically inhibit ER–Golgi transport, we purposefully classified individual cells according to three categories of fluorescence intensity and hence SOD1/TDP-43/FUS expression. The arbitrary values of low (~20) to high (~100) were calculated according to the pixel intensities, representing the levels of expression of SOD1, TDP-43 or FUS protein. Inhibition of transport correlated with protein expression level: cells with the highest expression of mSOD1 A4V, mSOD1 G85R, Q343R TDP-43 or R522G FUS inhibited ER–Golgi transport the greatest according to Mander’s coefficient, and transport inhibition decreased with decreasing expression of mTDP-43 or mFUS (Fig. 1e). Hence the degree of transport inhibition correlates with protein expression level, confirming that ER–Golgi transport is inhibited specifically by mTDP-43, mFUS and mSOD1.
COPII vesicles are not transported to the Golgi in cells expressing mTDP-43, mFUS and mSOD1
VSVG depends on an over-expressed, non-physiological marker; hence we next sought to validate these findings using alternative approaches. We first examined bulk protein secretion in Neuro2a cells by quantifying the levels of total protein secreted into conditioned medium. Bulk protein secretion was less in cells expressing mTDP-43, mFUS or mSOD1 compared to control cells expressing WT proteins or EGFP alone (Suppl. Fig. 2c). Hence, consistent with the VSVG assay results, bulk protein secretion was inhibited in cells expressing mTDP-43, mFUS and mSOD1. However, defects in bulk protein secretion could also result from post-cis Golgi trafficking defects or dysfunction in other secretory processes. Thus, to provide further evidence for inhibition of ER–Golgi transport, we next examined COPII function in cells, which is easily visualised by dense clustering of COPII subunits adjacent to the perinuclear Golgi [23]. In untransfected cells, and cells expressing EGFP, WTSOD1, WTTDP-43 or WTFUS, COPII (Sec31) displayed the characteristic perinuclear pattern (Suppl. Fig. 3). In cells expressing mTDP-43, this pattern was lost, leaving a scattered peripheral pool of COPII (Suppl. Fig. 3a). Similar results were obtained in mFUS or mSOD1 cells (Suppl. Fig. 3b, c). Quantification revealed that perinuclear Sec31 was significantly decreased from 83–95 % in WTTDP-43, EGFP or untransfected cells, to 60 % in cells expressing mTDP-43 (p < 0.001, Suppl. Fig. 3d); from 82–93 % in WTFUS expressing or untransfected cells to 40–65 % in cells expressing mFUS (p < 0.05, Suppl. Fig. 3e); and from 75–95 % in WTSOD1, EGFP or untransfected cells to 45–47 % in cells expressing mSOD1 (p < 0.01, Suppl. Fig. 3f). These findings suggest that the organisation of COPII vesicles and the Golgi complex are abnormal in cells expressing mSOD1, mTDP-43 or mFUS, consistent with the presence of a block in ER–Golgi transport.
Secretory cargo and COPII are depleted from ER-derived vesicles in cells expressing mTDP-43 and mFUS
We next investigated possible mechanisms responsible for inhibition of ER–Golgi transport in ALS. We examined the first stage of transport, the incorporation of secretory cargo into COPII vesicles and budding from the ER, using an in vitro ER budding assay [49]. Sec23 is a marker of ER-budded vesicles and ERGIC is a marker of the ERGIC compartment that also localises on budded COPII vesicles. Budded vesicles were recovered and the VSVG content was analysed by quantitative western blotting (Fig. 2a, b, c). In untransfected cells and cells expressing EGFP or WTTDP-43, a similar proportion of VSVG was recovered in the budded vesicle fraction, similar to COPII (Sec23) (Fig. 2a, d). In contrast, in cells expressing Q343R TDP-43, VSVG was almost depleted (9-fold decrease, p < 0.0001, Fig. 2a, d). Immunoblotting revealed similar levels of ERGIC53 in all fractions, implying that vesicle number was consistent in all populations. However immunoblotting for COPII (Sec23), normalised to the levels of ERGIC53, revealed that COPII was also significantly depleted in Q343R TDP-43 vesicle fractions compared to WTTDP-43 and controls (1.4-fold decrease, p < 0.001, Fig. 2a, d). Similar results were obtained in cells expressing mFUS. A comparable proportion of VSVG was present in budded vesicles obtained from untransfected cells and cells expressing WTFUS. However, this proportion was significantly reduced in cells expressing R522G mFUS (2-fold, p < 0.05, Fig. 2b, e). COPII levels on the budded vesicles were also significantly decreased in cells expressing R522G FUS (1.25-fold, p < 0.05, Fig. 2b, e). Immunoblotting of total cell lysates confirmed that there were no differences in the overall expression of VSVG and COPII between cell populations (Suppl. Fig. 1d). Hence these data suggest that defects in incorporation of membrane-associated cargo into budding ER vesicles inhibit ER–Golgi transport in cells expressing mTDP-43 or mFUS.

mTDP-43 and mFUS have an inhibitory effect on ER vesicle budding, but not mSOD1. VSVG-mCherry was trapped in the ER and ER-vesicles budding was reconstituted in vitro in perforated cells expressing a EGFP-TDP-43, b HA-FUS or c SOD1-EGFP. Budded vesicles were collected and subjected to western blotting using anti-VSVG and anti-COPII (Sec23) antibodies. ERGIC53 was used for control of equivalent budded vesicles. Relative intensities of VSVG and COPII (Sec23) were quantified in cells expressing d EGFP-TDP-43, e HA-FUS or f SOD1-EGFP, first normalised to ERGIC53 intensity for the relevant lane, and then normalised again to the control untransfected cells (Untr). Mean ± SEM (n = 3). **p < 0.01, *p < 0.05 difference with untransfected cells. Cells expressing g EGFP-TDP-43, h HA-FUS or i SOD1-EGFP were treated as in Fig. 1 and immunostained with ERGIC53. The degrees of co-localisation of VSVG with ERGIC53 in cells were quantified using Mander’s coefficient. j Cells expressing SOD1-EGFP were treated as in Fig. 1 and immunostained with ERGIC53 or cis-Golgi marker, GM130. The degrees of co-localisation of VSVG with ERGIC53 or GM130 in cells were quantified using Mander’s coefficient. Mean ± SEM, n = 3. ***p < 0.001, **p < 0.01, *p < 0.05 difference with WT protein transfected cells. One-way ANOVA with Tukey’s post hoc test
In contrast, in cells expressing mSOD1, there were no significant differences in the proportion of VSVG or COPII associated with budded vesicles compared to untransfected cells or cells expressing EGFP or WTSOD1 (Fig. 2c, f). The expression levels of VSVG and COPII were also similar between the cell populations (Suppl. Fig. 1d), suggesting that secretory cargo incorporates normally into ER-derived vesicles in mSOD1 cells.
To verify that less secretory cargo incorporates into vesicles in cells expressing mTDP-43 or mFUS, we examined incorporation of VSVG into COPII vesicles by immunocytochemistry (Suppl. Fig. 4). Mander’s coefficient between VSVG and ERGIC53 was similar in untransfected cells and cells expressing EGFP alone or WTTDP-43. However, significantly less VSVG co-localised with ERGIC53 in cells expressing mTDP-43 or mFUS compared to WTTDP-43, WTFUS or untransfected cells (p < 0.05, Fig. 2g, h and Suppl. Fig. 4a, b). Hence vesicular cargo does not incorporate normally into COPII vesicles in mTDP-43 or mFUS expressing cells. In contrast, there were no significant differences in Mander’s coefficient between cells expressing mSOD1 and WTSOD1, EGFP or untransfected cells (Fig. 2i and Suppl. Fig. 4c); however, less VSVG co-localised with GM130 in cells expressing mSOD1 compared to WTSOD1 or untransfected cells (Fig. 2j). This provides further evidence that incorporation of secretory cargo into COPII vesicles, and their budding from the ER and transport to ERGIC, is normal in cells expressing mSOD1. Hence this implies that inhibition of ER–Golgi transport by mSOD1 is downstream of the ERGIC compartment.
TDP-43 and FUS are associated with the ER
The depletion of cargo after budding from the ER suggests dysfunction to the ER in mTDP-43 and mFUS expressing cells. Hence we next examined whether TDP-43 and FUS are present within the ER. Using immunocytochemistry and z-stack series confocal imaging, we demonstrated that mTDP-43 and mFUS partially co-localised with ER marker, calnexin, suggesting that at least a proportion of mTDP-43 and mFUS are localised in the ER (Fig. 3a). Calculation of Mander’s coefficient also revealed increased co-localization between calnexin and mTDP-43 or mFUS, compared to WTTDP-43 or WTFUS (Fig. 3b, c), implying that the mutants associated more with the ER. Similarly, subcellular fractionation experiments demonstrated that endogeneous TDP-43 and FUS were present in the membrane fraction (containing the ER), as well as nuclear and cytoplasmic fractions, enriched in IRE1, Histone H3 and GAPDH, respectively (Fig. 3d). Both WT and mutant forms of TDP-43 and FUS were enriched in nuclear and membrane fractions but only slightly in the cytoplasmic fraction (Fig. 3d). To investigate this further, a fluorescence protease protection assay was performed, in which proteins contained within cellular membranes are protected from proteinase K after digitonin treatment [34] (Fig. 3e). In EGFP only expressing cells, the fluorescence disappeared following digitonin treatment, demonstrating that EGFP was expressed in the cytoplasm. In contrast, in cells expressing Dsred-tagged protein disulphide isomerase (PDI), a chaperone located in the ER of unstressed cells [82], the fluorescence was unchanged after extended proteinase K treatment (Fig. 3e). In cells expressing EGFP-TDP-43 (WT or Q343R), the fluorescence was retained after digitonin treatment, demonstrating that TDP-43 is associated with membranes, but it disappeared after addition of proteinase-K. Hence together with the calnexin immunocytochemistry results (Fig. 3a), these findings imply TDP-43 is present on the cytoplasmic face of the ER membrane. In contrast, in cells expressing GFP-FUS (WT or R521G), the fluorescence was retained after both digitonin and proteinase-K treatment (Fig. 3e). To confirm these findings, and to ascertain that the fluorescence was due to membrane bound protein, rather than misfolded cytosolic protein, Triton-x-100 was added as a final step. In cells expressing either WT or mFUS the fluorescence signal disappeared completely, similar to control cells expressing PDI-DsRed. Hence these data imply that both WT and mFUS are present within the ER lumen, similar to PDI (Fig. 3f).

TDP-43 is expressed on the ER membrane and FUS is located inside the ER. a Neuro2a cells expressing EGFP-TDP-43 (WT or Q343R, green) or HA-FUS (WT or P525L, green) were fixed and immunostained with anti-calnexin antibody (red). Nuclei were stained with Hoechst (blue). Z-series of merge images were shown. Scale bar 5 µm. White arrows indicate co-localization of calnexin with mTDP-43 or mFUS. Mander’s coefficients of b TDP-43 or c FUS with calnexin were analysed. Mean ± SEM, n = 3. ***p < 0.001. One-way ANOVA with Tukey’s post hoc test. d Subcellular fractionation of untransfected cells, cells expressing WTTDP-43, Q343R, WTFUS or P525L. Whole cell lysates (L), cytoplasmic (C), nuclear (N) and membrane (Mem) fractions from each sample were prepared, and western blotting for TDP-43, FUS, GAPDH, Histone H3 and IRE1 was performed. GADPH, Histone H3 and IRE1 were used as a marker of cytoplasm, nuclear and membrane, respectively. e Fluorescence protease protection assay demonstrated that TDP-43 is found on the ER membrane but FUS is found on the ER membrane and in the ER lumen. Neuro2a cells that expressed EGFP vector alone, PDI-dsred, EGFP-TDP-43 (WT or mutant) or GFP-FUS (WT or mutant) were subjected to the fluorescence protease protection assay. Individual images were taken before and after treatment with 60 µM digitonin and 50 µg/ml proteinase-K. f Fluorescence protease protection assay with 1 % Triton-x-100 treatment demonstrated that PDI and FUS (WT and mutant R521G) are present in the ER lumen. White arrows indicate the presence of FUS-GFP. Scale bars 50 µm
mSOD1 inhibits COPII vesicular transport to the Golgi
We next examined possible mechanisms whereby mSOD1 inhibits transport. ER-derived vesicles bud and transport cargo normally to the ERGIC but not to the Golgi in mSOD1 expressing cells (Fig. 2j), suggesting that ERGIC–Golgi transport, a microtubule dependent step, rather than ER–ERGIC transport, a microtubule independent step, is inhibited in these cells. mSOD1, unlike WTSOD1, also binds aberrantly to tubulin [31, 83]. Together these data suggest that mSOD1 may disrupt microtubule function, thus disrupting ERGIC–Golgi transport. We, therefore, examined the stability of microtubules in cells expressing mSOD1 by quantitating the levels of acetylated tubulin: a post-translational modification that stabilises and regulates microtubule function [45]. Whilst the levels of acetylated tubulin were similar in untransfected cells and cells expressing EGFP or WTSOD1, significantly decreased levels were detected in cells expressing mSOD1 (Fig. 4a–c), revealing that fewer stable microtubules are present in mSOD1 cells. Both tubulin and acetylated tubulin also co-localised with mSOD1 inclusions (Fig. 4d, e). In control experiments, neither mTDP-43 nor mFUS inhibited tubulin acetylation, and one mutant mTDP-43, A315T, slightly increased the levels of acetylated tubulin (Suppl. Fig. 5a), suggesting that inhibition of tubulin acetylation is specific for mSOD1.

COPII vesicles are unable to be transported to the Golgi complex in cells expressing mSOD1. a Neuro2a cells expressing SOD1-EGFP were fixed and immunostained with anti-acetylated tubulin antibodies. Merge images were shown. Thick white arrows indicate decreased fluorescence intensity of acetylated tubulin. b Quantified data for the average fluorescence intensities of acetylated tubulin in cells from a. c Immunoblotting of soluble cell lysates of untransfected (Untr), EGFP alone, WT or mSOD1 expressing cells. Blot was probed with anti-acetylated tubulin and anti-β-actin antibody was used for loading control. Graph represents the relative intensities of acetylated tubulin to β-actin. d Neuro2a cells co-expressing SOD1-EGFP (green) and tubulin-RFP (red) were fixed and visualised under confocal microscopy. Merge images were shown. e mSOD1 A4V or G85R transfected cells bearing inclusions were immunostained with anti-acetylated tubulin antibodies. Merge images were shown. Thin white arrows indicate co-localisation of mSOD1 inclusions and acetylated tubulin. f Immunoblotting of soluble cell lysates of cells expressing WT or mSOD1 upon Taxol and EpoD treatment. Blot was probed with anti-acetylated tubulin and anti-β-actin antibody was used for loading control. g Taxol or Epo D treated Neuro2a cells expressing WT or mSOD1 (A4V or G85R) were treated as in Fig. 1. The degrees of co-localisation of VSVG with calnexin or GM130 in cells were quantified using Mander’s coefficient. h Neuro2a cells expressing SOD1-EGFP for fixed and immunostained with anti-p115 and anti-GM130 antibodies. Merge images of p115 and GM130 were shown. i Quantified data of SOD1-EGFP transfected cells with co-localisation of p115 and GM130. Mean ± SEM, n = 3. ***p < 0.001, **p < 0.01, *p < 0.05 difference with control untransfected cells (Untr), ## p < 0.001 difference with WT transfected cells. One-way ANOVA with Tukey’s post hoc test. Scale bars 10 µm
We next examined whether microtubule stabilising agents, Taxol or Epothilone D (EpoD), rescue ER–Golgi transport in cells expressing mSOD1. As expected, the levels of acetylated tubulin were increased in Taxol/EpoD treated cells, confirming that both compounds stabilise microtubules, and EpoD was more effective than Taxol (Fig. 4f). Both compounds significantly increased ER–Golgi transport in cells expressing mSOD1 relative to control DMSO-treated cells (Fig. 4g). EpoD fully restored transport in both A4V and G85R cells, whereas Taxol fully restored transport in A4V, but only partially in G85R, expressing cells. In contrast, in control experiments, neither Taxol nor EpoD had any effect on the inhibition of VSVG transport from ER to Golgi, in cells expressing mTDP-43 or mFUS (Suppl. Fig. 5b). These findings reveal that stabilising microtubules rescues transport inhibition specifically in mSOD1 expressing cells. Furthermore, to provide further evidence that mutant SOD1 perturbs microtubule-based transport processes, we next examined the fusion of the autophagosome with the lysosome by co-expression with LC3 and LAMP1, as markers of each compartment, respectively. In cells expressing mSOD1 (A4V or G85R), significantly less CFP-LC3 co-localised with LAMP1-RFP, compared to in cells expressing WT or untransfected cells (p < 0.01, Suppl. Fig. 5c, d). Hence mSOD1 also inhibits autophagy-related trafficking, which is also microtubule-dependent. This was confirmed by treatment of mSOD1 expressing cells with EpoD, which restored the levels of autophagosome and lysosome fusion in cells expressing G85R to the levels found in control cells. For A4V, the levels were also increased but this was not statistically significant (Supp. Fig. 5e). These data, therefore, imply that unstable microtubules specifically impede ER–Golgi transport in cells expressing mSOD1.
To confirm these findings, we also examined tethering of COPII vesicles to the cis-Golgi using immunocytochemistry for p115 and GM130, where co-localisation indicated efficient vesicular tethering. In cells expressing WTSOD1, EGFP or untransfected cells (Fig. 4h, i), p115 and GM130 were co-localised in 80–90 % of cells, suggesting that COPII vesicles tether efficiently to the Golgi. However, the proportion of cells with co-localised p115 and GM130 was significantly decreased (1.5-fold, p < 0.01) in mSOD1 expressing cells. Hence tethering of COPII vesicles to the Golgi is antagonised by mSOD1, consistent with the presence of less stable microtubules in these cells.
Overexpression of Rab1 rescues inhibition of ER–Golgi transport in cells expressing mTDP-43, mFUS or mSOD1
We next looked for possible molecular targets of mSOD1, mTDP-43 and mFUS. Most proteins involved in ER–Golgi transport perform narrow, highly specific functions. Hence, dysfunction in their activity would not explain the diverse mechanisms of inhibition observed by mSOD1, mTDP-43 and mFUS. However, in contrast, Rab1 plays multiple roles in ER–Golgi transport, including vesicle budding, transport to the Golgi and microtubule stability. We, therefore, next investigated whether ALS-associated mutant proteins antagonise Rab1 function, and hence whether Rab1 overexpression could restore ER–Golgi trafficking. Rab1-tagged with FLAG or empty vector pCMV-FLAG as a control, was co-expressed in Neuro2a cells with EGFP-TDP-43, HA-FUS or SOD1-EGFP, and VSVG-mCherry. As expected, in cells co-expressing mTDP-43, mFUS or mSOD1 (with empty vector), more VSVG was retained within the ER and less was transported to the Golgi compared to controls (Fig. 5a–c). However, in cells co-expressing FLAG-Rab1, similar levels of VSVG were present in the Golgi in cells expressing mTDP-43, mFUS or mSOD1, as in controls expressing WT proteins, untransfected cells or EGFP alone (Fig. 5a–c). Immunoblotting using an anti-FLAG antibody confirmed that the expression levels of Rab1 were equivalent in cells expressing the ALS-mutants compared to controls (Suppl. Fig. 1e). Hence Rab1 rescues inhibition of ER–Golgi transport induced by mTDP-43, mFUS or mSOD1.

Over-expression of Rab1 rescues ER–Golgi transport inhibition, ER stress, apoptosis and inclusion formation induced by mTDP-43, mFUS and mSOD1. Neuro2a cells co-expressing a EGFP-TDP-43, b HA-FUS or c SOD1-EGFP and empty pCMV-FLAG vector (CMV) (top panels) or FLAG-Rab1 (bottom panels) were also co-expressed VSVG, Mander’s coefficients were quantified for transport of VSVG from ER to Golgi in transfected cells with/without Rab1 using calnexin and GM130. All other indications as in Fig. 1. Cells co-expressing TDP-43, FUS or SOD1 with d WTRab1, mutant Rab1 e S25N or f Q70L were fixed and immunostained with anti-XBP1 antibodies. Percentages of cells with XBP1 activation were quantified. Both inclusion formation and apoptotic nuclei were quantified for cells co-expressing SOD1 and g WT Rab1, mutant Rab1, h S25N or i Q70L. Mean ± SEM, n = 3. ***p < 0.001, **p < 0.01, *p < 0.05 difference with control untransfected cells (Untr). Unpaired student t test. Total 100 cells were scored from each group in each experiment. EGFP represents cells expressing EGFP vector alone
Overexpression of Rab1 rescues ER stress induced by mTDP-43, mFUS or mSOD1
We next examined whether overexpression of Rab1 can rescue ER stress in these cells, using activation of XBP1 as a marker of UPR induction [79]. XBP1 activation was significantly reduced in cells overexpressing Rab1 and mTDP-43, mFUS or mSOD1, compared to controls expressing empty vector (2-fold, p < 0.05), and it was restored to levels observed in cells expressing WT proteins, EGFP or untransfected cells (Fig. 5d). To rule out the possibility that the reduction of ER stress was due to restoration of Rab1 activity and not non-specific protein over-expression, two Rab1 mutants were examined; dominant negative Rab1S25N and constitutively active Rab1Q70L. Rab1S25N is maintained in an inactive GDP-bound state that cannot convert to its active GTP-bound form [42], whereas Rab1Q70L is constitutively active by remaining GTP-bound [18]. In contrast to WT Rab1, co-expression of Rab1S25N did not rescue ER stress in cells expressing mTDP-43, mFUS or mSOD1 (Fig. 5e). Furthermore, expression of GTP-bound Rab1Q70L decreased ER stress, indicated by XBP1 activation, induced by mTDP-43, mFUS or mSOD1 (2-fold, p < 0.05, Fig. 5f). To confirm the findings using XBP1, another marker of ER stress, nuclear immunoreactivity to CHOP [65], was examined in these cells, with similar findings (Suppl. Fig. 6). Hence the functional activity of Rab1 is protective against ER stress induced by mTDP-43, mFUS and mSOD1.
mSOD1 normally forms prominent inclusions in 15–18 % of cells and apoptosis in 10–15 % of cells [66]. Rab1 overexpression also significantly reduced the formation of inclusions (7–9 %, p < 0.001) and apoptosis (p < 0.05, 3-fold) in mSOD1 expressing cells (Fig. 5g). In control experiments, Rab1S25N did not affect mSOD1 inclusion formation and apoptosis (Fig. 5h), whereas Rab1Q70L further reduced mSOD1 inclusions (5–7 %, p < 0.05) and restored apoptosis to control levels (2.5 to 3-fold, p < 0.0001, Fig. 5i). These data thus link Rab1 functional activity to neurodegeneration and apoptosis in cells expressing mSOD1.
Rab1 is recruited to spinal motor neuron inclusions in patients with sALS and mis-localises in cells expressing ALS-associated SOD1, TDP-43 and FUS
We next examined the distribution of Rab1 in Neuro2a cells expressing TDP-43, FUS and SOD1 using immunocytochemistry. Rab1 was expressed diffusely in control cells and there was little co-localisation with WT TDP-43, FUS or SOD1. However, Rab1 co-localised extensively with mTDP-43 and mFUS (Fig. 6a, b). Analysis of Mander’s coefficient revealed significantly increased co-localization of Rab1 with mTDP-43 and mFUS, compared to WT TDP-43 or FUS (Fig. 6c, d). Furthermore, Rab1 co-located with mSOD1 inclusions in approximately 20 % of cells (Fig. 6e) implying that mSOD1, mTDP-43 and mFUS alter the cellular distribution of Rab1. In contrast, other proteins involved in ER–Golgi transport, including COPII subunit Sec23 did not bind to mTDP-43 or mFUS (Suppl. Fig. 7). Hence together these data suggest that Rab1 is associated with pathogenic mechanisms involving mTDP-43, mFUS and mSOD1. This was examined further in motor neurons of human spinal cord tissues from patients with sALS. For this analysis, large neurons located in the ventral horn region of spinal cord sections were identified as motor neurons (Suppl. Fig. 8a). Using immunohistochemistry, 80 % of motor neurons from sALS patients bore TDP-43 inclusions, all of which co-localised extensively with ubiquitin. Hence these motor neurons bear the typical ubiquitinated TDP-43 inclusions present in most cases of human ALS. Rab1 was expressed diffusely in control patients without neurological disease (Fig. 6f). However, in contrast, in an average of 50 % of sALS motor neurons, Rab1 formed prominent, inclusion-like structures (Fig. 6f, g). The presence of Rab1-positive inclusions in motor neurons was confirmed by performing immunohistochemistry using anti-Rab1 and anti-SMI32 antibodies (Suppl. Fig. 8b). Furthermore, approximately 40 % of the Rab1 inclusion-positive motor neurons co-localised with TDP-43 (Fig. 6f, h and Suppl. Fig. 8c). The Rab1-positive inclusions in sALS patients were intracytoplasmic punctate structures, some of which resembled Lewy-body-like or small Bunina body-like inclusions characteristic of ALS (Supp. Fig. 8d). In contrast, Rab1 was expressed diffusely in control motor neurons (Suppl. Fig. 8d). Hence, Rab1 is mis-localised and recruited into abnormal inclusions in motor neurons from sALS patients, thus implicating loss of Rab1 function in both sALS and familial ALS (fALS). Using immunoprecipitation, Rab1 also precipitated with phosphorylated TDP-43 in human sALS patient spinal cords (Suppl. Fig. 8e) and Rab1 and TDP-43 also co-precipitated more in cells expressing mTDP-43 (Suppl. Fig. 8f). Hence, Rab1 is mis-localised, recruited into abnormal inclusions and co-precipitates with pathological forms of TDP-43. This implies that a physical interaction exists between TDP-43 and Rab1, thus implicating loss of Rab1 function in both sALS and fALS.

Rab1 is mislocalised in cells expressing mTDP-43, mFUS and mSOD1 and forms inclusion-like structure in motor neurons from patients with sALS. Neuro2a cells expressing a EGFP-TDP-43 (WT or mutants) or EGFP vector alone; b HA-FUS (WT or mutants) were fixed and immunostained with anti-Rab1 antibodies. Representative images from each transfected cells were shown here. Nuclei were counter-stained with TOPRO-3 and merge images were shown. Mander’s coefficients of co-localisation of Rab1 with c TDP-43 and d FUS were analysed. Mean ± SEM, n = 3. ***p < 0.001 difference with WT transfected cells. One-way ANOVA with Tukey’s post hoc test. e SOD1-EGFP (WT or mutants) transfected cells were fixed and immunostained with anti-Rab1 antibodies. Representative images from each transfected cells were shown here. Nuclei were counter-stained with TOPRO-3 and merge images were shown. Graph represents percentage of inclusion-positive cells with co-localisation of Rab1 and mSOD1 inclusions. f Representative images of paraffin-fixed spinal cord sections from control individual and patients with sALS were dewaxed and immunostained with anti-Rab1, anti-TDP-43 and anti-ubiquitin antibodies. Images were fully reflective of the cells examined. g Graph represents percentage of motor neurons displayed Rab1 inclusions. h Graph represents percentage of Rab1-positive inclusion motor neurons with Rab1 and TDP-43 colocalization. Twenty to thirty motor neurons from each patient were scored. Merge images were shown. Scale bars 10 μm
ER–Golgi transport is inhibited in embryonic primary cortical and motor neurons from SOD1G93A transgenic mice
Finally, we examined transgenic mice overexpressing SOD1G93A for evidence of ER–Golgi transport inhibition in primary neurons. Primary embryonic cortical neurons and motor neurons isolated from SOD1G93A and non-transgenic controls were transfected with VSVG-mCherry and ER–Golgi transport was quantified as above. In both cortical neurons (Fig. 7a, c) and motor neurons (Fig. 7b, d), ER–Golgi transport was inhibited in SOD1G93A mice compared to controls (~2-fold, p < 0.001); more VSVG was retained in the ER and less transported to the Golgi. Hence these data provide further evidence that ER–Golgi transport is a pathogenic and early disease mechanism in ALS.

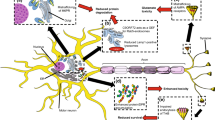
ER–Golgi transport is inhibited in primary cortical neurons and motor neurons isolated from SOD1G93A transgenic mice. Both a primary cortical neurons and b motor neurons are isolated from SOD1G93A transgenic (SOD) or non-transgenic (control) mice at E13.5. Primary neurons were then transfected with VSVG-mCherry for 24 h and then fixed and immunostained with calnexin for ER (green) and GM130 for cis-Golgi (blue). Merge images were shown. Scale bar 10 µm. The degree of co-localisation of VSVG with calnexin or GM130 in primary c cortical neurons and d motor neurons was quantified using Mander’s coefficient. Mean ± SEM, n = 40 cells. Total 4 mice were used in each group and 10 cells were scored from each mice. ***p < 0.001, **p < 0.01. Unpaired student t test. e Schematic diagram summarising how mTDP-43, mFUS and mSOD1 inhibit ER–Golgi transport. The primary response that ER–Golgi transport inhibited by mTDP-43 and mFUS is thought to be elicited by the depletion of Rab1 activity. One consequence is the cargo is unable to be sorted to COPII vesicles. In parallel, recruitment of COPII subunits to formation of vesicles could be antagonised. mSOD1 inhibits ER–Golgi transport could be resulting from a defect in the tethering process. This may be due to the interaction of mSOD1 with COPII vesicles or tubulin and dynein and thus leading to COPII vesicles are unable to be transported along microtubules to Golgi complex and thus decreasing the interaction of p115 with cis-Golgi marker, GM130
Discussion
Extensive evidence now implicates the failure of proteostasis as a trigger for neurodegeneration in ALS, and regulation of membrane trafficking is a key component of proteostasis. One third of all proteins transit through the ER–Golgi compartments destined for extracellular, transmembrane or other cellular locations [22]. ER–Golgi transport is, therefore, the most fundamental intracellular membrane trafficking system and it is a vital gateway to the endomembrane system. Disruption to ER–Golgi transport would therefore significantly impact on cellular function and viability. Here, we demonstrate that ALS-associated mSOD1, mTDP-43 and mFUS all inhibit ER–Golgi transport. We also detected inhibition of ER–Golgi transport in embryonic motor and cortical neurons obtained from SOD1G93A mice, validating this process in primary neurons and as an early disease mechanism. Moreover, the presence of Rab1-positive inclusions in sALS patients implies that secretory protein transport may also be inhibited in sporadic disease. Furthermore, restoration of ER–Golgi transport by Rab1 in our study prevented inclusion formation and apoptosis, thus linking this mechanism to neuronal cell death and degeneration. Inhibition of cellular trafficking may therefore be an important component of proteostasis in ALS.
We found that each protein inhibited ER–Golgi transport by distinct processes, but each mechanism was dependent on Rab1 function. In cells expressing mTDP-43, COPII vesicles budded normally, but they were almost completely depleted of cargo. Similarly in mFUS expressing cells, the vesicles were also depleted of cargo, but to a lesser extent than mTDP-43. Previous studies have suggested that the eventual size of COPII vesicles depends on the cargo loaded into these vesicles [29]. In preliminary studies, we found that COPII vesicles were reduced in size in cells expressing mTDP-43 or mFUS, consistent with this property. However, further experiments are required to confirm these findings. The level of COPII but not ERGIC53 on these vesicles was also reduced compared to control cells, suggesting that smaller vesicle diameter correlates with less vesicular COPII, which would result in atypical vesicles. Both mTDP-43 and mFUS inhibited ER–ERGIC rather than ERGIC-Golgi transport, and we detected TDP-43 on the cytoplasmic face of the ER, whereas FUS was present within the ER lumen. Using Mander’s coefficient, more mTDP-43 and mFUS co-localised with calnexin compared to WTTDP-43 or WTFUS. Hence, these data imply that mTDP-43 and mFUS inhibit ER–Golgi transport from either the cytoplasmic face of the ER or from within the ER lumen, respectively.
In contrast, in cells expressing mSOD1, COPII vesicles budded normally, were almost fully loaded with cargo, and were transported normally from ER to ERGIC. However, transport was inhibited between ERGIC and Golgi, and fewer acetylated microtubules were detected in mSOD1 expressing cells. Tubulin acetylation stabilises microtubules and promotes the recruitment of molecular motors kinesin-1 and cytoplasmic dynein [14, 53]. ERGIC–Golgi transport is a long-range, microtubule and dynein–dynactin-dependent step [1], in contrast to ER–ERGIC transport, which is short-range and microtubule independent. Our detection of inhibition of ERGIC–Golgi—but not ER–ERGIC—transport, and the loss of tethering of COPII vesicles to the Golgi in mSOD1 cells, is consistent with a microtubule-mediated defect. Previously, we could not detect SOD1 in the ER [3], similar to other groups [8, 41], suggesting that mSOD1 inhibits ER–Golgi transport from the cytoplasm. Dysfunction to microtubules would explain both inhibition of ER–Golgi transport and triggering of ER stress by cytoplasmic mSOD1. Consistent with our findings, previous studies have shown that tubulin and dynein both interact with mSOD1, and that mSOD1 modulates tubulin polymerisation [31, 83]. A recent study also reported that mutations in the gene encoding tubulin Alpha 4A, a component of microtubules, (TUBA4A) cause 1 % of fALS cases [64]. Furthermore, these TUBA4A mutants destabilised the microtubule network, diminishing its re-polymerization capability [64]. Taken together, these data implicate microtubule dysfunction in ALS pathology. Destabilisation of microtubules would impact on all microtubule-based processes, including axonal transport and autophagy-related trafficking, other mechanisms which are implicated in ALS [5, 32]. Indeed, we detected inhibition of autophagosome–lysosome fusion in cells expressing mSOD1, consistent with this notion. Rab1 is multi-tasking protein in ER–Golgi transport, mediating recruitment of cargo into COPII vesicles, regulation of COPII dynamics and function, transport between ER and ERGIC, and ERGIC and Golgi, recruitment of kinesin and dynein to microtubules, and microtubule organisation and function [27, 61, 63]. Previous studies have demonstrated that loss of Rab1 function prevents incorporation of secretory cargo into COPII vesicles [21] and leads to abnormal microtubules [61]. We observed that Rab1 overexpression rescued inhibition of ER–Golgi transport and ER stress triggered by mSOD1, mTDP-43 and mFUS, and apoptosis and inclusion formation triggered by mSOD1. However, the inactive mutant Rab1S25N did not rescue ER stress, and the constitutively active Rab1Q70L was more protective relative to WTRab1. This implies that the protective effects of Rab1 are driven specifically by its GTPase function, indicating that loss of Rab1 GTPase activity is associated with ER stress and neurodegeneration in ALS. These data, therefore, provide new evidence implying that restoring Rab1 function may be a novel therapeutic target in mSOD1, mTDP-43 and mFUS-associated ALS.
It should be noted that the studies using mSOD1 are less informative for the majority of ALS, because the TDP-43 pathology characteristic of most human ALS cases is not present in SOD1-associated ALS [20, 35]. FUS inclusions co-localise with TDP-43, p62 and ubiquitin and they are found in both FUS-associated fALS and also in some cases of sALS, but they are not associated with SOD1-fALS [13]. FUS also appears in TDP-43 inclusions in patients with TDP-43 mutations [13]. Since mSOD1 inclusions are distinct from both mTDP-43 and mFUS inclusions, in this study we focussed on recruitment of Rab1 to human motor neurons displaying the typical ubiquitinated TDP-43 inclusions found in most (97 %) ALS cases. Whilst Rab1 was distributed diffusely or punctate in control patients, it was recruited to these abnormal intracellular inclusions in sALS motor neurons. This implies that Rab1 is misfolded and loses its normal vesicular distribution in sALS, and hence is probably misfolded and non-functional in these patients. This suggests that similar pathological mechanisms are underway in sALS and fALS. In addition, previous studies have demonstrated that expression of Rab1 is upregulated in either lumbar spinal cords or blood from sALS patients [51, 57], providing further evidence that Rab1 is dysregulated in sALS. However, although glial pathology is implicated in both sALS and fALS [26, 46], in preliminary studies, Rab1 was not recruited to inclusions in either human astrocytes or microglial in sALS (data not shown). These data, therefore, imply that dysfunction to Rab1 is specific to motor neurons.
Inhibition of ER–Golgi transport is not specific to ALS. It has already been described in α-synuclein-associated Parkinson’s disease [11]. However, whilst disease-associated A53T mutant α-synuclein delays ER–Golgi transport, WT α-synuclein also impairs transport [11], so trafficking inhibition is not specific for mutant α-synuclein. Furthermore, distinct mechanisms are involved: α-synuclein appears to antagonise SNARE function and inhibit SNARE complex assembly [73], thus inhibiting tethering and docking of transport vesicles with the Golgi [11]. Furthermore, α-synuclein was not observed in association with the ER in previous studies [11].
Similarly, in Huntington’s disease post-Golgi cellular trafficking, rather than ER–Golgi trafficking, is inhibited by mutant huntingtin protein. This leads to less vesicular cargo leaving the trans-Golgi network, resulting in accumulation of protein in the Golgi, rather than the ER [12]. Whilst inhibition of post-Golgi transport in Huntington’s disease could eventually perturb ER–Golgi transport [12], ER–Golgi trafficking defects are not the primary event and have not been previously demonstrated directly. Disruptions in ER and Golgi homeostasis are also associated with Alzheimer’s and Prion diseases, but again involve distinct processes to ALS. Amyloid precursor protein has a signal peptide and hence it normally transits through the secretory pathway. However, whilst ER stress and Golgi fragmentation have been described previously in Alzheimer’s disease, surprisingly a recent report described an increase in ER–Golgi transport, rather than decreased ER–Golgi transport, in cells in which β-amyloid accumulates [30], implying that different processes are underway in ALS and in Alzheimer’s disease. Similarly, in prion-infected cells, VSVG is transported normally from ER to Golgi, but post-Golgi trafficking is significantly delayed. It is thought that prions may disturb post-Golgi trafficking of membrane proteins via accumulation in recycling endosomes [76]. Furthermore, Rab1 misfolding and recruitment to intracellular inclusions has not been described in patients with other neurodegenerative diseases. However, in our study, we found Rab1 recruitment to inclusions in sporadic patient motor neurons as well as in cells expressing mutant TDP-43, FUS or SOD1. Rab1 misfolding may, therefore, be a common process in ALS in contrast to other disorders. Hence whilst maintaining cellular proteostasis is fundamental in neurodegenerative conditions [24], there are clearly mechanisms specific to each disorder.
Maintaining proteostasis for post-mitotic cells presents specific challenges, and hence regulation of trafficking may be more critical for neurons than other cell types. Furthermore, the ER in neurons is poorly characterised but it is much more extensive than in other cells [52], extending throughout the neuritic processes, where its functions are largely uncharacterised. The reason why motor neurons are selectively targeted in ALS remains to be clarified. However, motor neurons are characterised by very long axons, up to 1 m in length in an adult human, and efficient anterograde (and retrograde) transport is essential to transport essential proteins to and from the synapse from the soma, along the axon. In other types of neurons, this distance is much shorter than in motor neurons where the axons are extremely long relative to the size of the cell body. The relationship between transport between the ER–Golgi and along the axon is poorly understood but these two processes are closely linked [2, 52]. In preliminary studies we have observed that after VSVG traffics from ER to Golgi, it subsequently is transported along the axon. Hence if transport between ER and Golgi is inhibited, it is likely that axonal transport is also inhibited. Thus, perturbations in ER–Golgi transport in motor neurons with long axons may present serious challenges to cellular function. Hence the long axons may, therefore, impose much stricter trafficking requirements and confer selective vulnerability on the motor neuron.
Consistent with this notion, previous studies have demonstrated that the fast-fatigable (FF) and fast-resistant (FR) motor neuron axons are already affected at before clinical symptoms (p48–p50) and late presymptomatic (p80–p90) SOD1 mice, respectively, whereas axons of slow (S) motor neurons are much more resistant to neurodegeneration [50, 58]. Compared to S motor neurons, FF motor neuron are larger cells, with larger axonal diameters, and the velocity of axonal transport is greater [6], which may impart selective vulnerability to axonal transport dysfunction on FF cells. Interestingly, the FF motor neurons also are the first to develop ER stress [58], thus linking ER–Golgi transport, ER stress and axonal transport to specific vulnerability of motor neuron subtypes to neurodegeneration in ALS. FF motor neurons also fire at higher rates than S motor neurons [6], consistent with the greater requirement for proteins necessary for synaptic function, supplied from the cell body via efficient axonal transport [62]. A recent study also demonstrated that the molecular motor dynein, which mediates both ER–Golgi transport and retrograde axonal transport, is upregulated in more vulnerable motor neurons, such as hypoglossal and spinal motor neurons, compared to oculomotor neurons, which are less vulnerable in ALS [10]. Hence these studies, together with our findings, link inhibition of ER–Golgi transport, ER stress and axonal transport, to specific vulnerability of motor neuron subtypes to neurodegeneration in ALS.
Mutations in other genes causing ALS, including alsin, vesicle-associated protein, dynactin, CHMP2B, optineurin, and valosin-containing protein, encode other proteins implicated in intracellular trafficking. Furthermore, we demonstrated recently that the normal function of C9ORF72, which contains a non-coding repeat expansion mutation in fALS, is to regulate endocytosis and autophagy, but this is dysregulated in ALS patient tissues [17]. Several ER–Golgi transport proteins are implicated in other motor neuron disorders, including atlastin [7] and seipin [28]. Disruption in ER–Golgi trafficking has also been described in spontaneous mouse mutants with motor phenotypes, pmn [59] and wobbler [60]. In addition, mice with a deletion of Scy1, implicated in COPI-mediated Golgi–ER transport, display a motor neuron degenerative phenotype [44]. We also recently showed that ALS mutations in optineurin disrupt its normal association to myosin VI, which inhibits intracellular trafficking [71]. The optineurin–myosin VI association was also disrupted in sALS patients, linking these defects to sporadic disease [71]. These findings suggest that disturbances to intracellular trafficking may be fundamental in neuronal degeneration and maintenance of proteostasis.
ER–Golgi transport is functionally related to other cellular processes implicated in ALS; hence, transport inhibition would impact on other closely related events. ER stress and Golgi fragmentation result from dysfunction to ER–Golgi transport [40] and we previously demonstrated that inhibition of ER–Golgi transport preceded both events in cells expressing mSOD1, implying that ER stress and Golgi fragmentation are consequences not causes of ER–Golgi transport inhibition [3]. Previous reports described ER stress and Golgi fragmentation in early, preclinical disease stages (p30) in SOD1G93A mice [39, 58], prior to neuromuscular denervation and axon retraction [77]. Our finding that ER–Golgi transport is inhibited in embryonic motor neurons in SOD1G93A mice implies that this mechanism precedes ER stress and Golgi fragmentation in vivo, as in vitro [3]. Similarly, ER–Golgi transport and autophagy, a major degradation pathway for intracytosolic aggregate-prone protein, are also functionally linked. Rab1 regulates autophagy and ER/Golgi membranes are required for autophagosome formation [80]. Autophagy dysfunction is implicated in ALS and in degradation of mSOD1 and mTDP-43 [9], but the underlying mechanisms remain unclear. RNA dysfunction is now widely implicated in ALS and ER-derived vesicles are involved in RNA trafficking [74]. Furthermore, Ypt1 binds to HAC1 RNA and modulates the UPR [75]. This indicates that ER–Golgi trafficking and the UPR communicate via RNA interaction with Rab1. Whilst both ER–Golgi transport and axonal transport are also functionally linked and COPII is implicated in both processes [4, 38], we could not detect an interaction between mTDP-43 and mFUS and COPII (Suppl. Fig. 7). Hence despite our previous finding that mSOD1 but not WTSOD1 interacts with COPII [3], COPII is not a common target of these proteins.
A schematic outlining possible mechanisms that inhibit ER–Golgi transport in ALS is presented in Fig. 7e. mTDP-43, mFUS and mSOD1 inhibit transport of ER-budded vesicles from ER to Golgi through antagonising Rab1 function, either via microtubule stability or COPII function. Modulation of disease processes common to multiple misfolded proteins in ALS has potential as a novel and effective therapeutic target: our data implicates Rab1 as a possible target. Description of the relationship between ER and Golgi transport to other pathogenic mechanisms is now warranted.
References
Appenzeller-Herzog C, Hauri HP (2006) The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci 119:2173–2183. doi:10.1242/jcs.03019
Aridor M, Fish KN (2009) Selective targeting of ER exit sites supports axon development. Traffic 10:1669–1684. doi:10.1111/j.1600-0854.2009.00974.x
Atkin JD, Farg MA, Soo KY, Walker AK, Halloran M, Turner BJ, Nagley P, Horne MK (2014) Mutant SOD1 inhibits ER-Golgi transport in amyotrophic lateral sclerosis. J Neurochem 129:190–204. doi:10.1111/jnc.12493
Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R (1994) COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77:895–907
Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G (2010) Deficits in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci USA 107:20523–20528. doi:10.1073/pnas.1006869107
Borg J, Grimby L, Hannerz J (1979) Motor neuron firing range, axonal conduction velocity, and muscle fiber histochemistry in neuromuscular diseases. Muscle Nerve 2:423–430. doi:10.1002/mus.880020603
Botzolakis EJ, Zhao J, Gurba KN, Macdonald RL, Hedera P (2011) The effect of HSP-causing mutations in SPG3A and NIPA1 on the assembly, trafficking, and interaction between atlastin-1 and NIPA1. Mol Cell Neurosci 46:122–135. doi:10.1016/j.mcn.2010.08.012
Chang LY, Slot JW, Geuze HJ, Crapo JD (1988) Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. J Cell Biol 107:2169–2179
Chen S, Zhang X, Song L, Le W (2012) Autophagy dysregulation in amyotrophic lateral sclerosis. Brain Pathol 22:110–116. doi:10.1111/j.1750-3639.2011.00546.x
Comley L, Allodi I, Nichterwitz S, Nizzardo M, Simone C, Corti S, Hedlund E (2015) Motor neurons with differential vulnerability to degeneration show distinct protein signatures in health and ALS. Neuroscience 291:216–229. doi:10.1016/j.neuroscience.2015.02.013
Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F et al (2006) Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313:324–328. doi:10.1126/science.1129462
del Toro D, Canals JM, Gines S, Kojima M, Egea G, Alberch J (2006) Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J Neurosci Off J Soc Neurosci 26:12748–12757. doi:10.1523/JNEUROSCI.3873-06.2006
Deng HX, Zhai H, Bigio EH, Yan J, Fecto F, Ajroud K, Mishra M, Ajroud-Driss S, Heller S, Sufit R et al (2010) FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol 67:739–748. doi:10.1002/ana.22051
Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, Saudou F (2007) Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J Neurosci Off J Soc Neurosci 27:3571–3583. doi:10.1523/JNEUROSCI.0037-07.2007
Emr S, Glick BS, Linstedt AD, Lippincott-Schwartz J, Luini A, Malhotra V, Marsh BJ, Nakano A, Pfeffer SR, Rabouille C et al (2009) Journeys through the Golgi—taking stock in a new era. J Cell Biol 187:449–453. doi:10.1083/jcb.200909011
Farg MA, Soo KY, Walker AK, Pham H, Orian J, Horne MK, Warraich ST, Williams KL, Blair IP, Atkin JD (2012) Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol Aging. doi:10.1016/j.neurobiolaging.2012.02.009
Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, Halloran MA, Gleeson P, Blair IP, Soo KY et al (2014) C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. doi:10.1093/hmg/ddu068
Filipeanu CM, Zhou F, Claycomb WC, Wu G (2004) Regulation of the cell surface expression and function of angiotensin II type 1 receptor by Rab1-mediated endoplasmic reticulum-to-Golgi transport in cardiac myocytes. J Biol Chem 279:41077–41084. doi:10.1074/jbc.M405988200
Forsberg K, Andersen PM, Marklund SL, Brannstrom T (2011) Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 121:623–634. doi:10.1007/s00401-011-0805-3
Forsberg K, Jonsson PA, Andersen PM, Bergemalm D, Graffmo KS, Hultdin M, Jacobsson J, Rosquist R, Marklund SL, Brannstrom T (2010) Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One 5:e11552. doi:10.1371/journal.pone.0011552
Garcia IA, Martinez HE, Alvarez C (2011) Rab1b regulates COPI and COPII dynamics in mammalian cells. Cell Logist 1:159–163. doi:10.4161/cl.1.4.18221
Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS (2003) Global analysis of protein expression in yeast. Nature 425:737–741. doi:10.1038/nature02046
Haas AK, Yoshimura S, Stephens DJ, Preisinger C, Fuchs E, Barr FA (2007) Analysis of GTPase-activating proteins: Rab1 and Rab43 are key Rabs required to maintain a functional Golgi complex in human cells. J Cell Sci 120:2997–3010. doi:10.1242/jcs.014225
Hetz C, Mollereau B (2014) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 15:233–249. doi:10.1038/nrn3689
Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD, Lippincott-Schwartz J (1998) Kinetic analysis of secretory protein traffic and characterization of golgi to plasma membrane transport intermediates in living cells. J Cell Biol 143:1485–1503
Ilieva H, Polymenidou M, Cleveland DW (2009) Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 187:761–772
Ishida M, Ohbayashi N, Maruta Y, Ebata Y, Fukuda M (2012) Functional involvement of Rab1A in microtubule-dependent anterograde melanosome transport in melanocytes. J Cell Sci 125:5177–5187. doi:10.1242/jcs.109314
Ito D, Suzuki N (2007) Molecular pathogenesis of seipin/BSCL2-related motor neuron diseases. Ann Neurol 61:237–250. doi:10.1002/ana.21070
Jin L, Pahuja KB, Wickliffe KE, Gorur A, Baumgartel C, Schekman R, Rape M (2012) Ubiquitin-dependent regulation of COPII coat size and function. Nature 482:495–500. doi:10.1038/nature10822
Joshi G, Chi Y, Huang Z, Wang Y (2014) Abeta-induced Golgi fragmentation in Alzheimer’s disease enhances Abeta production. Proc Natl Acad Sci USA 111:E1230–E1239. doi:10.1073/pnas.1320192111
Kabuta T, Kinugawa A, Tsuchiya Y, Kabuta C, Setsuie R, Tateno M, Araki T, Wada K (2009) Familial amyotrophic lateral sclerosis-linked mutant SOD1 aberrantly interacts with tubulin. Biochem Biophys Res Commun 387:121–126. doi:10.1016/j.bbrc.2009.06.138
Li L, Zhang X, Le W (2008) Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy 4:290–293
Lord C, Ferro-Novick S, Miller EA (2013) The highly conserved COPII coat complex sorts cargo from the endoplasmic reticulum and targets it to the golgi. Cold Spring Harb Perspect Biol 5(2), pii: a013367. doi:10.1101/cshperspect.a013367
Lorenz H, Hailey DW, Wunder C, Lippincott-Schwartz J (2006) The fluorescence protease protection (FPP) assay to determine protein localization and membrane topology. Nat Protoc 1:276–279. doi:10.1038/nprot.2006.42
Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H et al (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434. doi:10.1002/ana.21147
Manders EMM, Verbeek FJ, Atenm JA (1993) Measurement of co-localization of objects in dual-colour confocal images. J Microsc 169:375–382
Marie M, Sannerud R, Avsnes Dale H, Saraste J (2008) Take the ‘A’ train: on fast tracks to the cell surface. Cell Mol Life Sci CMLS 65:2859–2874. doi:10.1007/s00018-008-8355-0
Matsuoka K, Orci L, Amherdt M, Bednarek SY, Hamamoto S, Schekman R, Yeung T (1998) COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell 93:263–275
Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC (1996) The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu, Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci USA 93:5472–5477
Nakagomi S, Barsoum MJ, Bossy-Wetzel E, Sutterlin C, Malhotra V, Lipton SA (2008) A Golgi fragmentation pathway in neurodegeneration. Neurobiol Dis 29:221–231. doi:10.1016/j.nbd.2007.08.015
Nishitoh H, Kadowaki H, Nagai A, Maruyama T, Yokota T, Fukutomi H, Noguchi T, Matsuzawa A, Takeda K, Ichijo H (2008) ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev 22:1451–1464
Nuoffer C, Davidson HW, Matteson J, Meinkoth J, Balch WE (1994) A GDP-bound of rab1 inhibits protein export from the endoplasmic reticulum and transport between Golgi compartments. J Cell Biol 125:225–237
Orci L, Palmer DJ, Ravazzola M, Perrelet A, Amherdt M, Rothman JE (1993) Budding from Golgi membranes requires the coatomer complex of non-clathrin coat proteins. Nature 362:648–652. doi:10.1038/362648a0
Pelletier S, Gingras S, Howell S, Vogel P, Ihle JN (2012) An early onset progressive motor neuron disorder in Scyl1-deficient mice is associated with mislocalization of TDP-43. J Neurosci Off J Soc Neurosci 32:16560–16573. doi:10.1523/JNEUROSCI.1787-12.2012
Perdiz D, Mackeh R, Pous C, Baillet A (2011) The ins and outs of tubulin acetylation: more than just a post-translational modification? Cell Signal 23:763–771. doi:10.1016/j.cellsig.2010.10.014
Philips T, Rothstein JD (2014) Glial cells in amyotrophic lateral sclerosis. Exp Neurol 262(Pt B):111–120. doi:10.1016/j.expneurol.2014.05.015
Plutner H, Cox AD, Pind S, Khosravi-Far R, Bourne JR, Schwaninger R, Der CJ, Balch WE (1991) Rab1b regulates vesicular transport between the endoplasmic reticulum and successive Golgi compartments. J Cell Biol 115:31–43
Preston AM, Gurisik E, Bartley C, Laybutt DR, Biden TJ (2009) Reduced endoplasmic reticulum (ER)-to-Golgi protein trafficking contributes to ER stress in lipotoxic mouse beta cells by promoting protein overload. Diabetologia 52:2369–2373. doi:10.1007/s00125-009-1506-5
Prosser DC, Tran D, Gougeon PY, Verly C, Ngsee JK (2008) FFAT rescues VAPA-mediated inhibition of ER-to-Golgi transport and VAPB-mediated ER aggregation. J Cell Sci 121:3052–3061. doi:10.1242/jcs.028696
Pun S, Santos AF, Saxena S, Xu L, Caroni P (2006) Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci 9:408–419. doi:10.1038/nn1653
Rabin SJ, Kim JM, Baughn M, Libby RT, Kim YJ, Fan Y, La Spada A, Stone B, Ravits J (2010) Sporadic ALS has compartment-specific aberrant exon splicing and altered cell-matrix adhesion biology. Hum Mol Genet 19:313–328. doi:10.1093/hmg/ddp498
Ramirez OA, Couve A (2011) The endoplasmic reticulum and protein trafficking in dendrites and axons. Trends Cell Biol 21:219–227. doi:10.1016/j.tcb.2010.12.003
Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, Verhey KJ (2006) Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol CB 16:2166–2172. doi:10.1016/j.cub.2006.09.014
Robberecht W, Philips T (2013) The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 14:248–264. doi:10.1038/nrn3430
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JO, Deng HX (1993) Mutation in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Saraste J, Lahtinen U, Goud B (1995) Localization of the small GTP-binding protein rab1p to early compartments of the secretory pathway. J Cell Sci 108(Pt 4):1541–1552
Saris CG, Horvath S, van Vught PW, van Es MA, Blauw HM, Fuller TF, Langfelder P, DeYoung J, Wokke JH, Veldink JH et al (2009) Weighted gene co-expression network analysis of the peripheral blood from Amyotrophic Lateral Sclerosis patients. BMC Genom 10:405. doi:10.1186/1471-2164-10-405
Saxena S, Cabuy E, Caroni P (2009) A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci 12:627–636
Schaefer MK, Schmalbruch H, Buhler E, Lopez C, Martin N, Guenet JL, Haase G (2007) Progressive motor neuronopathy: a critical role of the tubulin chaperone TBCE in axonal tubulin routing from the Golgi apparatus. J Neurosci Off J Soc Neurosci 27:8779–8789. doi:10.1523/JNEUROSCI.1599-07.2007
Schmitt-John T, Drepper C, Mussmann A, Hahn P, Kuhlmann M, Thiel C, Hafner M, Lengeling A, Heimann P, Jones JM et al (2005) Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat Genet 37:1213–1215. doi:10.1038/ng1661
Schmitt HD, Wagner P, Pfaff E, Gallwitz D (1986) The ras-related YPT1 gene product in yeast: a GTP-binding protein that might be involved in microtubule organization. Cell 47:401–412
Shaw PJ, Eggett CJ (2000) Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J Neurol 247(Suppl 1):I17–I27
Slavin I, Garcia IA, Monetta P, Martinez H, Romero N, Alvarez C (2011) Role of Rab1b in COPII dynamics and function. Eur J Cell Biol 90:301–311. doi:10.1016/j.ejcb.2010.10.001
Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, Kenna KP, Scotter EL, Kost J, Keagle P, Miller JW et al (2014) Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84:324–331. doi:10.1016/j.neuron.2014.09.027
Soo KY, Atkin JD, Farg M, Walker AK, Horne MK, Nagley P (2012) Bim links ER stress and apoptosis in cells expressing mutant SOD1 associated with amyotrophic lateral sclerosis. PLoS One 7:e35413. doi:10.1371/journal.pone.0035413
Soo KY, Atkin JD, Horne MK, Nagley P (2009) Recruitment of mitochondria into apoptotic signaling correlates with the presence of inclusions formed by amyotrophic lateral sclerosis-associated SOD1 mutations. J Neurochem 108:578–590
Spang A, Matsuoka K, Hamamoto S, Schekman R, Orci L (1998) Coatomer, Arf1p, and nucleotide are required to bud coat protein complex I-coated vesicles from large synthetic liposomes. Proc Natl Acad Sci USA 95:11199–11204
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E et al (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672. doi:10.1126/science.1154584
Stenmark H (2009) Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 10:513–525. doi:10.1038/nrm2728
Stylli SS, Stacey TT, Verhagen AM, Xu SS, Pass I, Courtneidge SA, Lock P (2009) Nck adaptor proteins link Tks5 to invadopodia actin regulation and ECM degradation. J Cell Sci 122:2727–2740. doi:10.1242/jcs.046680
Sundaramoorthy V, Walker AK, Tan V, Fifita JA, McCann EP, Williams KL, Blair IP, Guillemin GJ, Farg MA, Atkin JD (2015) Defects in optineurin- and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum Mol Genet. doi:10.1093/hmg/ddv126
Sundaramoorthy V, Walker AK, Yerbury J, Soo KY, Farg MA, Hoang V, Zeineddine R, Spencer D, Atkin JD (2013) Extracellular wildtype and mutant SOD1 induces ER-Golgi pathology characteristic of amyotrophic lateral sclerosis in neuronal cells. Cellular Mol Life Sci CMLS. doi:10.1007/s00018-013-1385-2
Thayanidhi N, Helm JR, Nycz DC, Bentley M, Liang Y, Hay JC (2010) Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol Biol Cell 21:1850–1863. doi:10.1091/mbc.E09-09-0801
Todd AG, Lin H, Ebert AD, Liu Y, Androphy EJ (2013) COPI transport complexes bind to specific RNAs in neuronal cells. Hum Mol Genet 22:729–736. doi:10.1093/hmg/dds480
Tsvetanova NG, Riordan DP, Brown PO (2012) The yeast Rab GTPase Ypt1 modulates unfolded protein response dynamics by regulating the stability of HAC1 RNA. PLoS Genet 8:e1002862. doi:10.1371/journal.pgen.1002862
Uchiyama K, Miyata H, Sakaguchi S (2013) Disturbed vesicular trafficking of membrane proteins in prion disease. Prion 7:447–451
van Dis V, Kuijpers M, Haasdijk ED, Teuling E, Oakes SA, Hoogenraad CC, Jaarsma D (2014) Golgi fragmentation precedes neuromuscular denervation and is associated with endosome abnormalities in SOD1-ALS mouse motor neurons. Acta Neuropathol Commun 2:38. doi:10.1186/2051-5960-2-38
Vance C, Rogeli B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211
Walker AK, Soo KY, Sundaramoorthy V, Parakh S, Ma Y, Farg MA, Wallace RH, Crouch PJ, Turner BJ, Horne MK et al (2013) ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS One 8:e81170. doi:10.1371/journal.pone.0081170
Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S et al (2010) Alpha-synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol 190:1023–1037. doi:10.1083/jcb.201003122
Xu D, Hay JC (2004) Reconstitution of COPII vesicle fusion to generate a pre-Golgi intermediate compartment. J Cell Biol 167:997–1003. doi:10.1083/jcb.200408135
Yang YS, Harel NY, Strittmatter SM (2009) Reticulon-4A (Nogo-A) redistributes protein disulfide isomerase to protect mice from SOD1-dependent amyotrophic lateral sclerosis. J Neurosci Off J Soc Neurosci 29:13850–13859. doi:10.1523/JNEUROSCI.2312-09.2009
Zhang F, Strom AL, Fukada K, Lee S, Hayward LJ, Zhu H (2007) Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex. J Biol Chem 282:16691–16699
Acknowledgments
We thank Professor Malcolm Horne and Professor Phillip Nagley for helpful discussions. Human patient and control lumbar region tissues were received from the Victorian Brain Bank Network, supported by University of Melbourne, Mental Health Research Institute of Victoria, and Victorian Forensic Institute of Medicine and funded by Neurosciences Australia and the National Health and Medical Research Council of Australia (NHMRC). This work was supported by NHMRC Project grants [# 1006141, 1030513 to JA], Bethlehem Griffiths Research Foundation, and Angie Cunningham Laugh to Cure MND grant [to JDA and KYS].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. All applicable international, national and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Soo, K.Y., Halloran, M., Sundaramoorthy, V. et al. Rab1-dependent ER–Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP-43 and FUS-associated ALS. Acta Neuropathol 130, 679–697 (2015). https://doi.org/10.1007/s00401-015-1468-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1468-2