
Abstract
Ultrathin sectioning and solvent etching were used to assess microstructural changes in polyacrylonitrile (PAN) fibrils following a dry-jet wet spinning process. Morphologic assessment revealed the coexistence of fibrils and lamellae within the PAN fibers, with a frill-like structure found on the fibrils. The size of the lamella-like layers was consistent with a crystal size of 13.2 nm as characterized by X-ray diffraction and microfibrils were composed of alternate dark lamella-like crystal layers and surrounded by bright amorphous chains. Further, fibril arrangement followed a weave-like pattern. During the post-spinning process, break-rearrangement of chain-folded lamellae had an important role in fibril structure evolution. The high stream stretching and heat treatment induced the breaking of lamellae and the formation of new lamellae through the molecular chains rotating and slipping, resulting in a decrease in voids and the coalescence of microfibrils. Consequently, the fibrils were arranged in a more orderly manner and interlinked more tightly, a process conducive for the preparation of high quality PAN fibers.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Most commercial carbon fibers are produced using polyacrylonitrile (PAN) precursors due to their excellent mechanical stability [1,2,3,4]; therefore, their performance is largely dependent on the quality of PAN precursors [5,6,7]. Carbon fibers are commonly produced through a dry-jet wet spinning process, which involves the preparation of the spinning dope to manufacture the PAN precursors. Such processing encompasses a coagulation bath drawing, washing, steam stretching, and densifying [8,9,10]. In the spinning process, the fibril structure acting as the polymer backbone of the PAN fibers is formed by phase separation and coagulation, growing by the orientation of polymer chains during the spinning process. It plays an important role in fiber formation and development of the fiber microstructures, and is therefore worthy of further investigation.
Previous reports have described the fibril structure of PAN fibers. Craig et al. [11] highlighted the presence of fibril-void structures within PAN fibers, formed during coagulation and further developed through stretching and collapsing. Ogle et al. [12] observed dispersed fibrils of diameters of approximately 3.5, 0.4, 0.045, and 0.005 μm, and proposed that each stable fibril formed following the coalescence of smaller microfibrils. Warner et al. [13] showed that PAN fibrils were composed of partially ordered rods of approximately 8 nm in length, interspersed with amorphous regions of approximately 4 nm between the rods. Wang et al. [14, 15] adopted ultrasonic breakup and solvent etching to separate fibrils and found that the macrofibrils were composed of the bundling of microfibrils of approximately 200 nm in diameter parallel to the fiber axis and made up of periodic lamellae 30–45 nm thick.
Despite much research focusing on fibril structures, the underlying mechanisms of arrangement and development during the spinning process remain unclear due to the lack of the effective methods for their assessment. Fibril separation by a suitable solvent solution allows assessment of fibril morphology only on the fiber surface [14,15,16]. Thus, the arrangement and connection of the interior fibrils are difficult to observe. Ultrathin sectioning is commonly used in fiber microstructure analysis and high-resolution transmission electron microscopy (HRTEM) is an indispensable tool to determine these fine structures [17].
Herein, a new method was used to assess the fibril structures. The method involves the preparation of longitudinal ultrathin sections of PAN fibers, etching with dimethyl sulphoxide (DMSO) solvent to separate the fibrils, and examination by HRTEM. We expect that this novel method for the morphological assessment of fibrils will be of benefit in the understanding of PAN fiber microstructures and in optimizing the spinning technology for the fabrication of high-performance carbon fibers.
Experimental
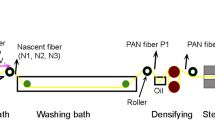
The spinning dope was prepared using 17.7 wt% of PAN (viscosity molecular weight of approximately 3 × 105 g/mol) dissolved in DMSO solvent. Spinning was conducted on a dry-jet wet spinning pilot plant at Shandong University, China. The spinning dope was passed through a spinneret into a coagulation bath, then stretched in drawing baths with draw ratio of 2.2. The PAN fibers were then washed in repeated water baths at 95 °C and successively immersed in an oil-agent bath, then densified at 155 °C. Finally, PAN fibers were treated in high-temperature water vapor by stretching, and a hot setting at 155 °C was applied to improve the fiber structures. In order to assess the fibril microstructural changes during the spinning process, two PAN fiber samples (S1 and S2) were collected (Fig. 1). PAN fiber S2 was obtained from densifying, steam stretching, and hot setting of the PAN S1.

Dry-jet wet spinning process
Several PAN fibers were embedded in epoxy resin solution. Following solidification, ultrathin (50 nm) longitudinal sections were cut with a diamond knife by an ultramicrotome (LKB-V, BROMMA Ltd., Sweden). The sections were separately processed in 70and 85 wt% aqueous DMSO solutions at 75 °C for 15 and 30 min. Finally, the sections were successively and rapidly washed with pure DMSO solvent, a low concentration DMSO solution and distilled water.
A X-ray diffractometer (D/max-c, Rigaku Co., Japan) with nickel-filtered CuKα radiation (wavelength λ = 0.1541 nm) at a scanning rate of 3°/min and a scanning step of 0.02° under 40 kV and 60 m [8] was used to determine the crystal structure of PAN fibers.
The microstructure of ultrathin sections was observed with scanning electron microscopy (SEM, SU-70, Hitachi Co., Japan) at an acceleration voltage of 3 kV following sputter coating with a thin platinum layer. Further, ultrathin sections were examined by HRTEM (JEM-2100, JEOL Ltd., Japan) at an acceleration voltage of 200 kV.
Results and discussion
The morphology of the PAN fiber longitudinal sections is shown in Fig. 2. PAN fiber S1 was primarily made up of lamellae perpendicular to the fiber axis with an average thickness of 45 ± 5 nm (evaluation of 84 lamellae) (Fig. 2a, b). The lamellae structures were similar to those of the fibers processed by ultrasonic etching [14, 15], likely formed by the folding of molecular chains into strip-like lamellar structures, surrounded by a minimally oriented amorphous material. Further, some lamellae were located across several fibrils, connected in a perpendicular direction to the fiber axis. Thus, lamellae were not only present within fibrils but also connected the adjacent fibrils, acting as an inter-fibril linking bridge. Further, a large number of voids were present between lamellae and fibrils. Following steam stretching and heat treatment of PAN fiber S1 to produce fiber S2, the size and number of voids decreased and the transverse strip-like lamellae of PAN fiber S1 were transformed into the node structures within PAN fiber S2 (Fig. 2c, d). Further, the node-like lamella structures with an average thickness of 48 ± 6 nm (evaluation of 76 lamellae) were likely protruding from the lateral of the fibrils, where several adjacent microfibrils were intersected. The changes in the lamellae indicated that the original lamellae (PAN fiber S1) were broken, and the fold-chain from the broken lamellae were rotated and slipped to form node structures (PAN fiber S1) following the post-spinning process. The fibrils were parallel to the fiber axis and had a diameter of 30 ± 6 nm (evaluation of 84 fibrils). The crystalline structure changes were observed by XRD (Fig. 2e). Transformation of the PAN fiber S1 to S2 led to an increase in crystallinity and crystal size from 10.3 to 13.2 nm. In addition, the size of lamellae was obviously larger than the crystal size.

HRTEM morphology of PAN fibers. a, b S1 longitudinal section. c, d S2 longitudinal section. e XRD patterns for fibers S1 and S2
In order to better understand the morphology and development mechanisms of the fibril structures, the morphology of PAN fibers S1 and S2 longitudinal sections following separate etching in 70 wt% DMSO solution for 15 min and 85 wt% DMSO solution for 30 min was observed (Fig. 3a, d). This method was employed to dissolve part of the fibril structures in DMSO, allowing the fibril morphology become visible (Fig. 3b, e). The microfibril diameters in PAN fiber S1 were observed to be within the range of 10 to 100 nm (Fig. 3c). The morphology of some fibrils (Fig. 3c, region Z1) looked like the frill-like structures, with individual fibrils aggregating to form larger fibrils (Fig. 3c, region Z2). The transverse lamellae-like layers, with a thickness of 14 nm, adhered to the lateral side of microfibrils linking the adjacent fibrils (Fig. 3c, region Z3).

HRTEM morphology of PAN fibers. a–c S1 longitudinal section etched in 70 wt% DMSO solution for 15 min. d–f S2 longitudinal section etched in 85 wt% DMSO solution for 30 min
In PAN fiber S2, the fibril diameter was of approximately 66 ± 26 nm (evaluation of 148 fibrils) (Fig. 3e). Similarly to PAN fiber S1, fibril bundles of approximately 110 nm in diameter (Fig. 3f, section A) were composed of fibrils of approximately 26 nm (Fig. 3f, section B). Fibril A showed an alternate dark and bright contrast, typical of lamella-like layers, and the lamellae-like layers of 13 nm observed in fibril B (yellow circle in Fig. 3f) were in good agreement with the lamellar crystal structure proposed by Warner [13]. Further, the size of the layers was consistent with the crystal size (13.2 nm) characterized by XRD (Fig. 2e). Thus, fibril formation occurs by the folding polymer chains leading to small crystallite lamella-like layers surrounded by amorphous chains easily dissolved by DMSO.
SEM images of PAN fiber S1 longitudinal section after etching in DMSO solution showed an interwoven and intersected arrangement and connection of the fibrils rather than the expected side by side packing in a plane (Fig. 4a). The average fibril length was of 4.0 ± 1.1 μm (evaluation of 76 fibrils), with an average diameter of 450 ± 93 nm (evaluation of 76 fibrils), which was higher than the value determined from HRTEM images. It is likely that, given insufficient contrast in the SEM images, the ultrathin nature of the sample, and the platinum sputter coating, several adjacent fibrils could have been falsely interpreted as one fibril. Additionally, a weave-like pattern can be clearly observed (Fig. 4b), wherein fibrils were inserted over each other (A, Fig. 4b) and intersected to form larger macrofibril (C and D to form B, Fig. 4b). Further, a rod-like molecular connection could be observed between adjacent fibrils. In PAN fiber S2, the fibrils were regularly packed with an average diameter of 68 ± 12 nm (evaluation of 136 fibrils) (Fig. 4c) and lamella-like layers of approximately 13 nm in thickness (yellow circle, Fig. 4c), in agreement with HRTEM results.

SEM morphology of PAN fibers. a, b S1 longitudinal section etched in 70 wt% DMSO solution for 15 min. c S2 longitudinal section etched in 85 wt% DMSO solution for 30 min
Generalizing the observations above (Fig. 2–4), the following underlying mechanism is suggested. Firstly, the transverse strip-like lamellae present in the PAN fiber S1 are transformed into the node-like structures observed in PAN fiber S2, with the shish-kebab fibrils developing into dense fibrils superimposed by lamella-like layer crystals. Thus, the break-rearrangement of chain-folded lamellae plays the greatest role in fibril structure evolution. During the heat treatment and steam stretching process from PAN fiber S1 to S2, heat and the high stretching ratio enhance the motion of the chains, leading slippage and inducing void collapse and lamellae breakage [18]. The folded chains from the broken lamellae then slip and rearrange to form novel and regular lamellar crystal layers, increasing the crystallinity of the structures. Furthermore, the folded chains in lamellae also connect the adjacent fibrils to act as an inter-fibril linking bridge, able to enhance the tensile strength of PAN fibers.
Conclusion
PAN fibers were successfully separated through ultrathin sectioning and solvent etching, allowing information on fibril morphology and arrangement to be obtained. The fibers were composed of coexisting fibrils and lamellae with the microfibrils superimposed by a lamella-like crystal layer formed by the folding of molecular chains and surrounded by amorphous chains. The fibrils were woven and intersected in the PAN fibers. During the post-spinning process, the rotation and slippage of molecular chains resulted in lamellae breaking and folding into regular lamellae. Consequently, the microfibrils were interwoven and self-assembled, leading to a more orderly and tightly interlinked fibril arrangement, conducive for the preparation of high-performance PAN fibers.
References
Fischer L, Ruland W (1980) The influence of graphitization on the mechanical properties of carbon fibers. Colloid Polym Sci 258(8):917–922. https://doi.org/10.1007/BF01584920
Chen R, Bin Y, Nakano Y, Kurata N, Matsuo M (2010) Effect of chemical crosslinking on mechanical and electrical properties of ultrahigh-molecular-weight polyethylene-carbon fiber blends prepared by gelation/crystallization from solutions. Colloid Polym Sci 288(3):307–316. https://doi.org/10.1007/s00396-009-2173-2
Xie Y, Lu L, Hou Z, Tang Y, Miao L, Liu X (2016) Fracture behavior of PAN-based carbon fiber tow in a chopping process on an elastic support. Fiber Polym 17(8):1262–1268. https://doi.org/10.1007/s12221-016-6306-1
Gulgunje PV, Newcomb BA, Gupta K, Han GC, Tsotsis TK, Kumar S (2015) Low-density and high-modulus carbon fibers from polyacrylonitrile with honeycomb structure. Carbon 95(2):710–714. https://doi.org/10.1016/j.carbon.2015.08.097
Akhlaghi O, Menceloglu YZ, Akbulut O (2016) Rheological behavior of poly(acrylonitrile) concentrated solutions: effect of Sb2O3 nanoparticles on shear and extensional flow. Colloid Polym Sci 294(9):1–11. https://doi.org/10.1007/s00396-016-3907-6
Liu X, Zhu C, Dong H, Wang B, Liu R, Zhao N, Li S, Xu J (2015) Effect of microgel content on the shear and extensional rheology of polyacrylonitrile solution. Colloid Polym Sci 293(2):587–596. https://doi.org/10.1007/s00396-014-3419-1
Tan L, Pan J, Wan A (2012) Shear and extensional rheology of polyacrylonitrile solution: effect of ultrahigh molecular weight polyacrylonitrile. Colloid Polym Sci 290(4):289–295. https://doi.org/10.1007/s00396-011-2546-1
Ouyang Q, Chen Y, Wang X, Ma H, Li D, Yang J (2015) Supramolecular structure of highly oriented wet-spun polyacrylonitrile fibers used in the preparation of high-performance carbon fibers. J Polym Res 22(12):229–238. https://doi.org/10.1007/s10965-015-0865-5
Chand S (2000) Review carbon fibers for composites. J Mater Sci 35(6):1303–1313. https://doi.org/10.1023/A:1004780301489
Chen JC, Harrison IR (2002) Modification of polyacrylonitrile (PAN) carbon fiber precursor via post-spinning plasticization and stretching in dimethyl formamide (DMF). Carbon 40(1):25–45. https://doi.org/10.1016/S0008-6223(01)00050-1
Craig JP, Knudsen JP, Holland VF (1962) Characterization of acrylic fiber structure. Text Res J 32(6):435–448. https://doi.org/10.1177/004051756203200601
Tucker P, George W (2010) Microfibers within fibers: a review. Polym Eng Sci 12(5):364–377. https://doi.org/10.1002/pen.760120509
Warner SB, Uhlmann DR, Jr LHP (1979) Oxidative stabilization of acrylic fibres. J Mater Sci 14(8):1893–1900. https://doi.org/10.1007/BF00551029
Wang Q, Wang C, Yu M, Ma J (2010) Microstructure of fibrils separated from polyacrylonitrile fibers by ultrasonic etching. Sci China Technol Sci 53(6):1489–1494. https://doi.org/10.1007/s11431-010-3142-1
Wang Q, Wang C, Bai Y, Yu M, Wang Y, Zhu B, Jing M, Ma J, Hu X, Zhao Y (2010) Fibrils separated from polyacrylonitrile fiber by ultrasonic etching in dimethylsulphoxide solution. J Appl Polym Sci Part B, Polym Phys 48:617–619. https://doi.org/10.1002/polb.21929
Kunzmann C, Schmidt-Bilkenroth G, Moosburger-Will J, Horn S (2018) Microscopic investigation of polyacrylonitrile fiber fibrils separated by ultrasonic etching. J Mater Sci 53(6):1–12. https://doi.org/10.1007/s10853-017-1858-z
Lednicky F, Pientka Z, Hromadkova J (2003) Ultrathin sectioning of polymeric materials for low-voltage transmission electron microscopy: relief on ultrathin sections. J Macromol Sci Part B, Phys 42(5):1039–1047. https://doi.org/10.1081/MB-120023556
Yu T, Zhu C, Gong J, Yang S, Ma J, Xu J (2014) Lamellae break induced formation of shish-kebab during hot stretching of ultra-high molecular weight polyethylene precursor fibers investigated by in situ small angle X-ray scattering. Polymer 55(16):4299–4306. https://doi.org/10.1016/j.polymer.2014.06.056
Funding
This work was supported by the National Natural Science Foundation, China (Grant Nos. 51773110 and 51573087), A Project of Shandong Province Higher Educational Science and Technology Program, China (Grant No. J14LA05), and the Natural Science Foundation of Shandong Province, China (Grant No. ZR2016EMM16).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Gao, Q., Jing, M., Wang, C. et al. Fibril microstructural changes of polyacrylonitrile fibers during the post-spinning process. Colloid Polym Sci 296, 1307–1311 (2018). https://doi.org/10.1007/s00396-018-4350-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-018-4350-7