
Abstract
Sparsely branched polyolefins often exhibit a thermorheological complexity, which was reported to be maskable by a modulus shift. However, the only physical background for a modulus shift is a density change, and this influence factor is only small in the relatively narrow temperature regime accessible by polyolefins. This paper deals with the question, how this modulus shift can be caused by experimental artifacts and real effects. The physical background of these two contributions to a vertical activation energy, as well as a meaningfulness of the application of a modulus shift, is found not to be given for polyolefins, when measuring only in a temperature range between 130 and 230 °C.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The thermorheological behavior of polymers has been of great interest since the first days of polymer science. One of the main reasons for that is its importance for processing operations, which are performed under non-isothermal conditions and, thus, can be greatly affected by the temperature dependence of the rheological behavior. The thermorheological behavior is determined by two processes in general. Close to the glass transition, the temperature dependence is determined by the free volume, which leads to a so-called Williams–Landel–Ferry (WLF)-temperature dependence (Williams et al. 1955).Footnote 1 Significantly above the glass transition, the temperature dependence is governed by a weaker Arrhenius type dependence. Usually the WLF dependence is valid at temperatures between Tg and about 100 K above Tg, while the Arrhenius dependence is valid at T > Tg + 200 K and below Tg. In the intermediate regime, the temperature dependence is dependent on the polymer and might be a mixture of both types of temperature dependencies (Münstedt and Schwarzl 2014). In solid state, i.e., below Tg, the time-temperature superposition principle (tTS-principle) often fails, and when it is fulfilled, Arrhenius dependences are observed (Capodagli and Lakes 2008; Hartwig 1994; Stadler et al. 2005).
Schwarzl and Staverman (1952) shaped the term thermorheological simplicity and thermorheological complexity for the cases, where the tTS-principle is valid or not, respectively. The thermorheological simplicity is nowadays considered to be rather the exception than the rule, considering the many sources of a thermorheological complexity—among them structural changes (crystallization (e.g., Hadinata et al. 2006), hydrodynamic volume (e.g., Hashmi et al. 2015), interactions between different phases (e.g., Bates 1990)), and different temperature dependencies of individual processes, e.g., of the relaxations of sparsely long-chain branched samples (e.g., Wood-Adams and Costeux 2001). Interestingly, the thermorheological complexity and changes to shift factors due to long-chain branches have only been reported for samples following an Arrhenius temperature dependence, but not a WLF one (e.g., Hepperle et al. (2005), Kapnistos et al. 2006). This, however, can be explained by the governing mechanisms of these temperature dependencies. It is obvious that the free volume of a polymer (WLF-temperature dependence) is not significantly influenced by the presence of branches as long as they are sparse (it is unknown what the maximum level of branching for “sparse” is), while for an Arrhenius type temperature dependence, the potential barrier is decisive, which is determined, among others, by the number of branches (e.g., Lohse et al. 2002), which directly leads to thermorheological complexity, when processes with different activation energies are present, which is the case for segmental and arm motions, e.g., (Carella et al. 1986).
The “vertical activation energy” postulated by Mavridis and Shroff (1992), cited frequently, is trying to mask the presence of a thermorheological complexity, i.e., the failure of the tTS-principle, in Ziegler-Natta catalyzed polyethylene (ZN-PE) and low-density polyethylene (LDPE). The concept of the “vertical activation energy” relies on the assumption that the shape of the material function remains unaltered, being not only shifted along the time t (or shear rate γ ̇ or frequency ω) domain (→ a T ) but also along the modulus (or viscosity) domain (→ b T ), which is therefore called a vertical shift factor. Because of the “modulus direction”, b T is also referred to as a modulus shift factor. The modulus shift can be used as an additional degree of freedom in the creation of a master curve and consequently create a better fit of the data determined at different temperatures. As the modulus is not necessarily plotted as the y-axis, it is not entirely correct to term b T a “vertical shift factor”, thus, the term “modulus shift factor” is more appropriate and used in the paper predominantly.
With the combination of the two shift parameters, Mavridis and Shroff (1992) determined not only a horizontal activation energy E a H, being equivalent to what is normally referred to as activation energy E a , but also a “vertical activation energy” E a V, calculated from the modulus shift factors b T . Unlike the normal Arrhenius argumentation, being the physical process behind the horizontal activation energy E a H, Mavridis and Shroff (1992) did not supply a physical explanation for the “vertical activation energy” E a V nor did they discuss any correlations between E a V and the molecular structure. In the following, E a will be used instead of E a H for the horizontal activation energy.
As an alternative to that concept, it was shown by Wood-Adams and Costeux (2001) before that sparse long-chain branches lead to an increase of the terminal activation energy E a 0 as well as to a failure of the tTS-principle, as the shape of the relaxation spectrum changes. Hence, the modulus shift alone cannot be applied for the creation of a perfect master curve. Wood-Adams and Costeux (2001) suggested using a plot of modulus (in linear scaling) vs. shifted frequency in logarithmic scaling to detect thermorheological complexity. Carella et al. (1986) reported a similar approach for hydrogenated polybutadiene (PBd) stars.
The intent of this comment is to discuss the possible sources of a modulus shift-density changes and thermorheological complexity. Furthermore, it is intended to show under which conditions a modulus shift makes physical sense.
Experimental
The measurements were performed on LB4, a commercially available LCB-mLLDPE (LCB—long-chain branch (ed), mLLDPE—metallocene catalyzed linear LDPE) with weight average molar mass M w = 85 kg/mol, polydispersity index M w/M n = 2.3 (determined from SEC-MALLS (size exclusion chromatography with coupled multi-angle laser light scattering) (see Stadler et al. (2006a) for experimental details) and on LCB-mHDPE B9 (mHDPE—metallocene catalyzed high-density polyethylene), a laboratory scale product synthesized by Piel et al. (2006) with M w = 65 kg/mol and M w/M n = 2—this product was also discussed with respect to thermorheological complexity elsewhere (Stadler et al. 2008).
The rheological data were measured by frequency sweeps in the range between 628 and 0.01 s−1 and—in the case of LB4—augmented by oscillatory data calculated from the retardation spectrum calculated from creep recovery tests (Kaschta and Schwarzl 1994a, b). All measurements were performed in the linear viscoelastic regime on a TA Instruments AR-G2 or ARES in nitrogen atmosphere. An in-depth description of the experimental methods for the rheological measurements is given elsewhere (Stadler and Münstedt 2009; Stadler et al. 2006b).
The data for assessing the gap dependence of the viscosity were measured using a Paar Physica MCR 52 with polydimethylsiloxane (AK1000000, Wacker Chemie, Burghausen, Germany) using a 25 mm parallel plate geometry by applying a constant shear rate experiments with a shear rate of 1 s−1 for 60 s at T = 40 °C. The presented data are the averages of the datapoints for the last 40 s of this test, although almost constant viscosities were obtained after 2 s using this setup. The sample was loaded and trimmed at a trim gap of 1.025 mm to make measurements at a default gap of 1 mm. The gap was set to values between 1.3 and 0.5 mm, leading to significantly distorted samples. Furthermore, the distorted sample at 0.5 mm was trimmed and measured again for checking the gap-independence of the rheological properties which proved that the setup used was in good shape.
Results
Modulus shift factors induced by thermal expansion of the geometry
Like (almost) every material, also stainless steel, usually used for rheometer geometries, exhibits a thermal expansion. The thermal expansion coefficient of a measurement system mainly depends on the construction of the geometry, primarily on the length of the geometry and on the material. One has to be aware that the oven also plays a role. If the same geometry is used in different ovens or ovens of different size, the thermal expansion coefficients do not necessarily have to agree, as a small Peltier oven heats up the geometry less than a forced convection oven, which is usually also larger.Footnote 2
Figure 1 shows the measurement of the thermal expansion performed for a 25 mm parallel plate geometry in a TA Instruments ARES. The data can be very well fitted with a linear correlation with a slope of 2.54 μm/°C, which is the thermal expansion coefficient of the geometry. As an experience value, a thermal expansion of 1–1.5 μm/°C per geometry is typically found—in case of the ARES, the overall expansion coefficient is higher because an upper and a lower geometry is used.

Thermal expansion of a standard 25 mm parallel plate geometry of a TA Instruments ARES
The typical measurement range of PE (for which the “vertical activation energy” has been mainly used so far) is between slightly above the melting point (for HDPE 150 °C, somewhat lower for LDPE and LLDPE) and 230 °C, because the thermal stability of PE usually is insufficient for reliable rheological measurements at higher temperature.
In this case, HDPE is measured in a temperature interval of 80 °C, which is equal to a thermal expansion gap change of 203 μm for an uncompensated thermal expansion coefficient of 2.54 μm/°C. When a gap of 1 mm is assumed, an apparent “vertical activation energy” of −5 kJ/mol is found, which will distinctly influence the horizontal shift factor a T , if not properly compensated. Doubling the gap will reduce the effect by 50 %, as the geometry expansion stays the same, while the effect on the modulus scales with 1/gap.
It is also obvious from Fig. 2 that the fit is not very good due to a slight curviness resulting from the reciprocal temperature in Arrhenius plots. However, this deviation from the linear reference is so small that it probably cannot be detected in real experiments.

Arrhenius plots of the apparent “vertical activation energy” being the consequence of the thermal expansion of the geometry
Modulus shift factors induced by changes of the sample density
Many of the state-of-the-art shear rheometers offer either a thermal expansion compensation by software or even a measurement system of the actual gap. If this system is properly used, these problems do not arise, but besides the tool expansion also the melt expansion has to be considered, which is caused by a change of the sample density. However, quite often, these automatic thermal expansion compensation systems do not work properly—e.g., due to incorrect configuration or calibration, thus making manual adjustments necessary.
For this reason, performing measurements with a cone and plate geometry proves much more difficult with respect to thermal gap compensation in comparison to a measurement with a parallel plate geometry, as the “gap” of these systems theoretically is 0.
For PE, this density change is very small in the range between 150 and 200 °C, and thus can be neglected for most applications. Olabisi and Simha (1975) established that for HDPE the density in the changes by 3.9 % in this interval. Kessner et al. (2009) found a similarly small effect for LDPE; their data are shown in Fig. 7.
Thus, the thermal compensation follows as: If a thermal expansion coefficient of 2.54 μm/°C, an initial gap of 1 mm at 150 °C, and the 3.9 % density change between 150 and 200 °C are used, the (apparent) gap has to be increased by 0.152 mm, i.e., 15.2 %. Additionally, a modulus shift b T of 0.87 due to the density change is physically required for LDPE (Kessner et al. 2009), in theory, which stems from the linear rubber elasticity according to
with ρ and ρ0 being the densities at the measuring temperature T and the reference temperature T0 on the absolute temperature scale, respectively.
However, the reproducibility of the experiments is in the same range as the theoretical b T so that this vertical shift is actually not necessary. If a significantly larger temperature range is accessed, the modulus shift induced by Eq. (1) becomes non-negligible.
Modulus shift factors induced by the sample shape
A polymer melt has a much higher thermal expansion than metals; hence, a change of the temperature leads to a significant change of the sample volume, which of course will change the shape of the sample outer side. This effect will become the stronger the larger a geometry is, as the sample cross section depends on the radius with a square power, while the circumference depends linearly on it. Hence, for example, decreasing the gap by 10 % will lead to the sample bending outward by ca. 5.4 % of the radius, when assuming that the disk-like shape is retained—which is not the case as the protruding shape will not be disk-like but rather more or less rounded (depending on viscosity). In case of a small 8 mm geometry (radius; R = 4 mm), the theoretical sample diameter will be 8.43 mm, which will have a rather small effect on the data, as a sample bulging out by about 1/4 to 1/3 of the gap (typically 1–2 mm) will have very little effect on the data. For a 50 mm geometry, the same situation will lead to a theoretical increase of sample diameter by 2.7 mm, which will have a significant effect on the data and will in case of lower viscous sample most likely lead to the sample dripping down. However, for inward bent samples, this geometry size dependence will not be present, as the data scale with viscosity η ~ 1/R4 for Newtonian fluids and, hence, the absolute deviation of radius of the sample (being smaller than the geometry) does not matter for the error introduced but only its relative deviation.
This will have a significant effect on the data, if the free sample surface bends inward (in this case, the actual radius of the sample is lower than that of the nominal radius of the geometry). However, also an outward bent sample surface has an effect on the data, too, although it is significantly smaller than an inward bent one (typically the maximum error of a bent-out surface is < +50 % in |G *|).Footnote 3
Figure 3 demonstrates this effect on the example of the viscosity of PDMS AK100000 at T = 40 °C and a shear rate of 1 s−1, when adjusting the gap without trimming the sample properly. This sample was loaded into a 25 mm parallel plate geometry and then properly trimmed. It is obvious that the change in viscosity is significantly steeper when the gap is too big than when it is too small. This is logical, as a too big gap leads to a reduced sample cross section, which as mentioned before is related to η ~ 1/R4. The outward bent part of the sample does not have a well-defined force connection and, hence, has a significantly smaller effect on the rheology, which, as can be seen below, will eventually lead to a leveling off of this artificial increase of viscosity at about 17 % for this material and the chosen geometry. Trimming the sample at 0.5 mm properly again leads to a the correct viscosity, proving that the geometry itself is ok. While it can be speculated how different viscosities influence the effect of outward bent samples, it is clear that the findings will be qualitatively the same.

Viscosity upon change of gap without proper trimming
For this reason, it is important to check the free sample surface before starting a test and after the test has finished—the latter becomes the more important the lower the sample is in viscosity and the more severe the applied deformations were (especially for high viscosity samples). In order to avoid geometry errors, the free sample surface should be approximately the same at all temperatures. As an experience value, it is the best to try to have the free surface bent outwards in the middle of the gap by about ¼ to 1/3 of the gap.Footnote 4
If all of those geometry-related issues are taken care of, the modulus shift factor should be very small and, thus, only detectable, when measuring over a large temperature scale, which is usually only possible for amorphous polymers. Should the sample be sufficiently thermostable and viscous, measurements in a temperature range of up to 200 °C are possible, which, however, can lead to problems with geometry compliance, when going to the onset of the glass transition (Liu et al. 2011; Stadler and van Ruymbeke 2010). The origin of this vertical shift lies in the density change of the melt (e.g., Colby et al. 1987; Liu et al. 2006).
Influence of a modulus/vertical shift on the horizontal shift factors
One of the main questions arising from the experimental artifacts and the density change discussed above is the question, how much they actually influence the horizontal shift factors and thus E a . Therefore, a way has to be found to assess the error in the horizontal shift factor a T induced by a modulus shift factor b T .
To perform such an estimation, a data set was shifted with a shift factor a T = 0.2 (which corresponds to E a = 35 kJ/mol for measurement temperatures of 150 and 230 °C) and b T = 0.9, 1, and 1.1. This was done to give the reader a better idea of what scale of effect on horizontal activation energy E a has to be expected.
The apparent a T was then determined and plotted as a function of the double logarithmic slope (i.e., dlog (rheological parameter)/dlog (frequency ω or time t), e.g., dlogG’/dlogω or dlogG”/dlogω) (Fig. 4), as neither the shape nor the quantity being shifted but only the local slope influences the b T -influence on a T . While a T = 0.2 is retained for b T = 1, a modulus shift factor b T = 1.1 apparently increases a T to 0.36 at a slope of 0.1 (reducing the apparent activation energy E a to ≈ 20 kJ/mol (for the set of input parameters in the previous paragraph)). For b T = 0.9, the apparent horizontal shift factor a T decreases to about 0.1 at a slope of 0.2 (apparent E a ≈ 50 kJ/mol (for the set of input parameters in the previous paragraph)).

Change of the horizontal shift factors aT by the introduction of a modulus shift factor bT as a function of the magnitude of the double logarithmic derivative
Thus, the influence of the vertical shift is most significant close to plateaus in the modulus, i.e., rubbery and glassy plateau, but in principle, this applies to all plateaus. Around dlogG’/dlogω = dlogG”/dlogω = 0.7 (Winter and Chambon 1986)—this is the crossover frequency for critical gels—the error of such a large modulus shift is ±15 %, which means that while the effect can be quite significant in the rubbery plateau (which is hard to access for semi-crystalline polymers), it will not influence the terminal regime very much.
The consequence of this is that the reliability of the activation energy E a increases with an increasing slope, i.e., in the terminal regime. In the rubbery or glassy plateau, however, the small slope of the different quantities makes a reliable determination of a T —to say the least—very difficult. Hence, modulus shifts are usually applied in the plateau regions (rubbery, glassy, …), while in regimes of higher slopes, namely in the terminal regime, it can be safely ignored in most cases (see also Fig. 4).
An important quality check is to determine the shift factors a T from “modulus free” quantities, i.e., phase angle δ or tanδ. If the shift factors a T determined from δ on the one hand and G’ and G” on the other hand match, a vertical shift is not necessary. However, one has to keep in mind that LDPE’s thermorheological complexity suggests the necessity of a modulus shift, while the phase angle can be shifted without finding this complexity (Kessner et al. 2009).
Hence, in general, the possibility to construct a very good master curve without using a modulus shift is a good indicator for a high experimental quality, while an insufficient master curve cannot be necessarily attributed to bad experimental quality but also to thermorheological complexity.
It seems to be a general rule that the smaller component of the complex moduli G’ (ω) and G” (ω) shows the features of the molecular structure more clearly than the bigger one. It was, for example, also established that the relaxation spectrum (plotted as 1/τ) and the smaller component of G’ (ω) and G” (ω) are quite similar for realistic spectra (Stadler 2010; Stadler and Bailly 2009). This statement is closely related to above argument of the double log. slope influence of the activation energy. Hence, using this lower, almost always steeper component of the modulus for checking the quality of master curves will lead to shift factors, which are less affected by vertical shift factors that are present in the system.
Detection of a thermorheological complexity
For narrowly distributed LCB-mPEs, a modulus shift does not work properly, as the different parts of the relaxation spectrum (fingerprint of the linear and long-chain branched chains) are well separated. This can be seen from the double bump shape of the spectra of LCB-mPE (Fig. 5), which is temperature dependent. The main one at low relaxation times is the trace of the linear chains, while the shoulder at high times is caused by the long-chain branches. However, this plot is not very suitable for detecting thermorheological complexity as well as G’ (ω) and G” (ω). Wood-Adams and Costeux (2001) suggested the plot complex modulus vs. the product of zero-shear viscosity and frequency (in linear-linear scaling), which like any change of scaling (linear, logarithmic, inverse, …) of data highlights insufficient matches of the best match in different scaling. As Kessner et al. (2009) and Resch et al. (2011) pointed out, for LDPE, a modulus shift is roughly sufficient to obtain a good master curve (see Resch et al. (2011) for spectra equivalent to Fig. 5).

Plot of the relaxation spectrum g (τ) (Kaschta and Stadler 2009) of LCB-mHDPE B9 at different temperatures
Therefore, a simple vertical shift might reduce the thermorheological complexity, but will definitely not lead to perfect TTS for LCB-mPE. This becomes evident when looking at the δ (|G *|)-plot of LCB-mHDPE B9, where it is not possible to resolve the issue of thermorheological complexity completely by modulus shifting as the shape changes. The δ (|G*|)-plot of B9 at 230 °C is almost a straight line between |G *| = 80,000 and 800 Pa, while the same modulus interval has a dent at 150 °C (Fig. 6a).

δ (|G *|)-plot of LCB-mHDPE B9, a without a vertical shift (b T = 1), b with a vertical shift b T . The solid thick line is the reference curve established for mPE with Mw/Mn ≈ 2 (Stadler and Mahmoudi 2011)
To demonstrate that, it was tried to find a “master curve” for B9 by modulus shifting in the δ (|G*|)-plot. If the material is thermorheologically simple, a modulus shift will lead to a master curve. However, as can be seen from Fig. 6b, unrealistically high values for b T are necessary for making this “master curve” (b T = 0.41 for a temperature difference of 80 °C). It is also visible that the “master curve” is only valid in the middle of the range of the range of complex moduli tested here. At the highest moduli, it clearly fails systematically (see arrow).
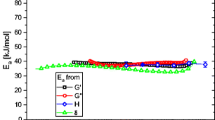
The modulus shift factors b T obtained from Fig. 6b (which are physically not meaningful) are given in Fig. 7 exhibiting an Arrhenius temperature dependence, but the “vertical activation energy” of 20 kJ/mol is too high to have a physical meaning. The “vertical activation energies” found by Mavridis and Shroff (1992) and Kessner et al. (2009) are much lower in comparison. While LDPE seems to have a thermorheological complexity that can be fully compensated for by a modulus shift, some evidence, which will be published by the author soon, suggests that this statement cannot be entirely correct, but that the thermorheological complexity can only be corrected to a certain extent.

While the data of B9 might still be not sufficiently convincing that a modulus shift does not make sense, the data of LB1 spanning a much wider range of frequencies (extended by creep recovery tests) clearly proves that a vertical shift is not possible at all (Fig. 8). While the data obtained at different temperatures collapse nicely for δ < 40 °C and δ > 87°, the intermediate regime clearly deviates. It would, therefore, be necessary to define a frequency (or modulus) dependent modulus shift factor b T , which of course means describing thermorheologically complex behavior in a way to mimic thermorheological simplicity.

δ (|G *|)-plot of LCB-mLLDPE LB4 at 110, 130, 150, 170, and 190 °C
While this example is very clear in terms of the physical meaningfulness of the vertical shift, obtaining such a broad frequency range requires a tremendous experimental effort and can only be done for samples, which are sufficiently stable for reaching the stationary linear regime (for the necessary procedures see, e.g., Gabriel et al. 1998) and whose terminal relaxation is within reasonable limits.
This is especially important as the terminal relaxation time is not given by the zero shear-rate viscosity η 0 , but by having reached the steady-state elastic recovery compliance, which is significantly longer (Stadler and Mahmoudi 2013). Hence, the number of samples, where this procedure has been achieved successfully, is only a fraction of the samples measured by conventional frequency sweeps.
It is not the intention of this paper to discuss the different evaluation methods for thermorheological complexities in detail. It has proven to be most successful to evaluate the apparent activation energies by slicewise finding the shift factors (at constant modulus, phase angle, or relaxation strength). In the next step, the activation energy spectrum is determined as the dependence of activation energy determined from any viscoelastic quantity plotted versus either relaxation time/frequency (Wood-Adams and Costeux 2001) or the quantity from which the activation energy spectrum is determined (Stadler et al. 2008; Resch et al. 2011).
Conclusions
The modulus/vertical shift has been used before to correct for modulus levels in order to obtain good master curves, which cannot be shifted into good master curves without such a modulus shift. However, no physical background for a modulus shift is provided except for the geometry thermal expansion and sample density compensation, which is significantly smaller than the shift factors required to obtain a “perfect master curve” of LCB-HDPE or an LDPE.
A possible candidate also contributing to the “vertical activation energy” might also lie in geometry errors (uncompensated thermal expansion or improper sample shape). It was shown that rather high “vertical activation energies” can be created by neglecting the thermal expansion of geometry (Ev ≈ 5 kJ/mol for a gap of 1 mm and an ARES geometry, smaller gaps will yield higher Ev) and sample (Fig. 3 shows that reaching a factor 3 lower viscosity by a 20 % too big gap is easily possible). Such a gap error might easily occur, when the gap control is insufficiently temperature compensated. Ev ≈ 8 kJ/mol, a value, which can be easily reached by these errors, is the apparent “vertical activation energy” necessary to make typical LDPEs appear thermorheologically simple. Thus, such geometry errors can have a significant effect on conclusions in terms of thermorheological behavior.
To determine whether a vertical shift is necessary is consequently very important. A good way to find out the necessity of vertical shifting is the use of the δ (|G *|)-plot (Fig. 6) and the comparison of the shift factors determined from δ (ω), G’ (ω), and G” (ω). While δ (ω) cannot be corrected by b T , G’ (ω) is not very susceptible to b T in the terminal regime, while being strongly influenced in the rubbery plateau. G” (ω) has a lower slope in the terminal regime and is thus more distinctly influenced by b T , but in the rubbery plateau, it is not constant and thus less influenced by b T in this regime.
Figures 6 and 8, however, clearly shows that the introduction of sparse long-chain branches leads to a distinctly different shape of δ (|G *|) (and also δ (ω), which is not shown in this manuscript) which means that the differences in the rheological behavior at different temperatures cannot be solved by a vertical shift.
If a material with a broad molar mass distribution (MMD)—such as LDPE—is characterized instead of the narrowly distributed LCB-mLLDPE LB4 or LCB-mHDPE B9, discussed here, the “flattened” shape of the material functions will lead to an apparent validity of the tTS-principle by the introduction of a “vertical activation energy”, while the same statement cannot be made for the more narrowly disperse LCB-mLLDPE LB4 or LCB-mHDPE B9. However, already the original paper of Mavridis and Shroff (1992) indirectly puts some doubt to this model, as no correlations between the “vertical activation energy” and the molecular structure were found. Instead, this quantity varies statistically for materials of comparable chemical composition.
Interestingly, the opposite signs and approximately equal magnitude of “vertical activation energy” that can be used to “hide” thermorheological complexity and corrections due to thermal expansion of geometry and sample can lead to an apparently thermorheologically simple behavior, although, e.g., an LDPE is clearly thermorheologically complex.
For the reasons mentioned above, it is concluded that the “vertical activation energy” basically is an empirical tool to “eliminate” the thermorheological complexity in broadly distributed samples without any physical meaning. From the physical point of view, the “vertical activation energy” is not a sensible concept, as an Arrhenius dependence is the physical expression of temperature dependence of the “speed”, at which a thermally activated process is taking place.
The modulus is—by definition—determined by the relaxation spectrum, which does not exhibit a temperature dependence in its relaxation strengths (except for the density effects)—only the relaxation times are changed, unless a thermorheological complexity is present.
Another important factor contributing to this conclusion is that for linear polymers measured under the same conditions using the exact same setup, no vertical shift was found to be necessary. This is a very important indicator that the differences in the behavior actually are linked to the molecular structure and not an experimental artifact. As a temperature dependence of the shape of the spectrum has been identified as the reason the thermorheological complexity of sparsely branched PE and PP (and also fluoropolymers (Stange et al. 2007), it physically reasonable to conclude that the thermorheological complexity is also present in other long-chain branched polymers, following an Arrhenius-type temperature, although at a varying degree. If the MMD is broadened, the different relaxation processes are smeared out and, thus, the detection of the thermorheological complexity becomes more complicated.
Suggested method
The following approach proved to be the most successful in determining the real shift factors. As a first step, the “time/frequency-independent” δ (|G*|)-plot is used to check for the necessity of a vertical shift or a thermorheological complexity. If a vertical shift is necessary, the experiments are carefully check to find out the reason for the vertical shift, mostly being related to geometry issues. However, in case of a very large temperature range (T > 100 K), a vertical shift caused by the density change becomes relevant as well. After the presence of a vertical shift is excluded (or, in the case of a very large temperature range, taken care of), the moduli G’ (ω) and G” (ω) are shifted along the frequency axis. Of course, one has to check, whether the modulus shift is in the right order of magnitude for a density effect (Kessner et al. 2009). After that the shift factors are checked by comparing the shift factors determined from G’, G”, and δ. This way it is barely possible to miss a thermorheological complexity.
This method can be improved further by firstly doing the complete procedure on a similar material without thermorheological complexity, e.g., on an mLLDPE when trying to measure LCB-mLLDPEs, and to optimize all parameter so that mLLDPE is truly thermorheologically simple. If the same parameters will lead to thermorheological complexity on the unknown or branched material, one can be reasonably sure that it is not an experimental artifact.
Summary
Missing a thermorheological complexity in thermorheologically complex samples with a broad MMD is rather easy, when a narrow frequency regime is used (Kessner et al. 2009). In this case, the data might apparently shift vertically to form the ideal master curve. However, this apparent “vertical activation energy” is either caused by neglecting the thermorheological complexity or by geometry errors.
It is, therefore, an auxiliary construct to unphysically avoid using a complicated activation energy spectrum.
In general, it can be stated that modulus shift factors are valid as density change correction, according to the Rouse-model (Eq. (1)). Several other sources of apparent modulus shifts were discussed what causes them and how they can be avoided.
Notes
Measuring the thermal expansion coefficient of a geometry is actually relatively easy. Most modern rheometers have a normal force control. All that has to be done is to zero the gap at a given temperature (in the case presented here room temperature) and set the normal force control to a low force (here: 0.5 N). Then the temperature is changed and the values of the apparent gap (the real gap is zero!) are recorded. It is important to wait until the gap has fully stabilized before recording the point at the respective temperature (this is usually later than when the temperature has stabilized, as stainless steel is a rather bad heat conductor). In case of the setup used, the required waiting time increased with temperature (because the temperature gradient inside the geometry increases) and was between 5 and 10 min. In order to be sure to be in equilibrium, the chosen waiting time was >30 min.
If a rheometer without a normal force controller is used, a thin steel bar should be put in the gap when zeroing it and removing it before increasing the temperature again – this avoids damage to the air bearing due to thermal expansion. At the desired temperature, the gap is to be determined, at which the geometry and the steel bar are locked to each other in the same way as during the initial zeroing. This way the thermal expansion coefficient can be measured with comparable accuracy to the method proposed for rheometers with normal force control.
Hashmi et al. (2012) found that it is possible to use very big samples (about factor 2 larger than necessary to fill the geometry) to use a constant correction factor for getting correct rheological data of hydrogels during swelling. Remmler T (personal communication, 2015) confirmed the small influence of outward bent surfaces, while inward bent surfaces will significantly reduce the viscosity.
The authors’ experience values on this matter are that for samples with acceptably short relaxation times (shorter than 1 min) and thus zero shear-rate viscosity η 0 (<106 Pas), achieving this is not a significant experimental challenge, but for very high viscosity melts achieving such an optimal sample surface proves difficult.
This is one of the reasons of the inferior reproducibility of such samples. Also the insufficient adhesion of these samples to the geometry and the possible presence of orientations and small fractures play a role (such high viscosity samples tend to be very brittle in the melt, ripping, in some cases, even at a Hencky strain εH below 1 in elongational tests).
In case of very low viscosity samples (η 0 < 100 Pas) the gravity prevents an outwards curvature of the free sample surface.
References
Bates FS (1990) Fluctuation effects in symmetric copolymers near the order–disorder transition. J Chem Phys 92(10):6255
Capodagli J, Lakes R (2008) Isothermal viscoelastic properties of PMMA and LDPE over 11 decades of frequency and time: a test of time-temperature superposition. Rheol Acta 47(7):777–786
Carella JM, Gotro JT, Graessley WW (1986) Thermorheological effects of long-chain branching in entangled polymer melts. Macromolecules 19(3):659–667
Colby RH, Fetters LJ, Graessley WW (1987) Melt viscocity-molecular weight relationship for linear polymers. Macromolecules 20(9):2226–2237
Fulcher GS (1925a) Analysis of recent measurements of the viscosity of glasses. J Am Ceram Soc 8:339–355
Fulcher GS (1925b) Analysis of recent measurements of the viscosity of glasses. II. J Am Ceram Soc 8:789–794
Gabriel C, Kaschta J, Münstedt H (1998) Influence of molecular structure on rheological properties of polyethylenes I. Creep recovery measurements in shear. Rheol Acta 37(1):7–20. doi:10.1007/s003970050086
Hadinata C, Gabriel C, Ruellmann M, Kao N, Laun HM (2006) Shear-induced crystallization of PB-1 up to processing-relevant shear rates. Rheol Acta 45(5):539–546
Hartwig G (1994) Polymer properties at room and cryogenic temperatures. Plenum Press, New York
Hashmi S, Obiweluozor F, Ghavaminejad A, Vatankhah-Varnoosfaderani M, Stadler FJ (2012) On-line Observation of Hydrogels during Swelling and LCST-induced changes. Korea-Aust Rheol J 24(3):191–198
Hashmi S, Vatankhah-Varnoosfaderani M, Ghavaminejad A, Obiweluozor FO, Du B, Stadler FJ (2015) Self-associations and temperature dependence of aqueous solutions of zwitterionically modified n-isopropylacrylamide copolymers. Rheologica Acta 54(6):501–516
Hepperle J, Münstedt H, Haug PK, Eisenbach CD (2005) Rheological properties of branched polystyrenes: linear viscoelastic behavior. Rheol Acta 45(2):151–163
Kapnistos M, Koutalas G, Hadjichristidis N, Roovers J, Lohse DJ, Vlassopoulos D (2006) Linear rheology of comb polymers with star-like backbones: melts and solutions. Rheol Acta 46(2):273–286
Kaschta J, Schwarzl FR (1994a) Calculation of discrete retardation spectra from creep data: 2. Analysis of measured creep curves. Rheol Acta 33(6):530–541. doi:10.1007/BF00366337
Kaschta J, Schwarzl FR (1994b) Calculation of discrete retardation spectra from creep data: 1. Method Rheol Acta 33(6):517–529. doi:10.1007/BF00366336
Kaschta J, Stadler FJ (2009) Avoiding waviness of relaxation spectra. Rheol Acta 48(6):709–713
Kessner U, Kaschta J, Münstedt H (2009) Determination of method-invariant activation energies of long-chain branched low-density polyethylenes. J Rheol 53(4):1001–1016
Liu C-Y, Yao M, Garritano RG, Franck AJ, Bailly C (2011) Instrument compliance effects revisited: linear viscoelastic measurements. Rheol Acta 50(5–6):537–546
Liu C-Y, Halasa AF, Keunings R, Bailly C (2006) Probe rheology: a simple method to test tube motion. Macromolecules 39(21):7415–7424
Lohse DJ, Milner ST, Fetters LJ, Xenidou M, Hadjichristidis N, Mendelson RA, Garcia-Franco CA, Lyon MK (2002) Well-defined, model long chain branched polyethylene. 2. Melt rheological behavior. Macromolecules 35(8):3066–3075
Mavridis H, Shroff RN (1992) Temperature-dependence of polyolefin melt rheology. Polym Eng Sci 32(23):1778–1791
Münstedt H, Schwarzl FR (2014) Deformation and flow of polymeric materials. Springer, Heidelberg
Olabisi O, Simha R (1975) Pressure-volume-temperature studies of amorphous and crystallizable polymers. I. experimental. Macromolecules 8:206–210
Piel C, Stadler FJ, Kaschta J, Rulhoff S, Münstedt H, Kaminsky W (2006) Structure–property Relationships of linear and long-chain branched metallocene high-density polyethylenes characterized by shear rheology and SEC-MALLS. Macromol Chem Phys 207(1):26–38
Resch JA, Keßner U, Stadler FJ (2011) Thermorheological behavior of polyethylene: a sensitive probe to molecular structure. Rheol Acta 50(5–6):559–575
Schwarzl F, Staverman AJ (1952) Time-temperature dependence of linear viscoelastic behavior. J Appl Phys 23:838–843
Stadler FJ, Kaschta J, Münstedt H (2005) Dynamic-mechanical behavior of polyethylenes and ethene-/α-olefin-copolymers. Part I. α′-Relaxation. Polymer 46(23):10311–10320
Stadler FJ, Piel C, Kaschta J, Rulhoff S, Kaminsky W, Münstedt H (2006a) Dependence of the zero shear-rate viscosity and the viscosity function of linear high-density polyethylenes on the mass-average molar mass and polydispersity. Rheol Acta 45(5):755–764
Stadler FJ, Piel C, Klimke K, Kaschta J, Parkinson M, Wilhelm M, Kaminsky W, Münstedt H (2006b) Influence of type and content of various comonomers on long-chain branching of ethene/α-olefin copolymers. Macromolecules 39(4):1474–1482
Stadler FJ, Kaschta J, Münstedt H (2008) Thermorheological behavior of various long-chain branched polyethylenes. Macromolecules 41(4):1328–1333
Stadler FJ, Münstedt H (2009) Correlations between the shape of viscosity functions and the molecular structure of long-chain branched polyethylenes. Macromol Mater Eng 294(1):25–34
Stadler FJ, Bailly C (2009) A new method for the calculation of continuous relaxation spectra from dynamic-mechanical data. Rheol Acta 48(1):33–49
Stadler FJ, van Ruymbeke E (2010) An improved method to obtain direct rheological evidence of monomer density reequilibration for entangled polymer melts. Macromolecules 43(21):9205–9209
Stadler FJ (2010) Effect of incomplete datasets on the calculation of continuous relaxation spectra from dynamic-mechanical data. Rheol Acta 49(10):1041–1057
Stadler FJ, Mahmoudi T (2011) Understanding the effect of short-chain branches by analyzing viscosity functions of linear and short-chain branched polyethylenes. Korea-Aust Rheol J 23(4):185–193
Stadler FJ, Mahmoudi T (2013) Evaluation of relaxation spectra of linear, short, and long-chain branched polyethylenes. Korea-Aust Rheol J 25(1):39–53
Stange J, Wächter S, Kaspar H, Münstedt H (2007) Linear rheological properties of the semi-fluorinated copolymer tetrafluoroethylene-hexafluoropropylene-vinylidenfluoride (THV) with controlled amounts of long-chain branching. Macromolecules 40(7):2409–2416. doi:10.1021/ma0626867
Tammann G, Hesse W (1926) The dependence of viscosity upon the temperature of supercooled liquids. Zeitschrift für Anorganische und Allgemeine Chemie 156:245–257
Vogel H (1921) The law of the relation between the viscosity of liquids and the temperature. Phys Z 22:645–646
Williams ML, Landel RF, Ferry JD (1955) The temperature dependence of relaxation mechanisms in amorphous polymers and other glass-forming liquids. J Am Chem Soc 77(14):3701–3707
Winter HH, Chambon F (1986) Analysis of linear viscoelasticity of a cross-linking polymer at the gel point. J Rheol 30(2):367–382
Wood-Adams P, Costeux S (2001) Thermorheological behavior of polyethylene: effects of microstructure and long chain branching. Macromolecules 34(18):6281–6290
Acknowledgments
The authors would like to thank the Nanshan District Key Lab for Biopolymers and Safety Evaluation (No. KC2014ZDZJ0001A) and Shenzhen City (JCYJ20140509172719311). This paper was inspired by several papers we reviewed and referees of our papers, explicitly asking us for using a modulus shift or questioning this concept.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Stadler, F.J., Chen, S. & Chen, S. On “modulus shift” and thermorheological complexity in polyolefins. Rheol Acta 54, 695–704 (2015). https://doi.org/10.1007/s00397-015-0864-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00397-015-0864-9