
Abstract
Accumulation of phosphorylated cytoplasmic TDP-43 inclusions accompanied by loss of normal nuclear TDP-43 in neurons and glia of the brain and spinal cord are the molecular hallmarks of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD-TDP). However, the role of cytoplasmic TDP-43 in the pathogenesis of these neurodegenerative TDP-43 proteinopathies remains unclear, due in part to a lack of valid mouse models. We therefore generated new mice with doxycycline (Dox)-suppressible expression of human TDP-43 (hTDP-43) harboring a defective nuclear localization signal (∆NLS) under the control of the neurofilament heavy chain promoter. Expression of hTDP-43∆NLS in these ‘regulatable NLS’ (rNLS) mice resulted in the accumulation of insoluble, phosphorylated cytoplasmic TDP-43 in brain and spinal cord, loss of endogenous nuclear mouse TDP-43 (mTDP-43), brain atrophy, muscle denervation, dramatic motor neuron loss, and progressive motor impairments leading to death. Notably, suppression of hTDP-43∆NLS expression by return of Dox to rNLS mice after disease onset caused a dramatic decrease in phosphorylated TDP-43 pathology, an increase in nuclear mTDP-43 to control levels, and the prevention of further motor neuron loss. rNLS mice back on Dox also showed a significant increase in muscle innervation, a rescue of motor impairments, and a dramatic extension of lifespan. Thus, the rNLS mice are new TDP-43 mouse models that delineate the timeline of pathology development, muscle denervation and neuron loss in ALS/FTLD-TDP. Importantly, even after neurodegeneration and onset of motor dysfunction, removal of cytoplasmic TDP-43 and the concomitant return of nuclear TDP-43 led to neuron preservation, muscle re-innervation and functional recovery.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease primarily defined by the presence of TAR DNA-binding protein of 43 kDa (TDP-43) pathology in the brain and spinal cord [31]. Moreover, ~50 % of frontotemporal lobar degeneration (FTLD) patients also develop TDP-43 pathology, with this form of disease known as FTLD-TDP [10, 31]. ALS and FTLD-TDP are therefore now considered to form a spectrum of disorders [12, 13]. Although only ~10 % of ALS and ~30 % of FTLD patients have a family history of disease, the majority of available mouse models rely on gene mutations identified in these small minority of ALS and FTLD patients. For example, mutant human SOD1 mice have been used to model ALS for >20 years and although they appear to have a clinical disease course that mirrors human ALS [17], they do not develop robust TDP-43 pathology [36, 41, 45]. Furthermore, no disease modifying treatments using the SOD1 mice have succeeded in clinical trials [14]. New valid mouse models that recapitulate TDP-43 pathology are therefore required for investigation of upstream disease pathogenesis and preclinical testing for both ALS and FTLD-TDP.
TDP-43 is a widely expressed and highly conserved protein with diverse functions in RNA metabolism, including RNA translation, splicing and transport [26]. Although primarily located in the nucleus, a small proportion of TDP-43 is normally present in the cytoplasm of cells [4, 47]. However, in postmortem tissue from patients with ALS, FTLD-TDP, or both, affected neurons and glia in the brain and spinal cord lose nuclear TDP-43 and accumulate insoluble phosphorylated TDP-43 in the cytoplasm [31]. Multiple attempts have been made to generate transgenic mouse models that recapitulate these neuropathological features of ALS and FTLD-TDP. For example, expression of human wild type, ALS-linked mutant, or cytoplasmic TDP-43 under the control of broad or brain-specific promoters, including PrP, Thy1, and Camk2a, resulted in rodent models which recapitulate some, but not all, features of ALS/FTLD-TDP [2, 20, 22, 44, 48, 49]. Notably, no or only rare TDP-43 inclusions were detected in these models and no or only modest loss of spinal cord motor neurons occurred. Moreover, many of these animals do not develop a progressive ALS-like phenotype, which is a requirement for future preclinical testing of ALS therapeutics [29]. In addition, since previous rodent models did not successfully recapitulate the key features of disease, the timeline for the progressive TDP-43 nuclear clearance, cytoplasmic accumulation, muscle denervation and neuron loss in vivo has been difficult to define.
Our previous efforts to generate transgenic mouse models that develop TDP-43 pathology included crossing doxycycline (Dox)-suppressible Camk2a promoter mice with transgenic mice expressing TDP-43 in which the nuclear localization sequence (NLS) was genetically ablated [22]. These mice accumulated cytoplasmic TDP-43 in the brain but with little pathological TDP-43 aggregates and did not develop an ALS-like phenotype due to lack of expression in the spinal cord [22]. To overcome these limitations, we generated a second generation of TDP-43 mouse models with cytoplasmic TDP-43 pathology and a progressive disease course similar to ALS, using newly generated mice with Dox-suppressible neurofilament heavy chain (NEFH) promoter-driven brain and spinal cord expression. Since it remains uncertain if modulating TDP-43 sub-cellular location and pathology is a valid avenue for therapeutic development, we further used these new mouse models of ALS/FTLD-TDP to investigate if decreasing cytoplasmic hTDP-43 levels, clearance of hTDP-43 pathology and return of nuclear mTDP-43 could ameliorate ALS-like phenotypes after the onset of neuron loss, muscle denervation and motor dysfunction.
Materials and methods
Generation of NEFH-tTA transgenic mice and bigenic rNLS mice
NEFH-tTA transgenic (Tg) mice were generated by injection of a plasmid with the human NEFH promoter isolated from BAC clone (CHORI: RP11-91J21) to drive expression of the tetracycline transactivator (tTA) gene [20] (kindly provided by Xu-Gang Xia, Thomas Jefferson University) into the pronucleus of fertilized eggs from C57BL/6J × C3HeJ F1 matings (Transgenic and Chimeric Mouse Facility of the University of Pennsylvania). Mice were maintained on a mixed C57BL/6J × C3HeJ F1 background. Monogenic NEFH-tTA mice were bred to previously described tetO-hTDP-43-∆NLS line 4 mice [22], which allow expression of K82A/R83A/K84A mutant human TDP-43 (hTDP-43∆NLS) and were also maintained on a mixed C57BL/6J × C3HeJ F1 background, to produce non-transgenic (nTg), NEFH-tTA monogenic, hTDP-43∆NLS monogenic and NEFH-tTA × hTDP-43∆NLS bigenic mice (referred to as rNLS mice). Similarly, monogenic NEFH-tTA mice were bred to previously described tetO-hTDP-43-WT line 4 and tetO-hTDP-43-WT line 12 mice [22]. Seven original NEFH-tTA Tg founders were produced but four lines were discontinued. The remaining three lines (designated 8, 9B and 43) were studied extensively as described here. Breeding mice and weaned mice on Dox were provided with chow containing 200 mg/kg Dox (Dox Diet #3888, Bio-Serv). To induce expression of hTDP-43∆NLS, mice were switched to standard chow lacking Dox (Rodent Diet 20 #5053, PicoLab). To re-suppress expression of hTDP-43∆NLS, Rodent Diet 20 was replaced with Dox Diet. Genotyping was performed using tail DNA [52] as described previously for tetO-hTDP-43∆NLS mice [22], and with the following primers for all three lines of NEFH-tTA mice: NEFH-tTA forward (5′-TCCACTTTGAGGGGTCTCTG-3′) and NEFH-tTA reverse (5′-AGCATCTCATCACTTCCCTG-3′) [20]. All procedures were performed in accordance with the NIH Guide for the Care and Use of Experimental Animals and studies were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Mouse phenotype monitoring and behavioral analysis
Mice were weighed and monitored for tremor and clasping phenotype three times per week. For observation of tremor, mice were held on their backs in the palm of the observer’s hand gripped gently between thumb and index finger and both forelimb and hindlimb movements were observed for ~30 s. The presence of either fast fine tremor at any point in this observation period or resting tremor when at rest in the home cage was recorded as a positive response. Tremor of this nature was never observed in nTg and monogenic littermate control mice. For observation of hindlimb clasping, mice were suspended by the tail ~30 cm above the cage and slowly lowered. The presence of both hindlimbs held together within 5 s of being raised and maintained for ~30 s was recorded as a positive response. Persistent hindlimb clasping of this nature was never observed in nTg and monogenic littermate control mice. Mice were tested once weekly for wirehang and rotarod performance. For the wirehang test of grip strength, mice were placed on a wire cage top and gently flipped to be suspended upside-down ~30 cm above a clean cage. The time to fall was recorded up to 180 s; if still hanging at the end of a session a time of 180 s was recorded. One training session and two test sessions were performed each with >5 min rest between sessions, with the final score being the average time of the two test sessions. For the rotarod test of motor coordination and balance, mice were placed on a rotarod apparatus (Model 7650, Ugo Basile) at a speed of 4 rpm with acceleration up to 40 rpm over 300 s. The time to fall was recorded up to 300 s; if still running at the end of the session a time of 300 s was recorded. One training session and two test sessions were performed each with >30 min rest between sessions, with the final score being the average time of the two test sessions. Upon signs of movement difficulty, mice were provided with moistened chow on the cage floor. Disease end stage in mice was defined as complete hindlimb paralysis, a loss of >30 % body weight from peak weight or the inability to right within 15 s when placed on their backs. Generally, rNLS bigenic mice did not develop complete hindlimb paralysis or persistent loss of righting reflex prior to reaching the weight cut-off for end stage using the simple supportive measures reported here.
Mouse tissue collection for NMJ and spinal cord motor neuron quantification
Mice were deeply anesthetized using ketamine/xylazine and intracardially perfused with ~15 mL room temperature phosphate-buffered saline (PBS) followed by ~30 mL of 10 % formalin. The tibialis anterior (TA) muscles were washed in PBS overnight, and spinal cords were post-fixed in 10 % formalin overnight and then all were processed for cryoprotective embedding. For visualization of neuromuscular junctions (NMJs) in whole TA muscles, BTX conjugated to AlexaFluor-488 (Invitrogen, Carlsbad, CA, USA) 1:500 was added with rabbit anti-VAChT 1:16,000 (gift of C. Henderson). The percentage of NMJ innervation was determined by dividing the total number of areas of overlap between VAChT and BTX signals (total number innervated endplates) by the number of areas of BTX signal (total number of endplates) on consecutive longitudinal 35 μm cryosections of muscles. We observed 700–1000 NMJs per TA. For the quantification of motor neuron numbers, motor neurons were counted in every fifth transverse 20 μm cryosections of the L4 and L5 levels of the spinal cord stained for VAChT and a mouse anti-human TDP-43 monoclonal antibody (MAb) (0.06 μg/mL, clone 5104, CNDR) [25].
Mouse tissue collection for biochemistry and IHC
Mice were deeply anesthetized using ketamine/xylazine and intracardially perfused with PBS as above. Tissue was rapidly dissected and weighed and either immediately frozen on dry ice and stored at −80 °C for use in biochemistry or post-fixed overnight in 10 % formalin. Post-fixed tissues were rinsed in TBS, pH 7.4 (50 mM Tris–HCl and 150 mM NaCl), embedded in paraffin, and sectioned at 6 μm. Generally, each mouse brain was bisected and the left hemisphere was dissected into different regions for biochemistry before freezing, and the right hemisphere used for immunohistochemistry (IHC) and immunofluorescence (IF).
Human tissue samples
Frozen and formalin-fixed, paraffin-embedded brain and spinal cord tissues were from the Center for Neurodegenerative Disease Research Brain Bank at the University of Pennsylvania. Human tissues were obtained at autopsy and processed for neuropathological assessment as described previously [43]. Informed consent was obtained in accordance with the University of Pennsylvania Institutional Review Board guidelines. Cortical and spinal cord tissues of ALS and FTLD patients with a high burden of TDP-43 pathology (FTLD-TDP) were used in experiments. Human tissue for biochemistry was sequentially extracted as described previously [37]. The urea-soluble fraction from frontal cortex of a 68 year-old female patient with a neuropathological diagnosis of FTLD-TDP was used for immunoblotting. Paraffin 6 μm sections of motor cortex and lumbar spinal cord from a 66 year-old female patient with a neuropathological diagnosis of ALS were used for IHC.
IHC and IF
For IHC and IF on paraffin-embedded sections, sections were deparaffinized in xylene and rehydrated through a series of decreasing concentration of ethanol. For IHC only, sections were incubated in 5 % H2O2 in methanol for 30 min to block endogenous peroxidases. Sections for all antibodies except anti-ubiquitin were subjected to antigen retrieval by microwaving at 95 °C for 15 min in 1 % antigen unmasking solution pH 6.0 (Vector Laboratories). Sections were allowed to cool and then blocked in 2 % FBS in 0.1 M Tris and incubated in primary antibodies for ~16 h. Primary antibodies used for IHC and IF were a rabbit anti-C-terminal TDP-43 polyclonal antibody (PAb) for detection of both human and mouse TDP-43 (0.15 μg/mL, CNDR C1039) [23], a rabbit anti-N-terminal TDP-43 PAb for detection of both human and mouse TDP-43 (0.22 μg/mL, CNDR N1065) [23], a mouse anti-internal TDP-43 MAb for detection of both human and mouse TDP-43 (0.03 μg/mL, clone 5117, CNDR) [25], a mouse anti-human TDP-43 MAb (0.06 μg/mL, clone 5104, CNDR) [25], a mouse anti-human TDP-43 MAb (0.06 μg/mL, clone 5123, CNDR) [25], a rabbit anti-mouse TDP-43 (0.10 μg/mL, CNDR 2340-181, affinity-purified IgG raised by immunizing rabbits with a peptide corresponding to amino acids 181-190 of mTDP-43), a rabbit anti-phospho-S403/404 TDP-43 PAb (1:2000, CosmoBio TIP-PTD-P05), a rat anti-phospho-S409/410 TDP-43 MAb (1:200, TAR5P-1D3, Ascenion, Munich, Germany) [30], a rabbit anti-phospho-S409/410 TDP-43 PAb (1:5000, CosmoBio TIP-PTD-P01), a rat anti-phospho-NFM + H MAb (1:200, clone TA51, CNDR) [42], a mouse anti-ubiquitin MAb (1:5000, clone Ubi-1, Millipore 1510), a rabbit anti-NeuN MAb (1:1000, clone EPR12763, Abcam 177487), a rabbit anti-GFAP PAb (1:2000, Dako z0334), a rat anti-GFAP MAb (1:2000, clone 2.2B10, CNDR) [28], a rabbit anti-Iba1 PAb (1:1000, Wako 019-19741), and a rabbit anti-Olig-2 PAb (1:250, Millipore 9610). For IHC, a Vectastain ABC kit was used with biotinylated anti-mouse, anti-rabbit or anti-rat secondary antibodies for detection with 0.05 % 3,3′-diaminobenzidine tetrahydrochloride hydrate (Sigma D5637) with 0.003 % H2O2 in 0.1 M phosphate buffer pH 7.4. Sections were counterstained with haemotoxylin, dehydrated through ethanol and xylenes, and mounted with Cytoseal 60 mounting medium (Vector Laboratories). For IF, sections were incubated with goat anti-mouse, anti-rabbit or anti-rat 488- or 594-conjugated AlexaFluor secondary antibodies (1:1000, Molecular Probes) and mounted using Fluoromount Gold plus DAPI (Southern Biotech). Images were acquired using a Nikon Eclipse Ni inverted microscope with DS-Fi2 camera (brightfield) or DS-Qi1Mc camera (fluorescence).
Preparation of mouse brain lysates
Tissues were thawed on ice and then sonicated in 5× v/w RIPA buffer (50 mM Tris, 150 mM NaCl, 1 % NP-40, 5 mM EDTA, 0.5 % sodium deoxycholate, and 0.1 % SDS, pH 8.0) containing 1 mM PMSF and protease and phosphatase inhibitor cocktails (Sigma). Samples were centrifuged at 4 °C, 100,000g for 30 min and the supernatant taken as the RIPA-soluble fraction. The pellet was washed by sonication with RIPA buffer as above. This supernatant was discarded and the pellet sonicated in 2× v/w urea buffer (7 M urea, 2 M thiourea, 4 % CHAPS, and 30 mM Tris, pH 8.5) and centrifuged at 22 °C, 100,000g for 30 min. This supernatant was taken as the RIPA-insoluble/urea-soluble fraction. Protein concentrations of the RIPA-soluble fractions were determined using the bicinchoninic acid protein assay (Pierce).
Immunoblotting
Twenty micrograms of RIPA-soluble protein and a volume equivalent to 7.5-times the amount of the corresponding urea-soluble fraction (normalized to non-specific bands detected by Ponceau S staining of membranes for urea-soluble spinal cord samples) were analyzed by 10 or 12 % SDS-PAGE with nitrocellulose membrane (Bio-Rad). Antibodies used for immunoblotting were C1039, 5104, 2340-181, anti-phospho-S403/404 TDP-43 PAb and TAR5P-1D3, a rabbit anti-neurofilament light chain PAb (1:500, phospho-independent NFL, CNDR) [5], a mouse anti-neurofilament medium chain MAb (1:500, phospho-independent NFM, clone RMO44, CNDR) [27] and a mouse anti-GAPDH MAb (1:10,000, clone 6C5, Advanced Immunochemical). Signals were detected using goat anti-mouse, anti-rabbit or anti-rat IRDye-680 or IRDye-800 conjugated secondary PAbs (Li-Cor or Rockland) with imaging using a Li-Cor Odyssey imaging system. Band infrared fluorescent signals were quantified using Li-Cor Image Studio Version 2.0.
Measurement of cortical thickness
Cortical thickness was measured from the pial surface to the dorsal corpus callosum from images of DAPI-labeled or haemotoxylin and eosin-stained brain sections at the level of the motor cortex (Bregma 1.10) according to the standard mouse brain atlas [33].
Statistical analyses and experimental design
Statistical significance between two values was determined using a two-tailed t test, analyses of three or more values were performed by one-way ANOVA with Bonferroni’s post hoc test using Prism 4 (GraphPad). P < 0.05 was considered statistically significant. All results are presented as mean ± standard error of the mean (SEM) and in figures *p < 0.05, **p < 0.01, and ***p < 0.001. The investigators were blinded for genotype for mouse wirehang and rotarod testing, and for NMJ and neuron counts. Mice were randomly allocated to groups for time point analysis and for Dox recovery experiments. Both sexes were analyzed. Littermate nTg and monogenic animals were used as controls. Littermate control and bigenic groups were matched to include equal sexes. Mice were excluded from all analyses when non-neurological disease was present: three rNLS8 mice and two rNLS9B mice developed skin lesions/fighting wounds, one rNLS9B mouse was born microcephalic, and one rNLS8 mouse died of unknown cause at 3 weeks off Dox.
Results
The NEFH promoter drives expression of hTDP-43∆NLS in brain and spinal cord
To drive the expression of hTDP-43 in neurons with large-caliber axons of the brain and spinal cord, we generated lines of mice in which the tetracycline transactivator protein (tTA) is expressed under the control of the human NEFH promoter [18, 20]. Three NEFH-tTA mouse lines (designated lines 8, 9B and 43) were crossed with our previously developed tetO-hTDP-43∆NLS line 4 mice, in which the nuclear localization sequence (NLS) is genetically ablated leading to cytoplasmic accumulation of hTDP-43 [22]. Therefore, in the resulting NEFH-tTA/tetO-hTDP-43∆NLS bigenic animals (designated as ‘regulatable’ or rNLS mice), hTDP-43∆NLS expression is controlled by the tTA gene product in a doxycycline (Dox)-dependent manner, such that hTDP-43∆NLS expression is suppressed in the presence of Dox (Fig. 1a). rNLS mice used in all experiments were bred and maintained in the presence of Dox-containing chow until approximately 5 weeks of age, when Dox was removed to allow hTDP-43∆NLS expression.

Expression of hTDP-43∆NLS in brain and spinal cord of rNLS8 and rNLS9B mice. a Schematic for Dox-regulatable expression of hTDP-43∆NLS in bigenic mice under the control of the NEFH promoter. Expression of hTDP-43 and total (h+m) TDP-43 protein in olfactory bulb (Ob), cerebellum (Cb), hippocampus (Hp), brainstem and remainder of the brain (Bs), cortex (Cx), spinal cord (Sc), spleen (Sp), kidney (Kd) and liver (Lv) of b rNLS8 and c rNLS9B mice at 4 weeks off Dox. Approximate molecular weight markers in kDa are shown on the left and GAPDH is a loading control. Representative immunoblots of n = 3. Representative images at 1 week off Dox show widespread expression of hTDP-43∆NLS in d Hp, Cx, and e Sc of rNLS8 mice. IHC in rNLS8 motor cortex showing cytoplasmic TDP-43 inclusions (arrows) detected with both f an N-terminal (Nt)-TDP-43 antibody, and g a C-terminal (Ct)-TDP-43 antibody, at 2 weeks off Dox. h, i There is widespread hTDP-43 expression (green) in NeuN+ neurons (red), shown at 4 weeks off Dox. Arrowhead in i indicates rare non-neuronal hTDP-43-positive glial cell, and nuclei are shown by DAPI (blue). Scale bars d 500 μm, e 200 μm, f, g 50 μm, h, i 50 μm
rNLS mice showed widespread, high levels of hTDP-43∆NLS expression in brain and spinal cord as early as 1 week off Dox, but not in peripheral tissues including spleen, kidney and liver (Fig. 1b–e). Line rNLS8 was chosen for the most thorough characterization due to the higher percentage of neurons expressing hTDP-43, although similar results were also confirmed in lines rNLS9B and rNLS43 (Fig. 1c; Fig. S1a). rNLS8 mice expressed full length hTDP-43∆NLS in the cytoplasm with frequent structures resembling pathological TDP-43 inclusions, as detected by antibodies against the N-terminus and C-terminus as well as human specific TDP-43 (Fig. 1f–h). We quantified expression in layer V of the motor cortex in rNLS8 mice, where greater than 95 % of NeuN-positive neurons were positive for hTDP-43 by 1 week off Dox, and this level of expression was maintained over time (Fig. S1b). Despite NEFH being a neuron-specific promoter, unexpectedly 5–10 % of GFAP-positive astrocytes also expressed hTDP-43∆NLS, although Iba1-positive microglia and Olig2-positive oligodendrocytes rarely expressed the transgene (Fig. 1i; Fig. S1c–e).
Insoluble phosphorylated TDP-43 accumulates in brain and spinal cord of rNLS mice
To confirm the presence of insoluble TDP-43 aggregates and to investigate the consequences of mislocalized cytoplasmic hTDP-43 in the rNLS mice, brain and spinal cord samples were separated into RIPA-soluble and RIPA-insoluble (urea-soluble) fractions for biochemical analysis. hTDP-43∆NLS was robustly detected in the RIPA-soluble protein fraction of cortex of rNLS8 mice after 1 week off Dox, with maximal expression 2–4 weeks after Dox removal (Fig. 2a). Quantitative analysis revealed total TDP-43 levels as more than 10-fold higher in rNLS mice at 2–4 weeks compared to non-transgenic (nTg) and monogenic littermates, with a concomitant ~50 % decrease in endogenous mTDP-43 levels (Fig. 2a; Fig. S2), indicating the previously described phenomenon of auto-regulation of endogenous TDP-43 upon TDP-43 over-expression [3, 22].

Accumulation of insoluble pTDP-43 over time in brain and spinal cord, and formation of pTDP-43 inclusions in rNLS mice. a RIPA-soluble hTDP-43∆NLS, mouse (m) TDP-43 and total (h+m) TDP-43 in rNLS8 cortex at various time points compared to non-transgenic (nTg) and monogenic (tTA only) controls at 1 week off Dox. Quantification is shown below each representative immunoblot with n = 3 mice for nTg, tTa and 1, 2, 4 and 6 weeks off Dox, and n = 9 for end stage mice (7–18 weeks); see also Fig. S2. Approximate molecular weight markers in kDa are shown on the left and GAPDH is shown as a loading control. b RIPA-insoluble hTDP-43∆NLS, mTDP-43 and h+mTDP-43 from 1 week off Dox in rNLS8 cortex. Higher molecular weight and phosphorylated (p) TDP-43 are detected from 2 weeks off Dox. Extract from a human FTLD patient is shown as a positive control. Asterisks indicate pTDP-43, arrowhead indicates 43 kDa TDP-43, and hash mark indicates 20–25 kDa C-terminal TDP-43 fragments. c RIPA-insoluble pTDP-43 in rNLS8 spinal cord from 4 weeks off Dox. Time points of 7–18 weeks in a–c are disease end stage. pTDP-43 pathology was detected with antibodies to both pS409/410- and pS403/404-TDP-43 in rNLS8 at 4 weeks off Dox in d motor cortex and f spinal cord (Sc), rNLS8 at 6 weeks off Dox in h striatum, in rNLS9B at 4 weeks off Dox in e motor cortex, g Sc, and i striatum, and in the j motor cortex and k Sc of human ALS patients. Arrows indicate examples of large cytoplasmic inclusions, and asterisks in e and i indicate small pTDP-43-positive puncta. Scale bar d–k 50 μm
Notably, hTDP-43 was readily detected in the RIPA-insoluble (urea-soluble) fraction from 1 week off Dox with a concurrent decrease in mTDP-43 levels, suggesting that the decrease in soluble mTDP-43 levels was indeed due to auto-regulatory down-regulation of the endogenous mTDP-43 gene rather than recruitment of mTDP-43 to the insoluble protein fraction (Fig. 2b). Likewise, RIPA-insoluble phosphorylated TDP-43 (pTDP-43; ~45 kDa) was also detected in rNLS8 mouse cortex from 2 weeks off Dox, which accumulated at high levels from 4 weeks off Dox, with both phosphorylated S403/404-TDP-43 (p403/404) and S409/410-TDP-43 (p409/410) specific antibodies. These findings indicate that accumulation of insoluble TDP-43 occurs prior to the phosphorylation of the protein. In addition, the detection of ~45 kDa pTDP-43 from 4 weeks accompanied by decreased levels of ~43 kDa TDP-43 after 2 weeks indicates a shift in apparent molecular mass and accumulation of the TDP-43 phosphorylated species, as described previously [53]. The RIPA-insoluble ~25 kDa C-terminal fragments typically found in FTLD-TDP patients were not detected in any rNLS mice with any of the anti-TDP-43 antibodies used here (Fig. 2b), suggesting a biochemical signature more akin to ALS, in which TDP-43 C-terminal fragments are less abundant. Furthermore, we also detected p403/404 and p409/410 TDP-43 in the RIPA-insoluble fraction of spinal cord of rNLS8, rNLS9B and rNLS43 mice (Fig. 2c; Figs. S3, S4). A decrease in both RIPA-soluble and -insoluble h, m and h+mTDP-43 with continued robust pTDP-43 levels at later time points off Dox in rNLS8 mice suggests potential neuron loss with disease progression.
In addition to the biochemical signature of pathological TDP-43 in rNLS mice, pTDP-43-positive inclusions were detected by IHC with both p403/404 and p409/410 antibodies (Fig. 2d–i). Numerous speckled and large pTDP-43 cytoplasmic inclusions were detected in many brain regions, including motor cortex (Fig. 2d, e), spinal cord (Fig. 2f, g; Fig. S5a), striatum (Fig. 2h, i), visual, entorhinal, and somatosensory cortex, cerebellum and hippocampus (Fig. S5a) in all rNLS lines, resembling those detected in motor cortex and spinal cord of ALS/FTLD-TDP patients (Fig. 2j, k) [30]. Rare cytoplasmic pTDP-43-positive inclusions were detected as early as 1 week off Dox in layer V of the motor cortex (Fig. S5b), and the percentage of NeuN-positive cells with hTDP-43-positive puncta increased over time to approximately 28 % of neurons at disease end stage (8–18 weeks off Dox) in rNLS8 mice (Fig. S5c), but TDP-43-positive inclusions were detected in fewer than 2 % of spinal cord motor neurons. As in ALS and FTLD-TDP [31], there was also an accumulation of cytoplasmic ubiquitin with pTDP-43-positive inclusions in the brains and spinal cords of rNLS mice (Fig. S6).
To further investigate whether or not the TDP-43 pathology resulted from high levels of cytoplasmic hTDP-43ΔNLS or was due simply to over-expression of hTDP-43 driven by the NEFH promoter, we crossed NEFH-tTA line 8 mice with previously reported tetO-hTDP-43-WT line 4 or line 12 mice [22]. As expected, nuclear hTDP-43-WT was detected in neurons of the brain and spinal cord of bigenic mice from both crosses (n = 7 NEFH-tTA line 8/tetO-hTDP-43-WT line 4 mice analyzed between 1 week and 6 months off Dox, and n = 6 NEFH-tTA line 8/tetO-hTDP-43-WT line 12 mice analyzed between 2 weeks and 6 months off Dox, Fig. S7). hTDP-43- or pTDP-43-positive cytoplasmic inclusions were not detected in any of the NEFH-tTA/tetO-hTDP-43-WT mice. Extremely rare small intranuclear pTDP-43 inclusions were occasionally detected, in several brain regions including the motor cortex and striatum but not the spinal cord (Fig. S7). These findings indicate that pathology in the rNLS mice is due to the combination of high levels of over-expression driven by the NEFH promoter and the presence of the cytoplasmic targeting of the hTDP-43ΔNLS.
Thus, similar to human FTLD-TDP and ALS cases, all three rNLS lines accumulated abundant TDP-43 cytoplasmic inclusions in cortex and spinal cord that correlated with the presence of RIPA-insoluble TDP-43. Our longitudinal analyses indicate that endogenous mTDP-43 is rapidly down-regulated, and the accumulation of insoluble TDP-43 occurs prior to robust TDP-43 phosphorylation in both brain and spinal cord in rNLS mice.
rNLS mice develop progressive cortical atrophy and neuromuscular junction denervation followed by spinal cord motor neuron loss with dramatic muscle atrophy
To sequentially characterize the ALS-like phenotype in the rNLS8 mice, we measured changes in cortical thickness, brain mass, tibialis anterior (TA) neuromuscular junction (NMJ) innervation, motor neuron (MN) number and muscle integrity. rNLS8 mice displayed decreased cortical thickness indicative of neuronal degeneration beginning at 4 weeks off Dox (Fig. 3a–c), which was accompanied by astrogliosis in many regions, including layer V of the motor cortex (Fig. S8). By disease end stage at 10–18 weeks off Dox, rNLS8 mice had significantly smaller brains when compared to littermate controls, indicating dramatic brain atrophy (Fig. 3d).

rNLS8 mice develop progressive cortical atrophy, muscle denervation, MN loss and muscle atrophy. a, b Representative brain sections from nTg and rNLS8 mice 18 weeks off Dox at Bregma 1.10, stained with haemotoxylin and eosin (H&E). Thickness is measured from the edge of the brain section to the white matter below (indicated with a bar). c Measurement of cortical thickness in nTg and rNLS8 mice at different ages, n = 3 per time point. d Brain mass of rNLS8 mice compared to littermate nTg or tTA monogenic controls at disease end stage of 10–18 weeks off Dox, n = 9 per group. e–h Representative images and quantification of the overlap of VAChT-positive motor terminals (red) with acetylcholine receptors stained using BTX (green) as an indicator of innervated motor endplates in the tibialis anterior (TA) muscle of rNLS8 mice. The TA muscle showed marked denervation at 6 and 8 weeks off Dox (f–g, vacated NMJs noted with asterisks), n = 4 mice per time point. i–k IF for MN marker VAChT (red) with nuclear marker DAPI (blue) revealed loss of MNs in the lumbar spinal cord at 6 and 8 weeks off Dox in rNLS8 mice. l Quantification of number of VAChT-positive lumbar MNs in rNLS8 mice at different times off Dox. n = 4 mice per time point. m, n H&E staining of TA muscle showed gross muscle atrophy in rNLS8 mice at disease end stage compared to littermate nTg control. Examples of central nuclei are shown by an arrow and atrophic fibers by an asterisk. o, p TA and gastrocnemius muscle masses of rNLS8 mice compared to littermate nTg or tTA monogenic controls at disease end stage of 10–18 weeks off Dox, n = 7 per group. Scale bars a, b 500 μm, e–g 50 μm, i–k, m–n 100 μm; *p < 0.05, **p < 0.01, ***p < 0.001 versus control by one-way ANOVA with Bonferroni’s post hoc test; ### p < 0.001 by paired two-tailed t test
To investigate the effect of hTDP-43∆NLS expression on innervation of NMJs, we analyzed hindlimb TA muscle using the cholinergic synaptic marker vesicular acetylcholine transporter (VAChT) to label motor terminals, and fluorophore-conjugated alpha-bungarotoxin (BTX) to mark motor endplates. Significant denervation occurred after only 4 weeks of transgene expression in rNLS8 mice, and by 6 weeks of transgene expression the TA showed massive denervation with only 39.6 ± 6.6 % (mean ± SEM, n = 4) of NMJs still intact (Fig. 3e–h). Interestingly, axonal dieback occurred before MN loss, since the number of spinal MNs at the lumbar L4-L5 level (innervating hindlimb muscles) were unchanged at this 4 week time point but were decreased by ~28 % after 6 weeks off Dox, and by 8 weeks off Dox only ~50 % of lumbar MNs remained (Fig. 3i–l). Finally, following loss of MNs at disease end stage, there was grouped fiber atrophy and centralized nuclei indicative of attempted regeneration in muscle, as well as dramatic decreases in the mass of isolated TA and gastrocnemius muscles (Fig. 3m–p).
rNLS mice develop a progressive motor phenotype leading to death
To determine if hTDP-43∆NLS expression also initiated motor dysfunction in rNLS mice, we monitored several behavioral phenotypes over time. rNLS8 mice developed early-onset hindlimb clasping and fine forelimb and/or hindlimb tremor (Fig. 4a, b; Video S1), progressive loss of grip strength as measured by wirehang test (Fig. 4c), and progressive decline in coordinated movement and balance as measured by impairment on the accelerating rotarod test (Fig. 4d). In addition, rNLS8 mice showed progressively decreasing body mass from a peak at 2 weeks off Dox (Fig. 4e), and early death with median survival of 10.3 weeks off Dox (Fig. 4f). Similar findings of brain and muscle atrophy, and progressive motor phenotype, were obtained in rNLS9B and rNLS43 mice, albeit with differences in the time of onset and length of survival (Figs. S9, S10). Since males and females of all lines developed consistent pathology and disease, they were both included in analyses. Notably, rNLS8 mice had a more protracted disease course than the rapid progression observed in rNLS9B and rNLS43 mice, consistent with their lower spinal cord hTDP-43 expression levels (Fig. 1b, c; Fig. S1a).

Expression of hTDP-43∆NLS results in dramatic motor impairments, weight loss, and death in rNLS8 mice. Onset of a hindlimb clasping, b tremor, and progressive declines in c wirehang and d rotarod performance in rNLS8 mice, n = 10 controls, n = 14 bigenic. e rNLS8 mice also showed significant weight loss relative to controls beginning 3 weeks after Dox removal, n = 19 per group. f Kaplan–Meier survival curve for rNLS8 mice and littermate controls shows a marked decrease in survival time in rNLS8 mice, n = 19 per group
Thus, the pathology and disease course in rNLS8 mice progressed with several ALS-relevant features, including early cytoplasmic accumulation of hTDP-43 along with down-regulation of endogenous mTDP-43 from 1 week off Dox, the formation of cytoplasmic pTDP-43 inclusions accompanied by onset of motor symptoms from 2 weeks off Dox, brain atrophy, astrogliosis and muscle NMJ denervation at 4 weeks off Dox, which were followed by lower spinal cord MN loss and an inability to perform several motor tasks from 6 weeks off Dox until disease end stage at a median of 10.3 weeks from initial Dox removal (Fig. 5). This sequence of events is likely the same for lines rNLS9B and rNLS43, although with compressed timing. Therefore, these rNLS mice faithfully recapitulate many of the pathological and phenotypic features of ALS.

Schematic showing the time course of major neurodegenerative events in the rNLS8 mouse model of ALS/FTLD-TDP. The earliest events are the accumulation of cytoplasmic hTDP-43 and down-regulation of mTDP-43. Next, mice begin to show motor impairments including clasping, tremor, and impairments in motor tasks, and in some cells hTDP-43 begins to aggregate and becomes phosphorylated at pathological sites. By 4 weeks after initiation of hTDP-43ΔNLS expression, there is a significant brain atrophy and marked axonal dieback from hindlimb muscle. Cell loss and axonal dieback, and their functional consequences, continue to progress until disease end stage
TDP-43 pathology is rapidly cleared and nuclear mTDP-43 returns following suppression of hTDP-43∆NLS expression in rNLS mice
Since most patients with ALS and/or FTLD are not diagnosed until well into the disease course when neuron loss has already occurred, we next asked whether suppression of hTDP-43∆NLS expression would eliminate existing TDP-43 pathology and allow functional recovery even after significant motor deficits had developed. We hypothesized that this paradigm would allow us to determine whether or not therapies which drive clearance of cytoplasmic TDP-43 and allow return of nuclear TDP-43 could be clinically relevant, and would establish the rNLS mice as a model for future preclinical testing of ALS and FTLD-TDP therapeutics.
After 6 weeks of transgene expression, when significant TDP-43 pathology was present with overt neuron loss and dramatic motor decline (Figs. 2d–i, 3, 4), we suppressed hTDP43∆NLS expression by re-introducing Dox (Fig. 6a). After only 2 weeks of Dox re-introduction, a dramatic decrease in accumulated hTDP-43∆NLS and pTDP-43 protein was observed in the cortex and spinal cord (Fig. 6b–d). Additionally, pTDP-43 pathology was no longer detected in the motor cortex or in any other brain region, including the thalamus and genu of the corpus callosum, of rNLS8 mice after 12+ weeks back on Dox (Fig. 6e; Fig. S11a–c).

Soluble and insoluble TDP-43 are rapidly cleared from brain and spinal cord of rNLS8 mice upon re-introduction of Dox. a Schematic of experimental design. b, c Levels of hTDP-43 and h+mTDP-43 in RIPA-soluble (R) and RIPA-insoluble/urea-soluble (U) protein fractions of rNLS8 mouse cortex and spinal cord at 6 weeks and at subsequent time points back on Dox (+Dox), with tetO-hTDP-43-∆NLS monogenic (Mono) littermate control. p403/404-TDP-43 and p409/410-TDP-43 were rapidly eliminated from the U fraction. Approximate molecular weight markers in kDa are shown on the left and GAPDH is the loading control. d hTDP-43 was eliminated from spinal cord MNs in rNLS8 mice +Dox, and e p409/410-TDP-43 inclusions were detected in the motor cortex in rNLS8 mice at 6 weeks, but were not detected in rNLS8 mice +Dox (images of 6 weeks off Dox +18 weeks back on Dox are shown). Scale bars d 100 μm, e 50 μm
As has been seen with previous TDP-43 transgenic mice [22], over-expression of hTDP-43∆NLS caused a loss of normal nuclear mTDP-43 protein (Fig. 7a; Fig. S11d). However, when mice were re-introduced to Dox at 6 weeks, nuclear mTDP-43 returned (Fig. 7a; Fig. S11d) and levels of mTDP-43 detected biochemically were significantly increased as early as 2 weeks back on Dox (Fig. 7b, c). Furthermore, re-introduction of Dox led to clearance of cytoplasmic TDP-43 (Fig. S11a, d) and diminished astrogliosis (Fig. S11e). Thus, suppression of hTDP-43∆NLS transgene expression resulted in elimination of cytoplasmic TDP-43, clearance of pTDP-43 pathology and return of endogenous mTDP-43 to the nucleus. Similar to our previous findings [22], we also detected a decrease in neurofilament light (NFL) chain but not neurofilament medium (NFM) chain protein in the cortex of rNLS8 mice at 6 weeks off Dox (Fig. 7d–f). Although NFL levels showed some increase upon re-introduction of Dox, these remained significantly decreased compared to controls after 18–32 weeks back on Dox (Fig. 7d–f). Furthermore, double labeling IF of pTDP-43 with phospho-NFM and neurofilament heavy chain proteins showed little or no co-localization of NF proteins with TDP-43 pathology in the cortex and spinal cord of rNLS8 mice, suggesting that mislocalization of NF proteins is not involved in driving neuronal death (Fig. S12).

Endogenous nuclear mTDP-43 rapidly returns in cortex of rNLS8 mice upon re-introduction of Dox. a Nuclear mTDP-43 levels in rNLS8 mice at 6 weeks off Dox and after Dox re-introduction (+18 weeks Dox shown), compared to tetO-hTDP-43-∆NLS monogenic (Mono) littermate control. b, c Endogenous mTDP-43 levels are decreased in rNLS8 mice at 6 weeks off Dox, but are significantly increased by +2 weeks Dox; ***p < 0.001 versus control by one-way ANOVA with Bonferroni’s post hoc test, n = 3 per group. d–f Levels of NFL protein, but not NFM, are significantly decreased in rNLS8 mice at 6 weeks off Dox. Representative immunoblots are shown; **p < 0.01 and ***p < 0.001 versus control by one-way ANOVA with Bonferroni’s post hoc test, n = 3 per group, +Dox indicates +18–32 weeks back on Dox. Scale bar a 50 μm
rNLS mice functionally recover following hTDP-43∆NLS transgene suppression
We next asked whether clearing hTDP-43∆NLS would lead to symptom alleviation and functional recovery in mice with overt ALS-like motor impairments. Remarkably, mice that had expressed hTDP-43∆NLS for 6 weeks, with associated pTDP-43 pathology, neuron loss and motor dysfunction, showed a rapid functional improvement as early as 2 weeks back on Dox, with partial recovery of hindlimb clasping phenotype and rapid weight gain (Fig. 8a, b; Video S1).

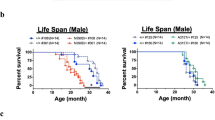
Suppression of hTDP-43∆NLS expression rescues motor phenotypes and restores lifespan in rNLS8 mice. a Hindlimb splaying phenotype of nTg control, rNLS8 mouse after 6 weeks of hTDP-43∆NLS expression and rNLS8 mouse back on Dox after 6 weeks of hTDP-43∆NLS expression (+Dox, 2 weeks back on Dox shown). b Weight loss reached a nadir at 6 weeks in rNLS8 mice back on Dox (n = 11 rNLS8 +Dox and n = 8 littermate nTg/monogenic controls). c MN number in lumbar SC, and d, e analysis of NMJ innervation in rNLS8 mice after 6 weeks of hTDP-43∆NLS expression and at 3–4 months back on Dox (n = 3–4 per group), *p < 0.05 versus 6 weeks off Dox, image shows rNLS8 mouse at 6 weeks off Dox +12 weeks back on Dox. f Wirehang performance of rNLS8 mice back on Dox at 6 weeks, p < 0.001 from 3 weeks back on Dox versus 6 weeks off Dox, and g rotarod performance of rNLS8 mice back on Dox at 6 weeks, p < 0.05 at 1 week back on Dox and p < 0.001 from 2 weeks back on Dox versus 6 weeks off Dox, by repeated measures ANOVA with Bonferroni’s post hoc test, n = 8. h Survival in rNLS8 mice back on Dox at 6 weeks (rNLS8 +Dox—blue line, n = 14, littermate nTg/monogenic controls—black line, n = 11) and in rNLS8 off Dox (rNLS8—dotted red line, n = 19, also included in Fig. 4f). Triangles/circles indicate censored animals either killed for analysis or remaining alive
Importantly, rNLS8 mice at 8 weeks off Dox had significantly fewer lumbar spinal cord MNs compared to those at 6 weeks off Dox (Fig. 3l), however there was no statistically significant difference in MN numbers between mice at 6 weeks off Dox and mice at 12–18 weeks back on Dox (Fig. 8c), suggesting prevention of continued MN loss in mice back on Dox. Likewise, there was no significant difference in motor cortex cortical thickness in rNLS8 mice at 6 weeks off Dox compared to rNLS8 mice back on Dox for 12–32 weeks (Fig. S11f, g). Indeed, despite the significant muscle denervation and even with the ~25 % reduction in the total number of MNs at the TA-innervation L4 level of the spinal cord (Fig. 3h), we detected a significantly higher percentage of innervated NMJs in mice back on Dox, suggesting that the remaining MNs were able to re-innervate the vacated TA motor endplates following suppression of hTDP-43∆NLS expression (Fig. 8d, e).
rNLS8 mice also displayed a remarkable return of grip strength and improvement in performance in the rotarod test beginning as early as 1 week back on Dox (Fig. 8f, g). Finally, although rNLS8 mice had a median survival of 10.3 weeks off Dox and a maximum lifespan of 18 weeks, rNLS8 mice returned to Dox at 6 weeks showed a dramatic extension of lifespan (Fig. 8h). Of n = 14 mice returned to Dox at 6 weeks, three were killed for early analysis, whereas 11 mice lived up to or past the maximum rNLS8 lifespan without sign of disease progression (Fig. 8h).
In addition to the phenotypic recovery in rNLS8 mice returned to Dox at 6 weeks, we also observed a similar functional recovery in rNLS9B mice returned to Dox at the time point of disease when these mice had lost 25 % of peak body weight (5.0–6.0 weeks off Dox). Similar to rNLS8 mice, rNLS9B mice back on Dox showed no change in MN number and a significantly higher percentage of innervated NMJs (Fig. S13a,b). In addition, although 3 mice progressed rapidly to disease end stage, 12 of 15 rNLS9B mice back on Dox showed a partial recovery of hindlimb clasping phenotype, improvement in the wirehang test, progressive weight gain, and extended survival (Fig. S13c–f). Notably, although 3 of the 12 recovered rNLS9B mice were killed for analysis after 4 months back on Dox, the 9 remaining recovered rNLS9B mice remained alive without progression of disease at between 5 and 8 months back on Dox, far past when all 17 of the rNLS9B littermate mice which were not returned to Dox had succumbed to disease (Fig. S13f).
Discussion
Our newly generated rNLS mice faithfully recapitulate many of the pathological features of ALS and FTLD-TDP. The previous development of numerous TDP-43 in vivo models has largely failed to recapitulate the combined key features of TDP-43 pathology including neuron loss, muscle denervation/atrophy and a progressive motor phenotype leading to premature death. The major reasons for our success include the use of the NEFH promoter to direct hTDP-43∆NLS expression not only to brain but also to spinal cord, accounting for the spinal motor neuron loss and muscle denervation leading to a more robust ALS-like phenotype. Our results also indicate that the NEFH promoter drives higher levels of expression than the Camk2a promoter (>10 fold versus 8 fold, respectively, in the cortex), which in turn increased hTDP-43 and resulted in the accumulation of abundant pathological pTDP-43 detected by both biochemical and histological methods [22]. Although high levels of over-expression of TDP-43 artificially targeted to the cytoplasm produces a model which diverges from the circumstances in human disease, given that the initial upstream initiation of disease has been hypothesized to require multiple pathogenic ‘hits’ [26, 34], this strategy has nevertheless allowed us to overcome the need for such multiple triggers and resulted in the most disease-relevant TDP-43 model produced to date. Moreover, our strategy of targeting hTDP-43 to the cytoplasm, the location of accumulation in human disease, instead of to the nucleus, allowed for higher hTDP-43∆NLS accumulation before the onset of neurotoxicity and brought about pathology more reminiscent of ALS/FTLD-TDP than was seen with expression of nuclear hTDP-43-WT.
It should be noted that although TDP-43 pathology accumulates in both brain and spinal cord in the rNLS mice, testing of cognitive function relevant to FTLD-TDP phenotypes is confounded by the early and robust progressive motor impairment. However, the common pathology in ALS and FTLD-TDP indicate that these diseases are syndromic variants of neurodegenerative TDP-43 proteinopathies, and suggest that our results could be generalized to both diseases. Indeed, our data provide insights into the role of neuron loss and TDP-43 phosphorylation in disease pathogenesis. Specifically, the motor phenotypes in the rNLS8 mice, which begin with hindlimb clasping, are detected at 1–2.5 weeks off Dox (Fig. 4a), prior to the detection of overt brain atrophy, NMJ denervation and neuron loss, thereby suggesting that the initial dysfunction seen in these animals is not due to simple neuron death but instead by an earlier disruption to neuronal function. Despite high levels of hTDP-43∆NLS expression from 1 week off Dox, pTDP-43 is biochemically detected at robust levels only after 4 weeks off Dox in rNLS8 mice, at the point when overt cell loss and muscle denervation begins, suggesting that phosphorylation of TDP-43 may be a signal in an attempt to clear the accumulated insoluble protein. Notably, although both clearance of TDP-43 pathology from the rNLS8 mice and recovery of motor function occurred rapidly following suppression of hTDP-43∆NLS expression, the halting of MN loss cannot be attributed to the removal of inclusions alone, since although they were readily detected, large pTDP-43 inclusions were observed in only a minority of MNs. Despite this, up to 50 % of MNs were lost by 8 weeks off Dox (Fig. 3l), indicating that although expressing cytoplasmic hTDP-43, most MNs that degenerated did not contain pTDP-43 inclusions. These findings suggest that large pTDP-43 inclusions, despite being hallmarks of disease, are not necessarily the cause of neuronal demise. Rather, our findings indicate a complex cascade of events leading to disease development and progression to death. It will be interesting to delineate the identity of the surviving neurons that allow for muscle re-innervation and recovery. Furthermore, it will be important to identify the molecular determinants of those neurons that succumb early in disease versus those that survive and are able to functionally recover following hTDP-43∆TDP-43 suppression.
Another key question that remains to be addressed is the contribution of loss of nuclear TDP-43 versus the accumulation of cytoplasmic TDP-43 pathology, which are both seen in rNLS mice, to the development of disease. Complete knockout of TDP-43 in mice is embryonic lethal [24, 40, 50], and ubiquitous inducible post-natal Cre recombinase-mediated deletion of TDP-43 in mice causes alterations in fat metabolism and death within 9 days [11]. In addition, the targeted depletion of TDP-43 from spinal cord motor neurons using an HB9 promoter-driven Cre-Lox system caused progressive motor phenotypes with motor neuron loss [51]. These studies highlight the importance of maintaining TDP-43 levels for neuronal viability and animal survival, and suggest that loss of normal nuclear TDP-43 function is detrimental to motor neurons and can cause ALS/FTLD phenotypes. However, loss of function is unlikely to be the sole cause of neurodegeneration because over-expression of wild-type TDP-43 resulted in elevated levels of nuclear TDP-43 but also caused disease phenotypes and neuron death in vivo [22, 49]. Thus, no single mechanism may be responsible for the full gamut of pathology and dysfunction in ALS/FTLD-TDP. To address this more specifically, future studies should distinguish the contributions to pathogenesis of a toxic gain of function or a loss of normal function by employing transgenic animals with genetic ablation of auto-regulatory sequences in the TDP-43 3′UTR to investigate the effect of cytoplasmic accumulation of TDP-43 separate from nuclear depletion.
Interestingly, in all three lines of NEFH-tTA mice analyzed, a small minority of astrocytes was found to express the hTDP-43ΔNLS transgene (Fig. 1; Fig. S1). The reason for this unexpected non-neuronal expression from the neuronal-specific NEFH promoter remains unclear, although this result is unlikely to be caused by the random chromosomal site of transgene insertion due to each line being derived from individual founder mice. However, since this is the first report of the use of the human NEFH promoter in transgenic mice, we speculate that there may be differences in the transcriptional recognition of the promoter in mouse compared to human. Regardless, the NEFH-tTA mice showed high levels of Dox-suppressible expression in neurons of the brain and spinal cord, and therefore offer a new model system to allow further investigation not only of TDP-43 but also other proteins involved in neurodegenerative diseases.
Recently, the role of cell-to-cell transmission and propagation of misfolded proteins through the central nervous system (CNS) has suggested a novel mechanism for neurodegenerative disease progression [15, 16, 19, 21, 38]. For example, misfolded tau and α-synuclein induce disease spread in vivo and cause pathology reminiscent of Alzheimer’s and Parkinson’s diseases, respectively [15, 16, 19, 21, 38]. Isolated pathological TDP-43 from ALS and FTLD-TDP patient tissue was also shown to seed aggregation of endogenous TDP-43 following direct transduction of cultured cells [32]. Furthermore, post mortem studies of TDP-43 pathology in ALS and FTLD-TDP patients demonstrate a progressive sequential spread of TDP-43 pathology in brain and spinal cord, suggesting focal initiation of disease with subsequent involvement of interconnected brain and spinal cord regions [7–9]. Our findings here in the rNLS mice indicate that intracellular TDP-43 pathology can be rapidly cleared in vivo, which halts further neurodegeneration, thus suggesting that the presence of intracellular pathological TDP-43 is insufficient for cell-to-cell spread and propagation of disease pathology throughout the neuraxis in the absence of continued cytoplasmic TDP-43 accumulation. It is possible, however, that in the extended disease course of human patients minute quantities of transmitted pathological TDP-43 may lead to amplification of disease over time. Further studies will be required in order to delineate the contribution of spreading of pathology in ALS and FTLD-TDP.
No effective therapy currently exists for either ALS or FTLD-TDP, and the most exciting aspect of this study is the demonstration of functional recovery following suppression of cytoplasmic TDP-43 expression even after the time point when TDP-43 pathology has formed, nuclear TDP-43 has decreased and neurodegeneration has begun. Although these findings in the rNLS mice cannot be directly applied to human ALS/FTLD-TDP therapy due to the reliance on an artificial genetic system in which transgene transcription can be rapidly prevented, they nevertheless show that functional recovery from TDP-43-related disease progression is possible after symptom onset in vivo. TDP-43 is an RNA/DNA binding protein that regulates its own expression via a direct auto-regulatory mechanism involving binding of TDP-43 to the 3′UTR of its own transcript [3, 22, 35]. For this reason, over-expression of exogenous TDP-43 (lacking the 3′UTR required for auto-regulation) causes down-regulation of endogenous TDP-43, and in the case of over-expression of cytoplasmically targeted hTDP-43 in rNLS mice, this leads to a loss of nuclear mTDP-43 (Fig. 2a; Fig. S11a, d) [22]. Therefore, in rNLS mice returned to Dox, suppression of cytoplasmic hTDP-43 expression would release the auto-regulatory inhibition of mTDP-43 transcription, leading to the return of nuclear mTDP-43 by renormalization of homeostatic TDP-43 expression. We cannot distinguish whether the clearance of cytoplasmic TDP-43 or the return of nuclear TDP-43 is responsible for the dramatic recovery seen, and indeed the combination of both phenomena may be required for this effect. However, accumulation of cytoplasmic TDP-43 occurs only in disease and directly leads to a decrease in nuclear TDP-43 levels. We therefore hypothesize that clearance of cytoplasmic TDP-43, which allows for the return of normal nuclear TDP-43, could be a therapeutic strategy for ALS and FTLD-TDP, even when disease is very advanced. Aggregated TDP-43 is cleared primarily by autophagy in cell culture [39], and thus autophagy is a likely mechanism by which the rapid clearance of hTDP-43ΔNLS occurs following transgene suppression. Indeed, small molecule enhancers of autophagy have shown promise in previous TDP-43 cell and mouse models [6, 46], suggesting that modulation of degradation may be one avenue for removal of insoluble and pathological TDP-43.
Previous attempts to delineate the effects of reversibility of phenotype due to hTDP-43 expression in vivo have produced conflicting results, likely due at least in part to the shortcomings of the previously available models. For example, although clearance of cytoplasmic hTDP-43∆NLS from the brain of Camk2a-tTA/tetO-hTDP-43∆NLS mice, which develop some forebrain neuron loss but lack a degenerative motor phenotype, allows for behavioral recovery when mice are returned to Dox at a young age, older mice showed no recovery at 2 weeks post Dox re-introduction [1]. Furthermore, NEFH-tTA/tetO-hTDP-43M337V rats showed a partial recovery of motor function and extension of lifespan upon transgene suppression, but due to the expression of primarily nuclear-localized mutant TDP-43M337V and the development of a very rapid disease course without notable TDP-43 accumulation or overt neuron loss in these animals, longitudinal analysis is difficult and their relevance to human disease is unclear [20]. Our findings now show marked muscle re-innervation in rNLS mice returned to Dox, likely as a result of axonal sprouting, even after significant neuron loss and muscle denervation had occurred (Fig. 8d, e). Thus, suppression of continued cytoplasmic hTDP-43 expression prevents further neuron loss, and surviving motor neurons can re-innervate muscle leading to functional recovery.
We here report new rNLS mouse models which for the first time recapitulate cytoplasmic TDP-43 pathology reminiscent of ALS and FTLD-TDP in brain and spinal cord, accompanied by a progressive neurodegenerative ALS-like phenotype. Our findings indicate that in symptomatic rNLS mice, clearance of cytoplasmic TDP-43 and normalization of nuclear TDP-43 levels prevents continued neurodegeneration and allows for muscle re-innervation, leading to functional recovery and dramatic extension of lifespan. Our results therefore highlight the exquisite ability of the CNS to recover from disease-associated dysfunction even at advanced stages of disease in new models of ALS/FTLD-TDP.
References
Alfieri JA, Pino NS, Igaz LM (2014) Reversible Behavioral Phenotypes in a Conditional Mouse Model of TDP-43 Proteinopathies. J Neurosci 34:15244–15259. doi:10.1523/jneurosci.1918-14.2014
Arnold ES, Ling SC, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D et al (2013) ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci 110:E736–E745. doi:10.1073/pnas.1222809110
Ayala YM, De Conti L, Avendano-Vazquez SE, Dhir A, Romano M, D’Ambrogio A et al (2011) TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J 30:277–288. doi:10.1038/emboj.2010.310
Ayala YM, Zago P, D’Ambrogio A, Xu YF, Petrucelli L, Buratti E et al (2008) Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 121:3778–3785. doi:10.1242/jcs.038950
Balin BJ, Clark EA, Trojanowski JQ, Lee VM (1991) Neurofilament reassembly in vitro: biochemical, morphological and immuno-electron microscopic studies employing monoclonal antibodies to defined epitopes. Brain Res 556:181–195
Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM et al (2014) Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol 10:677–685. doi:10.1038/nchembio.1563
Braak H, Brettschneider J, Ludolph AC, Lee VM, Trojanowski JQ, Del Tredici K (2013) Amyotrophic lateral sclerosis–a model of corticofugal axonal spread. Nat Rev Neurol 9:708–714. doi:10.1038/nrneurol.2013.221
Brettschneider J, Del Tredici K, Irwin DJ, Grossman M, Robinson JL, Toledo JB et al (2014) Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol 127:423–439. doi:10.1007/s00401-013-1238-y
Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M et al (2013) Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74:20–38. doi:10.1002/ana.23937
Chen-Plotkin AS, Lee VM, Trojanowski JQ (2010) TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol 6:211–220. doi:10.1038/nrneurol.2010.18
Chiang PM, Ling J, Jeong YH, Price DL, Aja SM, Wong PC (2010) Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci 107:16320–16324. doi:10.1073/pnas.1002176107
Geser F, Brandmeir NJ, Kwong LK, Martinez-Lage M, Elman L, McCluskey L et al (2008) Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol 65:636–641. doi:10.1001/archneur.65.5.636
Geser F, Martinez-Lage M, Robinson J, Uryu K, Neumann M, Brandmeir NJ et al (2009) Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol 66:180–189. doi:10.1001/archneurol.2008.558
Gordon PH, Meininger V (2011) How can we improve clinical trials in amyotrophic lateral sclerosis? Nat Rev Neurol 7:650–654. doi:10.1038/nrneurol.2011.147
Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B et al (2013) Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 154:103–117. doi:10.1016/j.cell.2013.05.057
Guo JL, Lee VM (2014) Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 20:130–138. doi:10.1038/nm.3457
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD et al (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264:1772–1775
Hirasawa M, Cho A, Sreenath T, Sauer B, Julien JP, Kulkarni AB (2001) Neuron-specific expression of Cre recombinase during the late phase of brain development. Neurosci Res 40:125–132
Holmes BB, Furman JL, Mahan TE, Yamasaki TR, Mirbaha H, Eades WC et al (2014) Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci 111:E4376–E4385. doi:10.1073/pnas.1411649111
Huang C, Tong J, Bi F, Zhou H, Xia XG (2012) Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J Clin Invest 122:107–118. doi:10.1172/jci59130
Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM (2013) Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 33:1024–1037. doi:10.1523/jneurosci.2642-12.2013
Igaz LM, Kwong LK, Lee EB, Chen-Plotkin A, Swanson E, Unger T et al (2011) Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest 121:726–738. doi:10.1172/jci44867
Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M et al (2008) Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol 173:182–194. doi:10.2353/ajpath.2008.080003
Kraemer BC, Schuck T, Wheeler JM, Robinson LC, Trojanowski JQ, Lee VM et al (2010) Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol 119:409–419. doi:10.1007/s00401-010-0659-0
Kwong LK, Irwin DJ, Walker AK, Xu Y, Riddle DM, Trojanowski JQ et al (2014) Novel monoclonal antibodies to normal and pathologically altered human TDP-43 proteins. Acta Neuropathol Commun 2:33. doi:10.1186/2051-5960-2-33
Lee EB, Lee VM, Trojanowski JQ (2012) Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci 13:38–50. doi:10.1038/nrn3121
Lee VM, Carden MJ, Schlaepfer WW, Trojanowski JQ (1987) Monoclonal antibodies distinguish several differentially phosphorylated states of the two largest rat neurofilament subunits (NF-H and NF-M) and demonstrate their existence in the normal nervous system of adult rats. J Neurosci 7:3474–3488
Lee VM, Page CD, Wu HL, Schlaepfer WW (1984) Monoclonal antibodies to gel-excised glial filament protein and their reactivities with other intermediate filament proteins. J Neurochem 42:25–32
Ling SC, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79:416–438. doi:10.1016/j.neuron.2013.07.033
Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y et al (2009) Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol 117:137–149. doi:10.1007/s00401-008-0477-9
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. doi:10.1126/science.1134108
Nonaka T, Masuda-Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T et al (2013) Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep 4:124–134. doi:10.1016/j.celrep.2013.06.007
Paxinos G, Franklin KBJ (2001) The mouse brain in stereotaxic coordinates, 3rd edn. Academic Press, San Diego
Pesiridis GS, Tripathy K, Tanik S, Trojanowski JQ, Lee VM (2011) A “two-hit” hypothesis for inclusion formation by carboxyl-terminal fragments of TDP-43 protein linked to RNA depletion and impaired microtubule-dependent transport. J Biol Chem 286:18845–18855. doi:10.1074/jbc.M111.231118
Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY et al (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14:459–468. doi:10.1038/nn.2779
Robertson J, Sanelli T, Xiao S, Yang W, Horne P, Hammond R et al (2007) Lack of TDP-43 abnormalities in mutant SOD1 transgenic mice shows disparity with ALS. Neurosci Lett 420:128–132. doi:10.1016/j.neulet.2007.03.066
Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A et al (2006) Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol 169:1343–1352. doi:10.2353/ajpath.2006.060438
Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A et al (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82:1271–1288. doi:10.1016/j.neuron.2014.04.047
Scotter EL, Vance C, Nishimura AL, Lee YB, Chen HJ, Urwin H et al (2014) Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J Cell Sci 127:1263–1278. doi:10.1242/jcs.140087
Sephton CF, Good SK, Atkin S, Dewey CM, Mayer P 3rd, Herz J et al (2010) TDP-43 is a developmentally regulated protein essential for early embryonic development. J Biol Chem 285:6826–6834. doi:10.1074/jbc.M109.061846
Shan X, Vocadlo D, Krieger C (2009) Mislocalization of TDP-43 in the G93A mutant SOD1 transgenic mouse model of ALS. Neurosci Lett 458:70–74. doi:10.1016/j.neulet.2009.04.031
Sobue G, Hashizume Y, Yasuda T, Mukai E, Kumagai T, Mitsuma T et al (1990) Phosphorylated high molecular weight neurofilament protein in lower motor neurons in amyotrophic lateral sclerosis and other neurodegenerative diseases involving ventral horn cells. Acta Neuropathol 79:402–408
Toledo JB, Van Deerlin VM, Lee EB, Suh E, Baek Y, Robinson JL et al (2014) A platform for discovery: the University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement 10(477–484):e471. doi:10.1016/j.jalz.2013.06.003
Tsai KJ, Yang CH, Fang YH, Cho KH, Chien WL, Wang WT et al (2010) Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J Exp Med 207:1661–1673. doi:10.1084/jem.20092164
Turner BJ, Baumer D, Parkinson NJ, Scaber J, Ansorge O, Talbot K (2008) TDP-43 expression in mouse models of amyotrophic lateral sclerosis and spinal muscular atrophy. BMC Neurosci 9:104. doi:10.1186/1471-2202-9-104
Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ et al (2012) Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci 109:15024–15029. doi:10.1073/pnas.1206362109
Wang IF, Reddy NM, Shen CK (2002) Higher order arrangement of the eukaryotic nuclear bodies. Proc Natl Acad Sci 99:13583–13588. doi:10.1073/pnas.212483099
Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH (2009) TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci 106:18809–18814. doi:10.1073/pnas.0908767106
Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I et al (2010) TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci 107:3858–3863. doi:10.1073/pnas.0912417107
Wu LS, Cheng WC, Hou SC, Yan YT, Jiang ST, Shen CK (2010) TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis 48:56–62. doi:10.1002/dvg.20584
Wu LS, Cheng WC, Shen CK (2012) Targeted depletion of TDP-43 expression in the spinal cord motor neurons leads to the development of amyotrophic lateral sclerosis-like phenotypes in mice. J Biol Chem 287:27335–27344. doi:10.1074/jbc.M112.359000
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC et al (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53:337–351. doi:10.1016/j.neuron.2007.01.010
Zhang YJ, Gendron TF, Xu YF, Ko LW, Yen SH, Petrucelli L (2010) Phosphorylation regulates proteasomal-mediated degradation and solubility of TAR DNA binding protein-43 C-terminal fragments. Mol Neurodegener 5:33. doi:10.1186/1750-1326-5-33
Acknowledgments
We thank Drs. Todd Cohen, Edward B. Lee, Sílvia Porta and Kurt Brunden for helpful discussion and input on the manuscript, Chi Li and Clark Restrepo for technical assistance, Drs. Manuela Neumann and Elizabeth Kremmer for providing the phosphorylation specific TDP-43 rat monoclonal antibody TAR5P-1D3, Dr. Xu-Gang Xia, Thomas Jefferson University for the NEFH-tTA construct, Dr. Chris Henderson, Columbia University for the VAChT antibody, and Dr. Jean Richa of the University of Pennsylvania Transgenic and Chimeric Mouse Facility for transgenic mouse production. This work was supported by NIH/NIA AG032953 and AG17586, and by Australian National Health & Medical Research Council C.J. Martin Biomedical Early Career Fellowship 1036835 (to A.W.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Ethical approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution at which the studies were conducted.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 2 (MP4 10923 kb)
Rights and permissions
About this article

Cite this article
Walker, A.K., Spiller, K.J., Ge, G. et al. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol 130, 643–660 (2015). https://doi.org/10.1007/s00401-015-1460-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1460-x