
Abstract
The design and synthesis of various polymer core–shell particles result from their distinct characteristics, which combine the properties of two or more components into one material. Many accessible synthetic strategies for obtaining polymer core–shell particles lead to the formation of particles for which the internal morphology differs from the ideal core–shell structure. Understanding the precise morphology characteristics is important for mechanistic studies of particle formation, which ultimately results in the design of particles for specific structures and properties. The detailed characteristics of complex polymer particle structures are complicated and require more than one method. This review focuses on imaging methods such as transmission electron microscopy (TEM), cryo-TEM, scanning transmission electron microscopy (STEM) and confocal fluorescence microscopy that reveal the radial redistribution of the components and methods for the quantitative analysis of individual phases (core, shell and interfacial layer), such as small-angle X-ray scattering (SAXS), small-angle neutron scattering (SANS), differential scanning calorimetry (DSC) and nuclear magnetic resonance (NMR). Methods that can determine the surface composition and makeup of the character of interfacial layer (gradient or containing small domains, etc.) were also reviewed.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
According to IUPAC terminology for polymers and polymerization processes in dispersed systems [1], a core–shell particle is defined as a “polymer particle comprising at least two phase domains, one of which (the core) lies within the other(s) that form the polymeric outer layer(s) (the shell(s)).” This definition indicates that the structure of polymer core–shell particles can be complicated and very often difficult to determine.
Polymer particles can be obtained through heterogeneous radical polymerization methods such as dispersion polymerization, emulsion polymerization, miniemulsion or microemulsion [2]. Polymer core–shell particle formation occurs in various ways. Generally, methods can be divided into one-, two- or more-step syntheses. A one-step process is based on the direct copolymerization of two comonomers, leading to a core–shell structure formation, which can be distinguished variants of two comonomers [3, 4] or a comonomer with a macromonomer [5–12]. The two-step method consists of, first, the synthesis of a seed and then the formation of a shell. In this case, the shell can be generated by various means. Usually, a polymer seed is swollen with a comonomer, which is polymerized [13, 14]. Shell formation can also be achieved by polymerization from reactive sites present on the surface of the seed (“grafting from” methods) [15, 16] or by binding various macromolecules to a polymer seed (“grafting to” methods) [17, 18]. The consequence of particle syntheses using two types of components is that the resulting particles can exhibit a diverse variety of morphologies. Landfester demonstrated particle morphologies formed by a two-step emulsion polymerization [19], indicating that an ideal core–shell morphology with a complete polymer phase separation, with the formation of a pure core and shell, is practically impossible. The exception is particles formed by grafting to method. In most cases of core–shell particles, an interphase region exists between the pure core and shell components.
The design of polymer core–shell particles results from the necessity to combine properties from two types of polymers into one material that requires specific characteristics for a particular application. Polymer core–shell particles are interesting objects that have many applications in a variety of areas such as durable exterior coatings [20], catalyst systems as supports for enzyme immobilization [21–24] adhesives [25, 26], diagnostics [27], drug delivery systems [28], etc. Advantageous characteristics of core–shell particles result from the contrast in the properties of the two or more types of polymers that the particles are made from. For example, one component could be a rigid material (core polymer) on which hydrophilic polymers with lower refractive index values are bound. Microspheres built solely from hydrophilic polymers would be prone to mechanical distortion, for instance, under the influence of centrifugation during the particle purification process. Moreover, hydrophilic polymers become highly swollen with water, which makes their refractive index values very similar to the refractive index value of water and means that particles composed solely of hydrophilic polymers cannot be conveniently analysed with light scattering methods. For example, poly(styrene/polyglycidol) microspheres are composed of a hard, hydrophobic core on which exist random copolymers containing a hydrophilic component with numerous hydroxyl groups. Such microspheres create many possibilities for surface modifications by covalently binding many compounds, in particular, biologically active systems such as enzymes [22]. The beneficial properties of P(S/PGL) are also related to the surface hydrophilicity, which demonstrates antifouling behaviour (i.e. reduction of protein adsorption). For poly(styrene/N-isopropylacrylamide) particles (P(S/NIPAM)), a thermosensitive PNIPAM shell (lower critical solution temperature (LCST)) of PNIPAM is 32 °C) creates many opportunities for the encapsulation of numerous components, such as noble metal nanoparticles (gold, silver, pallad [29]), enzymes [30], etc. As a result, the polymer shell of the core–shell structure can act as a reactor becoming a catalyst for various reactions. It is worth emphasizing that catalytic properties can be modulated by temperature changes. When the temperature is above LCST of PNIPAM, then shell shrinks and catalysis of reactions is not possible due to difficulties with substrate diffusion. A system with an anchored catalyst can be reused after washing by centrifuging of particles. A similar example of core–shell particles with thermosensitive shells is that of polystyrene/poly(N-vinylisobutyramide) (PS/PNVIBA) microspheres. LCST of PNVIBA is 40 °C [7]. A low cross-linked poly(N-isopropylacrylamide-acrylic acid) shell formed on a polystyrene core showed promise in lysozyme adsorption [30] with a biological activity in the adsorbed state that was three and five times higher than that in its native form.
The morphology of two or more component polymer particles has a significant influence on their properties and determines their further applications. Control of particle morphology is crucial meaning, particularly if a material needs to be prepared for a given application. Put another way, determination of the morphology of the synthesized particles composed of two or more polymer components is often necessary to understand the mechanisms governing particle formation in heterogeneous copolymerizations. It provides data on the mechanistic aspects in emulsion and dispersion polymerization and enables the design of future particles with specific characteristics. It is important to evaluate the dependence of the synthetic conditions on the final morphology of polymer particles, i.e. how chemical composition changes within the particle moving from the innermost to the outermost layer. The various morphologies that particles can exhibit are presented in Fig. 1. Changes in the synthesis parameters (temperature, solvent, molar ratio of comonomers, etc.) impact the distribution of components within a particle. Moreover, the determination of the shell structure is also essential. The shell structure of core–shell particles can be continuous or composed of small domains embedded in the core polymers or deposited onto the core’s surface.

Typical morphology variants of particles composed of two types of polymers. The final morphology of particles synthesised by seeded polymerization using polymer seed (black colour denotes polymer (1) swollen with monomer 2 with formation of polymer 2 (white colour) can be shown in a (ideal core–shell morphology) or in b—inverted core shell structure. Composite particle morphology can be composed of one type of a polymer (polymer 2) with embedded domains of polymer 1 (in case of particle c—in the whole particle, d domains present in the particles shell region), or vice versa. The favourable morphology of particles is one which exhibits the minimum interfacial free energy change. Shell chemical composition may also change gradually, i.e. particle’s shell is enriched with a latter component going from a particle’s core to its surface (in case of e)
Detailed morphological characteristics of particles can be defined by combining the results obtained from imaging methods and analytical techniques that provide data on a particle’s radial composition. Moreover, quantitative information about particles’ surface composition is required in case there is further surface modification.
This review focuses on the presentation of available methods that facilitate the determination of the morphology of analysed polymer particles made of complexes by revealing details such as the existence of an interfacial region between a particle’s core and shell. We concentrate specifically on imaging methods and techniques that provide a quantitative fraction for the various phases present in a particle’s morphology.
The first part of this review is devoted to methods specifically applied for examination of particle surface. The second part, though, is focused on techniques used for entire particles’ morphology characterization.
Methods of polymer core–shell particle characterization
Methods of particle surface analysis
The surface composition of complex polymer particles plays a crucial role in their further application, i.e. surface modification, and film formation [31]. Reactive functional groups facilitate covalent immobilization of various compounds on a particle’s surface. The presence of hydrophilic or hydrophobic [31] polymers on a particle’s surface creates opportunities for specific applications. Moreover, shell-building polymers can exhibit specific properties, such as thermosensitivity in the case of PNIPAM [30] and PNVIBA, antifouling behaviour of poly(ethylene glycol) (PEG) [32, 33] and polyglycidol [22] or conductivity in the case of polypyrrole [4, 34] and polyaniline [35]. To examine the surface composition, direct and indirect methods can be applied. Direct methods include X-ray photoelectron spectroscopy (XPS), static secondary ion mass spectroscopy (SSIMS) or scanning electron microscopy coupled with an energy-dispersive X-ray (SEM-EDX). Protein adsorption studies are indirect methods used to evaluate the character of a particle’s surface, as the hydrophobic surfaces, contrary to hydrophilic surfaces, exhibit high susceptibility towards biomolecules.
X-ray photoelectron spectroscopy
XPS, also known as electron spectroscopy for chemical analysis (ESCA), provides qualitative and quantitative data about the surface composition of particles. However, it should be noted that XPS analysis probes only the composition of the particles ca. 5 nm in depth, which makes this method invaluable in surface analyses [34]. The presence of a surface stabilizer [36] and the success of surface modifications can be confirmed by XPS [37]. Moreover, XPS can be used to identify an inverted core–shell structure.
In XPS analysis, the surface composition of each element in the analysed material is expressed as an atomic percentage (%) determined from the integrated peak area of all elements present in a sample and their experimental sensitivity factors.
For example, the fractional concentration of an element A, %A, is assessed as follows [36]:
where
I A is the integrated peak area attributed to its sensitivity factor (s A ) and I n and s n are the total integrated peak area and the sensitivity factors characteristic for each element.
The principle of XPS is as follows: at conditions of high vacuum, on the level of 10−9 Torr, a sample is irradiated by monochromatic X-rays which leads to emission of photoelectrons coming from the analysed material [38]. The emitted electrons are detected as a function of kinetic energy (E K), producing a spectrum. Knowing the values of irradiation energy and kinetic energy, the binding energy of electron emitted from a valence orbital can be calculated and attributed to the specific element. As each element is described by the specific binding energy, all elements except hydrogen can be identified. Moreover, quantitative information of different oxidation states for the elements existing in the analysed sample is acquired. For example, it is possible to distinguish carbon atoms in various environments, such as carbon atoms in aliphatic or aromatic systems or adjacent to heteroatoms [34]. Generally, carbon bound to another carbon or hydrogen atom, without regard for hybridization, yields C1s = 285 eV. However, heteroatoms and halogens cause the binding energy increase. For example, in an XPS spectrum of poly(N-vinylpyrrolidone) (PNVP), binding energy values of 285.9 and 287.7 eV correspond to carbon atoms in C–N and N–C=O groups, respectively [39]. A characteristic of polystyrene is a low-intensity feature at 291.7 eV because of the C(1s) π−π* shake-up satellite. There are differences in electron binding energies coming from nitrogen atom in different environments. For PNVP, one signal coming from N(1s) is observed in the spectrum, whereas polypyrrole is described by four signals located in the range of 396–406 eV with peaks at 398–399, 400–401, 402–403 and 403–404 eV, which are attributed to the N=C, N–H bonds and the two high oxidation states of nitrogen (NI ox and NII ox) in doped polypyrrole [39]. Quantitative analysis can be performed for a two-component surface provided that at least one type of element is different in each compound. Then, this atom acts as a unique elemental marker for one component. For example, a convenient quantitative analysis for the surface was discovered for hybrid systems such as polypyrrole-silica or polyaniline-silica particles. The surface composition was determined on the basis of the Si/N atomic ratio. Silica comes exclusively from silica oxide, whereas nitrogen is a specific elemental marker of a conducting polymer. This means that these elements are unique for each component, and therefore, the relative proportion of all components present in the outer layer of the surface can be easily calculated based on the elemental composition of the surface. A similar assessment of surface composition can be performed for polystyrene particles covered by poly(N-vinylpyrrolidone) [36]. Samples of bulk PS particles and bulk PNVP were analysed separately. Analysis of the particles’ surface composition relied on the simple assessment of the nitrogen signal (N(1s)) ratio registered for pristine PNVP and PS/PVNP particles, respectively (%PVNP = NPS/NPNVP) because the pure polystyrene surface did not contain any nitrogen atoms. On the other hand, the relative surface composition of PS/PNVP particles can also be calculated on the basis of carbon composition using data obtained for pure polystyrene particles (seed) and modified polystyrene particles [36].
The influence of different parameters, such as the composition of the polymerization mixture and polymerization conditions, on the final particle’s surface composition can be easily evaluated with XPS [40]. Moreover, XPS analysis can determine whether a particle’s core is efficiently covered by a shell-building polymer. Whether an increase in the amount of one comonomer during a particle’s synthesis causes the enrichment of a particle’s surface with this component can be also evaluated. For PEG-stabilised polystyrene particles covered with polypyrrole, the relative composition of all components in a particle’s surface was estimated. XPS spectra were recorded for each individual material (PEG, uncoated polystyrene latex and polypyrrole) and for the resulting composite materials (PS/PEG/polypyrrole). XPS data demonstrated that the polypyrrole shell is not continuous. The authors suspected that the polypyrrole layer is too thin to be observed using XPS because the XPS sampling depth is a maximum of 5 nm. In fact, transmission electron microscopy (TEM) images revealed the presence of nanosized polypyrrole particles (approximately 20–30 nm) on the polystyrene latex surface [34]. Basinska et al. used XPS to estimate the relative composition of the surface of particles’ synthesized by emulsion radical copolymerization of styrene and a hydrophilic macromonomer, α-t-butoxy-ω-vinylbenzyl-polyglycidol [41]. The particle’s shell was composed of polystyrene-poly(α-tert-butoxy-ω-vinylbenzylpolyglycidol) copolymer (Fig. 2).

Structure of polystyrene-poly(α-tert-butoxy-ω-vinylbenzylpolyglycidol) copolymer composing the poly(styrene-polyglycidol) shell [41].
The influence of the molecular weight of polyglycidol macromonomer and the initial polyglycidol macromonomer/styrene concentration ratio on surface composition was analysed. It has been shown that an increase in the molar fraction of polyglycidol macromonomer during synthesis resulted in the increased amount of polyglycidol fraction in the surface. The authors also examined the influence of changes in the particles’ synthesis conditions, namely from batch (all reagents introduced from the beginning of the process) to continuous (polyglycidol macromonomer was added in a continuous manner to the reaction mixture) polymerization [22].
Generally, for composite polymer particles synthesized by radical polymerization of comonomer A (styrene, methacrylate) with macromonomer B (poly(ethylene oxide), polyglycidol, etc. composed of z repeating units and containing a polymerizable group (for example, styryl or methacryloyl)), Fig. 3 shows the fraction of poly(macromonomer) building the particle’s surface, y, can be calculated according to the equation:

Schematic of the structure of a copolymer synthesized by radical copolymerization of a comonomer and a macromonomer carrying the same polymerizable entity. x denotes the number of repeating units of one type of comonomer; y denotes the number of repeating units of the macromonomer, and each of them carry a chain of the macromolecule
XPS analysis was used to study the surface layer of particles obtained by seeded dispersion polymerization of styrene in the presence of poly(methyl methacrylate) particles. It revealed that only polystyrene was present on the particles’ surface and confirmed the core–shell morphology of the particles [42].
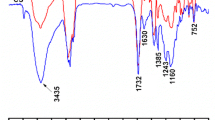
XPS facilitates depth profiling analysis. Zhang et al. investigated compositional changes by depositing a thin latex film prepared from polyacrylate core–shell particles containing fluorinated polyacrylates [43] on a glass plate, sputtering polymer from the film–air interface by bombardment with a 3 keV Ar+ ion beam. An extension of the sputtering time resulted in proceeding from the film–air interface towards the film–glass interface (etching) and revealed a gradient in the fluorine content. The highest fluorine concentration was found in the film–air interface and gradually decreased going towards the film–glass interface, where the lowest value was found. Cui et al. [44] noticed similar behaviour for a film formed on a glass plate composed of fluorinated core–shell particles containing a polyacrylate seed (methyl methacrylate (MMA), butyl acrylate (BA) and triethylene glycol dimethacrylate (TrEGDMA)) and a shell made of 2,4-di(trifluoromethyl)-2,3,4,5,5,5-hexafluoropentyl methacrylate (DFHMA), vinyltriethoxysilicone (VTES) and TrEGDMA. XPS analysis revealed that the composition differences in the film’s surface strictly depended on the type of interface, i.e. film-air or film-glass. Based on the XPS spectrum, the intensity of the signal coming from fluorine (F1s) was higher in the film–air interface than in film–glass interface. Migration of fluorine-containing groups from the copolymer structure to the film–air interface results from a decrease in the free energy in the surface of the latex film [45]. Examples of XPS spectra demonstrating the surface composition and deconvoluted C1s profile are presented in the Fig. 4.

The XPS spectra of latex film: a survey spectrum and b the high resolution of C1s spectrum. Reprinted with permission from [45] Copyright © 2010, ACA and OCCA
Static secondary ion mass spectroscopy
In SSIMS, [46] positive and negative secondary ions are emitted under the influence of primary ions interacting with the material being analysed. Primary ions can penetrate a sample up to several nanometres in depth (a typical ion’s energy is 2–4 keV) and deliver information about its composition within this range of thickness. Known m/z factors of the emitted ions facilitate the determination of the type of polymer present in the region being analysed. It is worth mentioning that the use of an intensive ion beam causes the removal of the outermost layer of the surface giving information about changes in composition with depth. This method is termed “static” because the gathered ion dose during spectral acquisition is sufficiently low enough for the surface to be essentially unperturbed during the measurement [46].
Brindley et al. [47] evaluated the degree of coverage of a polystyrene core with poly(ethylene oxide) (PEO) by combining XPS and static secondary ion mass spectroscopy (SSIMS) methods. Particles were obtained by emulsion polymerization of styrene and an acrylate macromonomer of PEO (M w = 2000). The authors noted that incomplete surface coverage by the hydrophilic macromonomer, as observed in the spectra of positive and negative ions, signals of secondary ions was present not only from PEO but also from polystyrene. In the spectrum of negative ions, signals were found that corresponded to the following m/z values: 43, 58, 59, 61 and 85, attributed to poly(ethylene oxide) fragments presented below:
CH2 = CHO−, CH2 = CHOCH3 −, HOCH = CHO−, HOCH2CH2O−, CH2 = CHOCH = CHO−
In the spectrum of positive ions, signals at m/z values of 43, 45, 59, 73, 87 and 89 corresponded to following structures, respectively: CH2 = CO+H, CH3CH = O+H, HOCH = C = O+H, CH3OCH = C = O+H, CH2 = CHOCH2CH = O+H and CH3CH2OCH2CH = O+H. The authors demonstrated that an increase in the PEO macromonomer concentration during particle synthesis causes an increase of this component in the surface. The spectra of positive ions provided information about changes of surface composition. The intensity of the signal m/z = 91, corresponding to the polystyrene fragment, diminished, whereas the signal of poly(ethylene oxide) (m/z = 45) increased. It should be emphasized that SSIMS is a semi-quantitative method. Despite its high sensitivity, this method needs to be supported by electron spectroscopic methods, such as XPS [47] to obtain precise fractions of components.
SEM-EDX
SEM-EDX can be used for quantitative elemental analysis of the particle’s shell surface. Simple SEM analysis yields information about surface topology; however, when combined with EDX, it can provide surface composition data. Under sample bombardment, X-rays are emitted. EDX consists of counting and sorting all characteristic X-rays according to their energy level. He et al. [48] successfully investigated complex particles composed of poly(butyl acrylate) core and a shell formed from methyl methylacrylate, methyl dodecafluorinacrylate and 3-methacryloxypropyltrimethoxysilane and methyltrimethoxysilane.
One must remember that EDX gathers the information from up to 2 μ of sample depth depending on sample composition and applied beam energy. It is much deeper in comparison with XPS and SSIMS methods. However, EDX is commonly coupled with microscopes working in a raster mode, which facilitates distinguishing particle’s surface from the rest of a particle.
Protein adsorption investigations
Evaluation of the degree of hydrophobic seed coverage (complete, partial or none) with a hydrophilic polymer can be performed indirectly using protein adsorption studies. In such an experiment, the amount of adsorbed proteins on the particle’ seed is compared to the surface concentration value of adsorbed proteins on the modified seed surface [49]. Protein adsorption studies deliver qualitative information about surface composition, which is useful in the evaluation of post-modification processes, antifouling properties and interactions with various surfaces.
Protein adsorption studies were carried out to confirm the presence of PEO on the surface of polystyrene particles. PEO is a commonly used hydrophilic polymer that reduces non-specific interactions [32, 33, 50]. Similar behaviour is displayed by polysaccharides, polybetains, polyglycidol, etc. The polyglycidol structure resembles poly(ethylene oxide). The difference lies in the presence of a methylhydroxyl group in each repeating unit of polyglycidol. Surfaces composed of hydrophilic polymers result in antifouling behaviour due to the high mobility of their chains and/or formation of a water layer that acts as an effective barrier, preventing the protein from landing. For polyglycidol, water does not exclusively interact with the polyether polymer main chain but with numerous other accessible hydroxyl groups. The free enthalpy of hydration calculated for polyglycidol and poly(ethylene oxide) conformers is −26.14 and −16.05 kcal/mol [51], respectively, which undoubtedly indicates the higher effectiveness of polyglycidol hydration.
In addition to the XPS method, the presence of polyglycidol on the surface of poly(styrene/polyglycidol) particles was also demonstrated through protein adsorption studies [22, 49]. Particles were obtained by emulsion polymerization of styrene and macromonomer α-tert-butoxy-ω-vinylbenzyl-polyglycidol in water. The hydrophilic property of polyglycidol determines its location in the outermost region of the core–shell particles during their synthesis. However, massive hydrophilic segments of polyglycidol do not completely cover the polystyrene domains. The authors noticed that as the fraction of polyglycidol in the particle’s surface increases, non-specific protein adsorption is distinctly reduced [22]. A similar trend was observed for particles built from polystyrene and poly(ethylene oxide) shells [52]. For example, the surface concentration of human serum albumin on poly(styrene/polyglycidol) particles with a 27.8 mol% fraction of polyglycidol (value from XPS analysis) was equal to 0.22 mg/m2, whereas on a polystyrene particle’s surface, the amount of HSA exceeded 1.50 mg/m2 [22].
Visualization methods of the whole core–shell particle morphology
Microscopic methods such as atomic force microscopy (AFM), SEM and TEM can be used to determine a particle’s shape and size [53]. In AFM, particles deposited on a flat surface are scanned using the probe tip of the cantilever. Interatomic forces, which occur between the tip and the analysed sample’s surface, cause the deflection of the cantilever. The degree of deflection results from changes in the sample’s surface topography or material properties such as chemical, magnetic, etc. In the case of SEM, polymer particles are deposited on a flat surface and are usually covered with a thin layer of conducting material (e.g. gold) and are then scanned with an electron beam. Electrons interacting with a sample mainly cause the generation of secondary electrons (emitted from atoms present in the surface layer), which are collected with a detector, and the signal is then transformed into an image. It should be emphasized that AFM and SEM methods provide data about only the surface of the examined sample. In contrast to SEM, during a TEM measurement, the electrons transmitted through a sample are collected. Elements of high atomic number scatter more strongly than those of low atomic number and appear as dark regions in the image. In addition to the shape and size of polymer objects, TEM can also provide information, to some extent, about the radial internal distribution of polymer components in complex particles.
Satisfactory differentiation of polymer components by TEM frequently requires the application of a staining agent to change the contrast between components. However, there are examples of TEM images showing complex particles where staining is not needed to see the particle’s cross sections. The structure of polysilsesquioxane-poly(styrene-butyl acrylate-fluorinated acrylate) hybrid particles was determined without staining treatment [54]; distinct variation in electron penetrability of a particle’s core and shell was sufficient. Dark and light regions were visible within the particles, which represent the core and shell, respectively. TEM images of poly(styrene/polyglycidol) particles without a staining treatment showed a core–shell structure with a polystyrene core and poly(styrene/polyglycidol) shell [55]. The core–shell morphology of particles composed of a poly(methyl methacrylate) core and polystyrene shell embedded in epoxy resin and sectioned was easily visualized without any staining agents, as PMMA decomposes in the electron beam [56]. For the same reason, the morphology of poly(styrene/tert-butyl acrylate) (PS/PTBA) and poly(tert-butyl acrylate/styrene) (PTBA/PS) particles could be resolved owing to high polystyrene and low PTBA contrast, respectively [57], as PTBA also degradated under electron beam.
Nevertheless, it is rare that TEM can unambiguously determine a particle’s morphology and point out the location of each polymer’s counterparts within the sphere, without staining. Usually, it is difficult to see a clear barrier separating the core and shell because of the weak contrast between various phases. In such situations, sample staining is required for proper determination of the particle’s morphology. Generally, there are positive and negative staining methods [58]. Negative staining relies on sample preparation by embedding the polymer particles in an electron-dense surrounding medium or sample deposition on a stained substrate. This type of staining is popular in the analyses of biological samples. Positive staining consists of a chemical reaction between the staining agent and specific parts of the sample, which causes a visible difference between the stained regions (with accumulated staining agent) and the regions that did not react with the staining agent.
The most popular staining agents are oxides of metals such as ruthenium and osmium (RuO4 and OsO4, respectively) or compounds such as phosphotungstic acid or its sodium or potassium salts, uranyl acetate and caesium hydroxide. Phosphotungstic acid and uranyl acetate are examples of negative staining agents. The heavy metal oxides RuO4 and OsO4, which have oxidative properties, are used as positive staining agents. Rhutenium oxide exhibits stronger oxidative properties in comparison to osmium oxide [59]. It is able to stain most polymers, excluding poly(methyl methacrylate) and polyacrylonitrile. As a result, RuO4 is a more versatile staining agent than OsO4. RuO4 can be used to stain both saturated and unsaturated areas, whereas osmium tetraoxide is useful for the analysis of polymers containing unsaturated moieties [60]. RuO4 successfully stains saturated/unsaturated hydrocarbons, aromatics, polymers containing hydroxyl, aldehyde, amine and ester groups [61]. Due to the fact that osmium tetraoxide displays a preferential affinity for amines, hydroxyl and ether groups through a coordination reaction, the morphology of core–shell particles composed of a polystyrene core and poly(ethylene oxide) shell (PS/PEO) was able to be determined. TEM-stained images allowed for the differentiation of individual components within the particle structure [62], making it possible to estimate shell thickness; a dark PEO shell and light polystyrene core were distinguishable. Moreover, it was noted that the application of a hydrophilic polymer with a higher molecular weight resulted in an increase in the shell’s thickness. The thickness of a poly(ethylene oxide) shell covering a polystyrene core was also successfully determined using selective RuO4 staining [63]. The dark region of the particles was attributed to the poly(ethylene oxide), whereas the light region was attributed to polystyrene. Ito et al. [63] noted that the thickness of the PEO shell (h) on the polystyrene core can also be roughly determined based on the hydrodynamic diameter value (D DLS) determined by dynamic light scattering in water and with the diameter corresponding to dried polystyrene core (D TEM) determined on the basis of TEM images (h = (D DLS − D TEM) / 2. Dynamic light scattering, also called photon correlation spectroscopy (PCS), is a method used for the routine determination of a particle’s diameter, which is suspended in a dispersing medium. A sample of the diluted suspension is illuminated with a laser beam. The scattered light on the dispersed particles is registered at an angle θ in relation to the incident beam [64]. Because particles in a suspension undergo continuous Brownian motion, the intensity of scattered light changes over time. Analysis of these fluctuations as a function of time provides information about the particle’s motion. The diffusion coefficient of a particle is then determined. Using the Stokes–Einstein equation [64], the average hydrodynamic diameter of the particle can be calculated.
A representative procedure of sample staining with RuO4 or OsO4 vapour treatment for TEM analysis is as follows: A dry sample of the particles is embedded in resin and then placed in a microtome, where ultrathin sections are cut on water surface; then, the sections are put on a copper grid and dried under reduced pressure conditions; after drying, the samples are treated with a vapour of metal oxide; the samples are then analysed. It is worth noting that imaging of the entire particle without microtoming can lead to a mistaken evaluation of the particles’ morphology, due to the difference in the thickness of various regions of a spherical particle, i.e. the difference in thickness of the middle and the periphery of a particle.
Selective visualization of a region containing poly(acrylic acid) areas in the structure of poly(butyl acrylate)/poly(methyl methacrylate) particles was achieved by positive staining with caesium hydroxide (CsOH), which did not react with the other components of the particle [65]. A diluted suspension of particles was mixed with a 3 wt% aqueous solution of CsOH. A drop of this prepared suspension was placed on a copper grid, dried and analysed.
The morphology of particles composed of a polystyrene core with a shell of polyacrylate containing fluorine was successfully determined by depositing of a diluted particle suspension on a carbon-coated copper grid, staining it with 1.0 wt% phosphotungstic acid (PTA) for 3 min and drying it [45]. The use of phosphotungstic acid also allowed for successful determination of the structure of particles composed of a poly(methyl methacrylate) core and a hydrophilic shell made of biopolymers or polymers containing amine groups, such as casein, gelatin, chitosan, branched poly(ethylene imine), poly(allylamine) [66] or poly(vinylamine) [67]. A diluted sample of analysed latex was put on a Formvar-coated or a carbon-coated grid and dried. Then, samples were stained with a 2 wt% solution of phosphotungstic acid for 30 min and dried. Cui et al. also used a 2 % phosphotungstic acid solution to stain the polyacrylate latex containing fluorine and silicon atoms in the shell to achieve a clear, significant contrast between core and shell [44]. A solution of phosphotungstic acid was also used to investigate the morphology of polymer particles composed of a poly(n-butyl acrylate) core and poly(methyl methacrylate-1,2,2,6,6-pentamethylpiperidin-4-yl acrylate) shell [68]. The core and shell regions were efficiently distinguished as white and grey areas, respectively. The core–shell structure of particles with a polyacrylate core (the white part) and a polydimethoxysilane shell (the dark circle) was successfully confirmed with TEM using a phosphotungstic acid solution as a staining agent [69]. It was possible to estimate the thickness of shell using TEM pictures to show the polymer particle. The shell thickness varied depending on the weight ratio of the monomers used during their syntheses.
Core–shell morphology was also confirmed using a combination of positive and negative staining agents. For example, poly(n-butyl acrylate)/poly(methyl methacrylate) (P(n-Bu/MMA)) composite microspheres were contrasted using negative staining with uranyl acetate and positive staining with rhuthenium tetraoxide [65]. The PMMA regions of the particle were not stained; a virtual difference in chemical reactivity of two particle components towards the used staining agent, P(n-Bu), were visible as darker areas. The morphology of these particles was also determined with TEM using a Pt-shadowing technique. This revealed a light grey area that corresponded to the hard PMMA part of the particle and more highly contrasted dark inner area representing the P(n-Bu) regions. The procedure of double staining applied to the poly(styrene/pyrrole) core–shell particles [4] assured a distinguishable contrast between the core and shell regions. First, the sample was deposited on a copper grid and treated with an RuO4 vapour of 0.1 wt% RuO4 aqueous solution at 40 °C for 1 h. Then, the grid was immersed in a 0.4 wt% phosphotungstic acid aqueous solution.
The sample preparation procedure for TEM analysis can vary and should be adjusted for the specific system being examined. It is known that registration of TEM images for whole particles without a microtomed section can lead to mistakes. However, sample preparation by microtoming can also lead to some artefacts due to particle plasticization by the epoxy resins or an overstaining effect [70]. Sometimes, it is difficult to find a proper staining agent or a set of staining agents to ensure enough contrast between the core and the shell of a particle. Moreover, analyses of TEM images of a particle’s structure are not precise due to a lack of clear information about the existence of the interfacial region between the core and the shell. Complete information about the composite particle’s structure cannot be obtained by TEM analysis alone and must be supported by another method.
Scanning transmission electron microscopy (STEM) is a specific type of TEM that combines features of SEM and TEM. A thin sample (like in TEM) is scanned with a focused beam of electrons in a raster pattern (like in SEM). The beam of electrons transmitted by the sample is recorded. STEM supported by an EDX [71] delivered the elemental composition of shell and core parts of polypyrrole/poly(methyl methacrylate) particles, revealing the existence of a continuous shell composed of pure PMMA.
It is worth mentioning that particle images obtained from conventional TEM do not reflect the actual spatial component distribution in a particle’s native environment (in an aqueous suspension). The structure of the soft shell component is lost during drying, which is required for suitable sample preparation, because polymer chains collapse on the hard polymer core [72]. Thus, conventional TEM is not useful in studies of volume transition changes for thermosensitive polymers. A TEM technique that enables the determination of a particle’s actual structure in liquid suspension, excluding reorganizational changes of particle structure as a result of the sample preparation technique, is cryogenic TEM (cryo-TEM). Cryo-TEM is a useful technique to visualize a particle’s structure in an aqueous suspension (in the dispersed state) by rapid vitrification. Generally, a thin aqueous film is prepared on the microscopy grid; then, the grid is immersed in a cooling medium (usually ethane, just above its freezing point). At these temperature conditions, a sample vitrifies immediately, avoiding the effects of crystallization. Then, the grid with the prepared sample is transferred to the microscope and analysed at the temperature of liquid nitrogen [73]. Cryo-TEM is a good tool for obtaining information about the spatial distribution of the shell component attached to a polymer core and to evaluate whether the shell is closely attached to the core. Ballauff et al. [74] demonstrated that seeded emulsion polymerization of polystyrene particles with N-isopropylacrylamide and N,N-methylenebisacrylamide, whose fractions varied from 1.25 to 5 mol%, leads to the formation of core–shell particles, whose cross-linked poly(N-isopropylacrylamide) shells are not fully attached to the surface of the polystyrene core (Fig. 5). Cryo-TEM images were recorded to visualize the appearance of PS/PNIPAM particles at room temperature, in which the PNIPAM shell is swollen. It is worth noting that in images recorded by conventional TEM, this swelling is not visible for a sample in the dried state, where a PNIPAM shell releases water and the shell component collapses onto the polystyrene core [75]. This observation forced the authors to look for new methods of poly(styrene/NIPAM) particle synthesis.

Cryo-TEM images showing differences in PS/PNIPAM particle morphology depending on the cross-linking degree of PNIPAM shell by N,N-methylenebisacrylamide, 1.25, 2.5 and 5.0 %, respectively. Blue and red circles signify hydrodynamic radii of polystyrene core and PS/PNIPAM core–shell particles determined by DLS [74]. With kind permission from Springer Science and Business Media
Ballauff et al. [76] investigated the thermosensitive properties of a PNIPAM microgel on a polystyrene core at 25 and 45 °C. A decrease in the particle’s diameter at 45 °C (above LCSTPNIPAM = 32 °C) was consistent with the DLS measurements. At this temperature, the cryo-TEM data for a particle’s average diameter matched the DLS data, due to the compact shell morphology that resulted from shrinkage by water expulsion. However, a small difference in the values of particles’ radii was observed in two methods. The average diameter obtained by DLS was 109 nm, whereas with cryo-TEM, it was 103 nm. The reason for the diameter difference may still be a result of the fact that TEM is not sensitive to single PNIPAM chains. To prepare the sample at 45 °C, a drop of the particle suspension was deposited onto a copper TEM grid; the grid was kept in a humidity chamber at this temperature to prevent water evaporation. After blotting a drop onto a thin film, the grid was immersed in liquid ethane at its freezing point.
Cryo-TEM was applied to visualize the core–shell particles with a soft shell composed of a zwitterionic spherical polyelectrolyte brush made of poly(2-(methacryloyloxy) ethyl dimethyl-(3-sulfopropyl)ammonium hydroxide) (pMEDSAH), covering a hard polystyrene core [72]. The formation of the soft polymer shell was analysed with contrast enhancement using the prior particles contrasting with caesium iodide (CsI). Caesium and iodine ions act as counterions towards ions present in the polymer particle shell, improving the electron contrast. In cryo-TEM images, pMEDSAH chains displayed a stretched conformation on the grid’s surface, which facilitated the estimation of the particle’s shell thickness. Interestingly, the authors noticed a clear difference between the shell thickness estimated by cryo-TEM images (LcryoTEM) and the measurements taken with DLS (LDLS). This inconsistency resulted from insufficient contrast of the single polymer chains on cryo-TEM images, which are noticeable in DLS experiments (Fig. 6). LDLS was calculated using the difference between the hydrodynamic radius (R h, DLS) of the core–shell particles and the radius determined for the uncovered polystyrene core.

Image representing the structure of particles composed of a polystyrene core covered by a polyelectrolyte brush (pMEDSAH) using the cryo-TEM (dashed green line) and DLS (dashed red line) methods. Reprinted with permission from [72]. Copyright 2011 American Chemical Society
A complex formed between spherical poly(styrene sulfonate), the SPB brush, and the cationic surfactant cetyltrimethylammonium bromide (CTAB) in a dilute solution was investigated using cryo-TEM [77]. This was possible because interactions taking place between the anionic particle’s shell brush and the surfactant leads the shell to contract. When the ratio of the charge on the SPB to the charge of CTAB (r) was 0.6, the surface layer composed of poly(styrene sulfonate) chains and the adsorbed CTAB molecules partially collapsed. Moreover, the complex formed in the solution of NaBr (0.05 M) resulted in the attachment of globular structures to the poly(styrene sulfonate) surface (see Fig. 7). In this situation, when r = 1, the complex formed resulted in a fully collapsed layer formed by the polymer brush and surfactant molecules.

Cryo-TEM images of particles obtained as a result of poly(styrene sulfonate) interactions with cetyltrimethylammonium bromide. The image on the left demonstrates a PSS-CTAB complex (r = 0.6) formed at a charge in salt-free solution. The image on the right side shows a complex formation at a charge ratio of 0.6 in 0.05 M NaBr. Dashed white and black lines denote the hydrodynamic diameters of the core and shell, respectively. Reprinted with permission from [77]. Copyright 2007 American Chemical Society
Polymer core–shell particles that contain a fluorophore can be analysed using a confocal fluorescent microscopy. For example, this method allowed confirmation of a particle’s morphology synthesized from a poly(styrene/divinylbenzene) seed covered by poly(p-phenylenevinylene) (PPV) [78], which is a fluorophore. Particles were embedded in epoxy resin, and microtomy was used to create a thin film containing particle cross sections, which were later analysed. Proper determination of the particle shell thickness relied on finding the disk corresponding to the middle of the sphere (the thinnest thickness value taken from all shell cross sections) and rejecting those that deviated from this value.
Analysis of a particle’s morphology based only on visual methods, such as TEM or confocal microscopies, is not comprehensive. In fact, precise determination of a particle’s structure is a complex matter and requires the use of additional methods besides imaging to provide detailed information about the weight fractions of distinct regions within the particle [70]. It is essential to determine whether an interfacial region exists between pure polymers composing the core and the shell, along with the region’s thickness and structure (gradient or small domains).
Non-microscopical, quantitative methods of core–shell morphology studies
Differential scanning calorimetry
Differential scanning calorimetry (DSC) can be used to determine the precise size of interphase region—the region existing between the core and shell composed of pure polymers. DSC provides quantitative information about the distribution of the components. Temperature-modulated differential scanning calorimetry (TM-DSC) is especially sensitive to differences in polymer composition. In this technique, also known as modulated DSC (MDSC) and alternating DSC (ADSC), a periodic temperature modulation of small amplitude is superimposed on the underlying rate of conventional DSC [79].
Generally, for blends composed of two polymers, thermal analysis reveals three cases [80, 81]:
-
1.
If two polymer components are well-miscible, then one single signal of dCp/dT in the function of temperature is observed,
-
2.
If two components are partially miscible, then peaks attributed to T g of both polymers are smaller than measured for pure components and do not return to baseline in between the two T g,
-
3.
If two components are completely immiscible, then two signals are present and are ascribed to both types of polymers, in dependence of dCp/dT in function of temperature.
A similar situation exists for polymer particles. The degree of separation of two polymer components can differ from one system to another. The final two-component particle morphology depends on the mutual miscibility of two polymer phases as well as reaction conditions such as temperature, type of continuous phase, etc.
Quantitative analysis of two-component polymer core–shell particles by DSC is performed under the assumption that in particle morphology, the core, shell and diffuse interphase between them can be distinguished (Fig. 8). These investigations, however, require an understanding of the differential of heat capacity dependence with respect to temperature at the glass transition of individual pure polymers.

The model of the core–shell particles taken into consideration in DSC studies assuming the existence of the core, shell and interfacial region. R c, R s and R i denote the thickness of the core, shell and interphase, respectively. Reprinted with permission from [82]. Copyright 2012 Elsevier B.V
The heat capacity of the whole latex particle (ΔC pw) can be expressed as follows [82]:
where ΔC pc, ΔC pi and ΔC ps are heat capacities corresponding to the core, interfacial region and shell, respectively.
Based on the known heat capacity for the pure polymers that form the particle’s core (ΔC pc0) and shell (ΔC ps0), heat capacity values measured for a composite particle can be used to calculate the weight fraction of individual phases and their corresponding radii according to following expressions [82]:
where ω c, ω i and ω s are the weight fractions of the core, interfacial region and shell, respectively, and
where R c, R i and R s are the radii of the core, interfacial region and shell, respectively.
MDSC was used to characterize the composite sulfonated-polystyrene-poly(methyl methacrylate) (S-PS/PMMA) particles, obtained by polymerization of methyl methacrylate introduced to emulsified S-PS in various oil/water systems (toluene/ethanol; cyclohexanone/acetone; 1,2-dichloroethane/ethanol) [83]. The authors obtained three types of particles. TEM images showed no difference in morphology between particles. However, MDSC results indicated that the compatibility of two polymers varied from complete to partial miscibility. The particles exhibited various T g values. On the basis of dCp/dT dependence in the function of temperature, it was possible to estimate the degree of separation. In fact, one type of particle was formed from completely miscible polymers as the dependence of dCp/dT = f(T) showed one signal located between the signals that are attributed to pure sulfonated-polystyrene and poly(methyl methacrylate)—T g = ω1 T g1 + ω2 T g2, respectively (ω 1, ω 2—weight fraction of component 1 and component 2, respectively). For the two other types of synthesized S-PS/PMMA particles, building polymers were partially miscible. However, there is a noticeable difference in the glass transition temperature values of the two other types of particles. One of them exhibited a lower T g value, which suggests that in situ formed PMMA has a lower molecular mass. As a result, a poly(methyl methacrylate) of a lower mass causes the plasticization of the S-PS system.
MDSC was used to investigate the effect of the cross-link density of a polystyrene seed on the thickness of the interfacial region in the polystyrene/poly(methyl acrylate) (PS/PMA) particles synthesized by seeded polymerization [84]. The weight fraction of interfacial region was evaluated on the basis of depletion of dCp/dT for PS/PMA compared to dCp/dT values for pure components. The weight fraction of the interfacial region fluctuated from approximately 10 % for a 1 mol% cross-linking agent (divinylbenzene (DVB)) to above 15 % for 10 mol% of DVB. The calculation of weight fractions was performed because ΔC p is proportional to the weight fraction of pure components.
TMDSC experiments are time-consuming due to the need for sample scanning at different frequencies during independent runs. The solution to this inconvenience is to use the TOPEM-DSC method (multi-frequency temperature-modulated differential scanning calorimetry) introduced by Mettler–Tolledo in 2005 [79], which facilitates the evaluation of complex heat flow at various frequencies. In this method, stochastic dependence of temperature modulation over time (heating and cooling rate) is used. The solution is guaranteed by taking random temperature pulses of various durations and superimposing them on the underlying heating and cooling rates. Such an approach delivers a wide frequency spectrum, which facilitates the estimation of both “quasi-static” (frequency independent) and “dynamic” heat capacity (frequency dependent). Transitions, either frequency-dependent, like the glass transition, or frequency-independent (crystallization, melting), can be distinguished by this method in a single scan. TOPEM-DSC facilitates the separation of temperature-dependent and time-dependent thermal effects, which means that additional signals can be observed that are not observed through typical DSC.
In the last 10 years, interest in TOPEM DSC for the analysis of core–shell polymer particles has grown. In practice, this method turned out to be a useful tool to explore the impact of the hard/soft characteristics of the core on the thickness of the interfacial region.
Quantitative estimation of phase separation within the core–shell microspheres, based on the heat capacity data, was done for different complex, three-component polymer particles obtained by seeded polymerization [82]. Based on data concerning heat capacity (C p) attributed to pure components (the core and shell of particles), real weight fractions (ω) and thickness R of all present regions within the composite particle structure (core (ω c, R c), shell (ω s, R s) and interfacial region (ω i , R i)) was calculated assuming a rigid global shape and the existence of the interfacial region exactly between the particle’s core and shell [82, 85]. Morphological investigations of particles by DSC are possible provided that we know the dCp/dT values for the pure core and shell, respectively. Particles’ core and shell were all formed from the same comonomers—styrene, methyl methacrylate, butyl acrylate; however, they are mixed at various molar ratios, resulting in the formation of soft or hard regions. The hard region was formed from a higher amount of styrene and methyl methacrylate, whereas the soft region was formed from a molar excess of butyl acrylate. A series of composite polymer particles, which were, respectively, composed of a hard core and soft shell (T g,c > T g,s) or vice versa (T g,c < T g,s), was synthesized. The authors noticed that in the case of a hard core/soft shell particle formation, a decrease in T g,c resulted in core–shell particles with a greater interfacial region (see Table 1, particles denoted as A1, B1 and C1). This means that a lower T g of the core, especially below polymerization temperature, leads to an increase in the phase miscibility and in the interfacial region thickness as the mobility of polymer chains is higher.
During the formation of a hard shell (T g,s > T synthesis) on a soft core, a soft core–hard shell particle morphology was observed due to restricted core penetration by the oligomeric radicals of the growing chains of the shell polymer. Lower values of T g,s favoured an increase in the interfacial region thickness (see Table 1, particles A2 and B2). Moreover, C2 particles exhibited a homogenous morphology rather than a core–shell structure. TOPEM-DSC curves recorded for A1, B1 and C1 and A2, B2 and C2 are presented in Fig. 9.

TOPEM-DSC curves (temperature-dependent heat capacity curves (a–c) and temperature-dependent dCp/dT curves (d–f) for hard core–soft shell particles on the left side (A1, B1, C1 particles) and soft core–hard shell particles on the right side (A2, B2, C2 particles). On the left side, P0 indicates a homogeneous shell particles phase; P1, P2 and P3 denote homogeneous core polymer latex particles. On the right side, P0 indicates a homogeneous core particles phase; P1, P2 and P3 denote homogeneous shell polymer latex particles. Reprinted with permission from [82]. Copyright 2012 Elsevier B.V
The impact of the hydrophilicity of the shell mixture composition on the final morphology of particles synthesized starting from a hard core was also analysed (see Table 1, particles denotes as A1*, B1* and C1*). Acrylic acid (besides the styrene, methyl methacrylate and butyl acrylate used to build particles’ shell) caused depletion of the latex interfacial region. The final particle morphology was consistent with the known behaviour of hydrophilic components, which tend to exist in the outermost layer between the particle and the water phase. As a result, within composite particles, a better core/shell separation was observed.
It is worth noting that TOPEM-DSC allowed for the estimation of the influence of the mobility of macromolecules building a particle’ core on the radial composition of polymer particles obtained by seeded polymerization.
Based on the data presented in Table 1, it is apparent that TOPEM-DSC was a sensitive tool for the determination of the presence and thickness of an interfacial region between the core and shell of a particle. An interfacial layer as thin as 0.6 nm can be recognized using TOPEM-DSC, which means that this technique is useful for the precise analysis of particle’s radial composition changes.
Mu et al. used TOPEM-DSC to demonstrate a multi-layered structure for core–shell particles composed of five types of copolymers by revealing the presence of interfaces in a single scan over a range of frequencies without quantitative details [86]. Apart from the signals attributed to each component, two additional signals were present that related to the presence of the two interfacial regions. It was possible to confirm the particle’s morphology due to sufficient differences in the T g values for each layer.
Small-angle scattering methods
Small-angle scattering methods such as small-angle X-ray scattering (SAXS) and small-angle neutron scattering (SANS) were applied in structural studies of polymer core–shell colloids. They provide information on the radial structure of particles. The difference between these methods lies in the type of radiation used. In SAXS, samples are treated by monochromatic X-ray, which is scattered by electrons of atoms that are present in analysed material. The higher the atomic number of element present in a sample, the stronger the scattering of the X-ray. In SANS, a neutron beam is used, which is scattered as a result of neutrons colliding with the nuclei of the atoms in experimental material. The intensity of neutron interactions with the atoms’ nuclei depends on their mass and differs for various isotopes of the same element.
SANS and SAXS are sensitive to the excess of scattering length density or scattering electron density \( \left(\rho \left(\overrightarrow{r}\right)-{\rho}_m\right) \) of the particles, where ρ m is ascribed to the solvent in which particles are suspended. Generally, the scattering intensity is measured as a function of the magnitude of the scattering vector q (q = (4π/λ)sin(Θ/2)), where λ is the wavelength of radiation and Θ is the scattering angle) [87].
In the total measured scattering intensity (I(q)), for a suspension of non-interacting core–shell particles, distinguished contributions can come from three types of structures:
-
Core–shell structure (ICS(q)),
-
Polymer shell displaying static inhomogeneities (IS(q)),
-
A solid polymer core (IC(q)).
To use small-angle scattering methods for particle investigation, there must be sufficient electron density (for SAXS) or scattering length density (for SANS) between the particle and the dispersing medium. Satisfactory analyses of core–shell particles are obtained provided that the core and shell differ in the scattering intensity of electrons or neutrons. A convenient situation is when one of small-angle scattering methods facilitates the observation of only a particles’ core, whereas the latter is sensitive to a particles’ shell. As an example, Ballauff et al. [75, 88] examined particles built from a polystyrene core with shell composed of poly(N-isopropylacrylamide). The polystyrene core was invisible in SAXS analysis (6.4 e/nm3), whereas in SANS, it exhibited the significant ability of neutron scattering. In contrast, PNIPAM emitted a small signal in SANS, whereas in SAXS, the shell built from PNIPAM was explicitly evident (45.8 e/nm3) [75] (see Fig. 10). Therefore, such analyses of core–shell particles can deliver complementary information about the particle structure.

Analysis of PS/PNIPAM core–shell morphology by SAXS. Reprinted with permission from [75]. Copyright 1998 American Chemical Society
SANS plays the important role of easily manipulating the contrast of the components. It facilitates the selective scattering enhancement or reduction coming from the sample or its parts, which is important during analysis of multi-component samples. Generally, the strategy of contrast modulation relies on the exchange of isotopes of elements composing parts of the particle, for example, using deuterated monomers, which exhibit a more significant scattering length density. Another method of sample contrasting is dispersion of polymer particles in a mixture of H2O and D2O, which causes contrast enhancement due to stronger scattering of neutrons on the deuter nucleus. This procedure was used to estimate the morphology of polymer particles composed of a polystyrene core and PNIPAM shell [88]. The hydrophobic character of polystyrene prevents water from penetrating the particles’ core. In contrast to polystyrene, the PNIPAM shell was highly swollen with water containing D2O. It is worth noting that SANS and SAXS turned out to be appropriate tools in the investigations of the radial structure of PS/PNIPAM particles at different temperatures focusing on the PNIPAM shell volume changes. The PNIPAM shell microgel deposited onto a solid polystyrene core exhibited swollen and shrunken states, i.e. below or above the lower critical solution temperature (LCST) of PNIPAM, respectively [88]. Figure 11 displays the results obtained from SAXS measurements carried out at 25 and 40 °C, revealing thermoresponsive properties of a cross-linked PNIPAM shell on a polystyrene core.

SAXS analysis of PS/PNIPAM core–shell particles at 25 and 40 °C revealing changes of thermosensitive shell thickness. Reproduced from Ref. [88] with permission from the PCCP Owner Societies
SAXS was applied in studies of the adsorption of non-ionic and ionic surfactants, such as Triton X-405 and sodium dodecyl sulfate, on the surface of polystyrene particles [89–91]. The impact of the surfactant concentration on the thickness of the surfactant coverage was determined. Moreover, SAXS was used to analyse the structure of core–shell particles made from two immiscible polymers. A polystyrene core and shell formed from poly(methyl methacrylate) was synthesized by seeded polymerization [92, 93]. To obtain the radial structure of the particles, a sucrose solution was used to ensure a sufficient contrast variation of electron density. The ρ m value of electrons/nm3 of the sucrose solution was determined (332.79 + 1.2827c, where c denotes the weight percentage of sucrose in the respective solution) [92]. SAXS analysis provided the quantitative information concerning the interface between the polystyrene core and poly(methyl methacrylate) shell of particles prepared by seeded polymerization in two ways: Latex I and Latex II [92]. Latex I was synthesized in monomer-starved conditions, whereas Latex II prepared by first swelling polystyrene seed particles with monomeric methyl methacrylate. SAXS analyses revealed a difference in the characteristics of the interfacial region in the two types of particles (a very sharp or a diffuse interface between the two incompatible polymers). The thickness of the interface was estimated by determining the region of the differentiated electron density between the core and shell (electron density of this region was more than 2 % above the value attributed to polystyrene core and 98 % below the value of the electron density corresponding to the PMMA shell) [92].
Chen et al. used SAXS to determine with high accuracy the radial structure of core–shell polystyrene/poly(butyl acrylate) particles, using various concentrations of sucrose to assure appropriate contrast of the polymers’ electron density [94]. The estimated shell thickness was equal to 2 nm. Moreover, the authors noted a lack of structural impact of contrast on the particles’ morphology.
It is worth noting that small-angle scattering of X-ray or neutron methods are sensitive to particle regions that display a relevant difference in electron or neutron density compared to the continuous phase (dispersant). Therefore, SANS and SAXS do not allow for the observation of single polymer chains. Structural results obtained by small-angle scattering methods can be supplemented with dynamic light scattering, which delivers information on the total hydrodynamic particle size. The application of efficient contrast variation does not assure the visualization of single chains by SAXS and SANS, which are involved in the particle diameter obtained by DLS (Fig. 12) [95].

The range of particle diameter revealed by various scattering methods
Expressions describing the intensity of scattering (I(q)) for polymer core–shell particles in SAXS, which include the influence of the densities of individual parts of a particle and the density of the surrounding medium can be found in ref. [94].
Solid-state NMR studies
Solid-state NMR has been applied as a tool to determine the structure of core and shell particles with detailed evaluation of the substructures of the interphase region, which exists between the core and the shell. The spin-diffusion technique has great significance. Such an experiment is based on the difference in proton mobility between phases and is composed of three parts: a selection period, a spin-diffusion process called the mixing period of duration (t m) and a detection period [57]. The selection period is based on proton magnetization of one component, which is made using a dipolar filter (magnetization filter) that is derived from the original Goldman–Shen experiment [96]. The dipolar filter consists of a pulse sequence separated by a certain delay time (t d) in the range of 10–100 μs. The higher the delay time applied, the stronger the dipolar filter is. Therefore, a short t d means a weak filter, whereas a long t d means strong filter. The strength of the dipolar filter can also be enhanced by increasing the number of cycles to keep the delay time constant [57]. A strong filter facilitated the detection of only very mobile regions, excluding the rigid component phase and interphase region whose mobilities are reduced. Generally, the rigid components display strong dipolar couplings, whereas soft (mobile) phases exhibit weak dipolar couplings. Filters of differentiated strength used to investigate composite core–shell particles enable regions of different molecular mobilities exhibiting various spin-spin relaxation times, T 2 to be revealed. In the case of the interphase, the situation is complicated. When applying a weak filter, mobilized parts of the rigid element involved in the mobile phase can be still observed, whereas the fraction of the mobile component, including the interphase, can be reduced due to its decreased mobility and can be suppressed by applying the dipolar filter. Experiments carried out without filters enable the identification of all of the components that comprise the whole particle. It is interesting to note that the spin-diffusion technique is a suitable tool to use to define the type of heterogeneities present within a particle’s morphology (microdomains, concentration fluctuations on the length scale of several nanometres) and to determine its size. The basic requirement for an analysed sample is the difference in the NMR parameters of the two components that allow for their distinction. Chemical shifts in the proton spectrum or a large enough difference in the mobility of the two components comprising the particle is required. Mobility is associated with the glass temperature (T g) of polymer elements. The component is regarded as mobile if its T g is lower than the temperature conditions of NMR analysis. In contrast, a polymer for which the T g is higher than the temperature of the spin-diffusion experiment is defined as rigid because its motion is very slow.
In practice, for core–shell particles composed of two types of polymers, between the two immiscible or partially miscible components, there exists a gradient region, which is characterized by gradually changing dipolar couplings in NMR experiments.
A spin-diffusion curve expressed as signal intensity versus square root of mixing time (t m 1/2) is obtained in the form of a monotonical decay to a plateau value. The initial slope of the spin-diffusion decay and later obtained plateau (called the final value) provides crucial data about the structure of the whole particles and their interphase region, including the precise measurement of its thickness. In Fig. 13, a typical spin-diffusion curve is presented. The final value and t*m 1/2 can be used to calculate the volume to interface ratio (V tot/S tot) ϕ A, on the assumption that the proton densities of phases A and B are identical [19, 65], according to following expression:

Typical spin-diffusion curve with an illustration of how to determine the final value and t m*1/2. Reproduced with permission from [65]. Copyright Wiley-VCH Verlag GmbH&Co. KGaA
where V tot is the total volume of the particle; S tot denotes the total interface area between the two phases A and B; ϕ A and ϕ B denote the proton fraction of phases A and B, respectively, and D A and D B denote spin-diffusion coefficients of phases A and B.
For particles with perfect core–shell morphology, slow spin-diffusion decay is expected [19] (Fig. 14, the highest curve). For particles containing small regions in the interphase of the core–shell structure, fast spin-diffusion decay is observed on the spin-diffusion curve (Fig. 14, intermediate curve). For particles composed of a core–shell morphology with small domains, a superposition of the abovementioned curves is observed (Fig. 14, the lowest spin-diffusion curve). The t*m 1/2 value can be estimated by extrapolating a straight line in the region of the initial spin-diffusion crossing the abscissa presenting mixing time.

Schematic of spin-diffusion curves demonstrating the sensitivity to different structures in the latex particles. Reproduced with permission [19] Copyright Wiley-VCH Verlag GmbH&Co. KGaA
The rate of spin-diffusion decay (initial slope) can be used to estimate the thickness of the interphase region [57].
On the basis of spin-diffusion analysis of the core–shell particles, it is possible to elucidate whether the interphase displays a gradient character (gradient dipolar couplings change). This means that the mobile polymer component is immobilized by rigid component as its mobility is decreased in comparison with a related pure mobile component. Based on the known stoichiometric ratio of two comonomers used during particle synthesis and the final value of the spin-diffusion curve corresponding to the real mobile fraction, it is possible to evaluate the fraction of the mobile phase engaged in the interphase region and the fraction of the suppressed rigid component.
Using filters of different strength can help precisely analyse the interphase region. For example, spin-diffusion experiments carried out on particles built from a soft core and hard shell and applying a weak dipolar filter revealed the particles’ fractions for the core and interphase regions, whereas a strong filter showed only the particles’ core. Representative studies of such systems using core–shell particles composed of a poly(n-butyl acrylate) core and a poly(methyl methacrylate) shell were conducted by Landfester et al. [19].
It is worth noting that the temperature of the spin-diffusion experiment is essential to assure the relevant mobility of components and to distinguish all individual components, specifically the interfacial region. If the temperature of the experiment is too high, it will yield information about the whole core–shell structure, without detecting the aspects in the interfacial region, due to the lack of mobility difference. On the other hand, at low temperatures, the experiment will not provide detailed data about the presence of small regions, due to an excessively fast spin-diffusion process [97] The morphology of poly((tert-butyl acrylate)/styrene) (PTBA/PS) inverse core–shell particles and PS/PTBA particles was successfully analysed using 1H spin-diffusion solid NMR [57]. For proper analysis, the temperature of the experiment was adjusted to 60 °C to facilitate the observation of PTBA magnetization causing suppression the magnetization of rigid component (PS), as the temperature of glass transition of uncross-linked PTBA and PS equal 40 and 108 °C, respectively.
The spin-diffusion curve for PTBA/PS particles revealed a two-step signal decay, which indicated the presence of two structures. A fast initial slope indicated the existence of small regions in the interface between the soft and hard phases. The second step of magnetization decay for PTBA was slow at longer mixing times and expresses the whole structure of particles. Experiments based on a gradual increase of filter strength (Fig. 15), simultaneously maintaining a constant delay time, caused a decrease in the fraction of the mobile phase at the final plateau, which indicated a gradient region between the two pure components.

1H spin-diffusion curves assigned for PTBA/PS inverse core–shell particles obtained with varying filter strength n cycle = 1–5. Reprinted with permission from [57]. Copyright 1999 American Chemical Society
Spin-diffusion data supported by numerical simulation provided information about the thickness of the interfacial region. For PS/PTBA particles, the interfacial region was quite thin (<5 nm) and was assessed base on the rapid decrease of PTBA magnetization (initial slope) and high fraction of mobile phase estimated from the final value of the magnetization plateau.
Landfester et al. [65] demonstrated the influence of the appropriate t d value on the magnetization suppression of the rigid component (poly(methyl methacrylate)) for the core–shell poly(n-butyl acrylate)/poly(methyl methacrylate) (PBA/PMMA) particles. The temperature of the experiment was set at 60 °C to obtain a magnetization of PBA acting as a mobile phase. The mobile component exhibited a high mobility, with a line width in the 1H NMR spectra of 400 Hz. In these conditions, the spin-diffusion coefficient of PBA was approximately 0.1 nm2 ms−1, whereas for a rigid polymer, it was equal to 0.8 nm2 ms−1.
For PBA/PMMA particles synthesized from an equimolar mixture of comonomers by semi-batch emulsion polymerization, when the t d was below 30 μs, no spin-diffusion of the soft core was observed. An increase in filter strength by adjustment of t d to 30 μs revealed the existence of two different structures. The authors demonstrated the effect of a delay time value on the shape of spin-diffusion curves. For series of PBA/PMMA particles differentiated with the molar ratio of two components, spin-diffusion experiments were conducted at two t d values, i.e. 2 and 80 μs. For the weaker filter, PMMA magnetization was not entirely suppressed. The final value corresponding to the mobile phase was not consistent with the stoichiometric ratio as it exceeded the expected value. The situation was completely different when a stronger filter was used. Investigations uncovered differences in the particles’ structures that were strictly dependent on the mutual molar ratio of the two components used. It was also confirmed that with the increase in the rigid shell fraction, the mobility of the core phase gradually decreased, as evident from the decrease in the final values. Spin-diffusion experiments carried out for various PBA/PMMA particles showed that an increase in the amount of rigid shell-building component during the particle synthesis exerts a better phase separation, causing depletion of the interphase region.
At this point, it is worth mentioning that solid-state NMR analysis of composite core–shell polymer particles can deliver information about the whole structure of particles and detailed information about the present heterogeneities. It strongly depends on the experiment parameter, i.e. the filter strength in a spin-diffusion experiment. Usage of a weak filter strength results in spin-diffusion sensitivity to the entire structure of composite polymer particle, indicating the existence of core–shell morphology. An increase in the filter strength causes the enhancement of spin-diffusion sensitivity exclusively to the heterogeneities formed from the mobile copolymer, containing also rigid type of component. Summarizing, the small substructures of the particle core can be detected when the strength of the spin-diffusion filter is increased. Moreover, Kirsch et al. indicated the sensitivity limitations of spin-diffusion experiments in determination of entire particle morphology which diameter was as big as 400 nm [57].
Ishida et al. [98] used solid-state13C NMR spectroscopy to identify the impact of the cross-linking agent (allyl methacrylate) concentration on the final morphology of core–shell particles (PBA/PMMA) prepared by two-step emulsion polymerization of butyl methacryle and methyl methacrylate with the molar ratio of 4:1. In the first step, allyl methacrylate was introduced. The resulting microspheres did not display a typical core–shell morphology, but rather, poly(methyl methacrylate) areas were embedded in the PBA phase. The application of a lower concentration of allyl methacrylate resulted in phase separation favouring the core–shell structure. Evaluation of the particles’ morphology was performed using 1H spin-lattice relaxation times (T 1ρH) and 13C spin-lattice relaxation times (T 1ρC), which revealed phase separation in the range of several nanometres.
Conclusions
We have discussed methods that enable the resolution of the morphology of polymer particles composed of two or more components. Overview of these methods is presented in Table 2.
Of the imaging methods presented, the most popular one is transmission electron microscopy. Appropriate sample preparation facilitates the successful visualization of the particles’ core and shell. However, precise data concerning the particles’ morphology can be obtained only using methods that provide a quantitative analysis of all phases, including the interfacial region, which is not visible using imaging methods. Amongst the methods used in detailed morphological studies of core–shell particles were DSC, NMR and small-angle scattering methods (SANS, SAXS). We have also highlighted the potential of direct and indirect methods that can be used for determination of surface composition.
Control of a particle’s morphology can play a decisive role not only from a scientific point of view but also when deciding about the potential application of the product. In terms of future applications, it is essential to determine the composition of the outermost layer of the particles.
It is worth noting that a full analysis of polymer particles’ morphology synthesized from two or more components usually requires the application of at least two methods (Table 3).
References
Slomkowski S, Aleman JV, Gilbert RG, Hess M, Horie K, Jones RG, Kubisa P, Meisel I, Mormann W, Penczek S, Stepto RFT (2011) Terminology of polymers and polymerization processes in dispersed systems (IUPAC Recommendations 2011). Pure Appl Chem 83:2229–2259. doi:10.1351/PAC-REC-10-06-03
Arshady R (1999) Definitions, nomenclature and terminology. In: Arshady R (ed) Microspheres microcapsules and liposomes. Preparation and chemical applications. Citus Books, London, pp 55–81
Jin JM, Lee JM, Ha MH, Lee K, Choe S (2007) Highly crosslinked poly(glycidyl methacrylate-co-divinyl benzene) particles by precipitation polymerization. Polymer 48:3107–3115. doi:10.1016/j. polymer .2007.03.068
Lee JM, Lee DG, Lee SJ, Kim JH, Cheong IW (2009) One-step synthetic route for conducting core–shell poly(styrene/pyrrole) nanoparticles. Macromolecules 42:4511–4519. doi:10.1021/ma9001897
Liu J, Chew CH, Gan LM, Teo WK, Gan LH (1997) Synthesis of monodisperse polystyrene microlatexes by emulsion polymerization using a polymerizable surfactant. Langmuir 13:4988–4994. doi:10.1021/la970252m
Tuncel A (2000) Emulsion copolymerization of styrene and poly(ethylene glycol) ethyl ether methacrylate. Polymer 41:1257–1267. doi:10.1016/S0032-3861(99)00287-6
Serizawa T, Chen M-Q, Akashi M (1998) Graft copolymers having hydrophobic backbone and hydrophilic branches. XVIII. Poly(styrene) nanospheres with novel thermosensitive poly(N-vinylisobutyramide)s on their surfaces. J Polym Sci A Polym Chem 36:2581–2587. doi:10.1002/(SICI)1099-0518(199810)36:14<2581::AID-POLA17>3.0.CO;2-E
Ishizu K, Tahara N (1996) Microsphere synthesis by emulsion copolymerization of methyl methacrylate with poly(methacrylic acid) macromonomers. Polymer 37:2853–2856. doi:10.1016/0032-3861(96)87650-6
Thompson KL, Armes SP, York DW, Burdis JA (2010) Synthesis of sterically-stabilized latexes using well-defined poly(glycerol monomethacrylate) macromonomers. Macromolecules 43:2169–2177. doi:10.1021/ma9027799
Dupin D, Thompson KL, Armes SP (2011) Preparation of stimulus-responsive liquid marbles using a polyacid-stabilised polystyrene latex. Soft Matter 7:6797–6800. doi:10.1039/c1sm05889d
Thompson KL, Bannister I, Armes SP, Lewis AL (2010) Preparation of biocompatible sterically stabilized latexes using well-defined poly(2-(methacryloyloxy)ethyl phosphorylcholine) macromonomers. Langmuir 26:4693–4702. doi:10.1021/la903567q
Gosecka M, Basinska T, Slomkowski S, Tracz A, Chehimi MM (2014) Mechanism of particle formation in radical emulsion copolymerization of styrene with α-tert-butoxy-ω-vinylbenzyl-polyglycidol macromonomer. Polymer 55:788–797. doi:10.1016/j. polymer .2013.12.008
Ahmad H, Miah MAJ, Pervin MS, Rahman MM (2003) Composite polymer particles prepared with different amounts of acrylic acid and their usefulness as carriers for biomolecules. Colloid Polym Sci 281:897–901. doi:10.1007/s00396-002-0856-z
Ahmad H, Miah MAJ, Rahman MM (2003) Preparation of micron-sized composite polymer particles containing hydrophilic 2-hydroxyethyl methacrylate and their biomedical applications. Colloid Polym Sci 281:988–992. doi:10.1007/s00396-003-0862-9
Guo X, Weiss A, Ballauff M (1999) Synthesis of spherical polyelectrolyte brushes by photoemulsion polymerization. Macromolecules 32:6043–6046. doi:10.1021/ma990609o
Lu Y, Ballauff M (2011) Thermosensitive core–shell microgels: from colloidal model systems to nanoreactors. Prog Polym Sci 36:767–792. doi:10.1016/j.progpolymsci.2010.12.003
Jayachandran KN, Chatterji PR (1998) Grafted polymeric microspheres: a synthetic approach to structurally well-defined brush polymers. J Macromol Sci Pure Appl Chem A 35:1971–1986. doi:10.1080/10601329808000991
Meng F, Engbers GHM, Feijen J (2004) Polyethylene glycol–grafted polystyrene particles. J Biomed Mater Res A 70:49–58. doi:10.1002/jbm.a.30056
Landfester K, Spiess HW (1998) Characterization of interphases in core–shell latexes by solid-state NMR. Acta Polym 49:451–464. doi:10.1002/(SICI)1521-4044(199809)49:9<451::AID-APOL451>3.0.CO;2-U
Christopher KR, Pal A, Mirchandani G, Dhar T (2014) Synthesis and characterization of polystyrene-acrylate/polysiloxane (PSA/PSi) core shell polymers and evaluation of their properties for high durable exterior coatings. Prog Org Coat 77:1063–1068. doi:10.1016/j.porgcoat.2014.03.008
Besteti MD, Souza FG Jr, Freire GMG, Pinto JC (2014) Production of core-shell polymer particles-containing cardanol by semibatch combined suspension/emulsion polymerization. Polym Eng Sci 54:1222–1229. doi:10.1002/pen.23660
Gosecka M, Slomkowski S, Basinska T (2014) Interactions of serum proteins and alkaline phosphatase with poly(styrene/α-tert-butoxy-ω-vinylbenzyl-polyglycidol) microspheres with various surface concentrations of polyglycidol. Polym Adv Technol 25:1264–1272. doi:10.1002/pat.3311
Mao X, Huang J, Leung MF, Du Z, Ma L, Huang Z, Li P, Gua L (2006) Appl Biochem Biotechnol 135:229–239
Hou X, Liu B, Deng X, Zhang B, Chen H, Luo R (2007) Covalent immobilization of glucose oxidase onto poly(styrene-co-glycidyl methacrylate) monodisperse fluorescent microspheres synthesized by dispersion polymerization. Anal Biochem 368:100–110. doi:10.1016/j.ab.2007.04.034
Zhou C, Che R, Zhong L, Xu W, Guo G, Lei J (2011) Effect of particle structure on the peel strength and heat resistance properties of vinyl acetate/acrylate latexes laminating adhesives. J Appl Polym Sci 119:2857–2865. doi:10.1002/app.32953
Mishra S, Singh J, Choudhary V (2010) Synthesis and characterization of butyl acrylate/ methyl methacrylate/glycidyl methacrylate latexes. J Appl Polym Sci 115:549–557. doi:10.1002/app.30963
Basinska T, Wisniewska M, Chmiela M (2005) Principle of a New immunoassay based on electrophoretic mobility of poly(styrene/a-tert-butoxy-o-vinylbenzyl-polyglycidol) microspheres:application for the determination of helicobacter pylori IgG in blood serum. Macromol Biosci 5:70–77. doi:10.1002/mabi.200400112
Lee C-F, Hsu M-L, Chu C-H, Wu T-Y (2014) Synthesis and characteristics of poly(methyl methacrylate-co-methacrylic acid)/poly(methacrylic acid-co-N-isopropylacrylamide) thermosensitive semi-hollow latex particles and their application to drug carriers. J Polym Sci A Polym Chem 52:3441–3451. doi:10.1002/pola.27411
Ballauff M, Lu Y (2007) “Smart” nanoparticles: preparation, characterization and applications. Polymer 48:1815–1823. doi:10.1016/j.polymer.2007.02.004
Welsch N, Wittemann A, Ballauff M (2009) Enhanced activity of enzymes immobilized in thermoresponsive core − shell microgels. J Phys Chem B 113:16039–16045. doi:10.1021/jp907508w
Chang G, He L, Liang J, Wang N, Cao R, Zhao X (2014) Polysiloxane/poly(fluorinated acrylate) core–shell latexes and surface wettability of films. J Fluor Chem 158:21–28. doi:10.1016/j.jfluchem.2013.11.015
Wang RLC, Kreuzer HJ, Grunze M (1997) Molecular conformation and solvation of oligo(ethylene glycol)-terminated self-assembled monolayers and their resistance to protein adsorption. J Phys Chem B 101:9767–9773. doi:10.1021/jp9716952
Stein MJ, Weidner T, McCrea KR, Castner DG, Ratner BD (2009) Hydration of sulphobetaine and tetra(ethylene glycol)-terminated self-assembled monolayers studied by sum frequency generation vibrational spectroscopy. J Phys Chem B 113:11550–11556. doi:10.1021/jp9015867
Cairns DB, Armes SP, Chehimi MM, Peruchot C, Delamar M (1999) X-ray photoelectron spectroscopy characterization of submicrometer-sized polypyrrole-polystyrene composites. Langmuir 15:8059–8066. doi:10.1021/la990443k
Zhang WL, Piao SH, Choi HJ (2013) Facile and fast synthesis of polyaniline-coated poly(glycidyl methacrylate) core–shell microspheres and their electro-responsive characteristics. J Colloid Interface Sci 402:100–106. doi:10.1016/j.jcis.2013.04.011
Barthet C, Armes SP, Chehimi MM, Bilem C, Omastova M (1998) Surface characterization of polyaniline-coated polystyrene latexes. Langmuir 14:5032–5038. doi:10.1021/la980102r
Gosecka M, Griffete N, Mangeney C, Chehimi MM, Slomkowski S, Basinska T (2011) Preparation and optical properties of novel bioactive photonic crystals obtained from core-shell poly(styrene/α-tert-butoxy-ω-vinylbenzyl-polyglycidol) microspheres. Colloid Polym Sci 289:1511–1518. doi:10.1007/s00396-011-2447-3
Allen G, Bevington JC, Booth C, Price C (1989) Comprehensive polymer science, the synthesis, characterization, reactions and applications of polymers. Pergamon Press, Oxford
Perruchot C, Chehimi MM, Delamar M, Lascelles SF, Armes SP (1996) Surface characterization of polypyrrole-coated polystyrene latex by X-ray photoelectron spectroscopy. Langmuir 12:3245–3251. doi:10.1021/la960057s
Kirsch S, Pfau A, Landfester K, Shaffer O, El-AAsser MS (2000) Particle morphology of carboxylated poly(n-butyl acrylate)/poly(methyl methacrylate) composite latex particles. Macromol Symp 151:413–418. doi:10.1002/1521-3900(200002)151:1<413::AID-MASY413>3.0.CO;2-G
Basinska T, Slomkowski S, Dworak A, Panchev I, Chehimi MM (2001) Synthesis and characterization of poly(styrene/α-t-butoxy-ω-vinylbenzyl-polyglycidol) microspheres. Colloid Polym Sci 279:916–924. doi:10.1007/s003960100517
Takahashia K, Uyama H, Kobayashi S (1998) Seed dispersion polymerization to micron-size monodisperse polymer particles. J Macromol Sci Pure Appl Chem 35:1473–1487. doi:10.1080/10601329808007311
Zhang C, Chen Y (2005) Investigation of fluorinated polyacrylate latex with core–shell structure. Polym Int 54:1027–1033. doi:10.1002/pi.1803
Cui X, Zhong S, Wang H (2007) Emulsifier-free core–shell polyacrylate latex nanoparticles containing fluorine and silicon in shell. Polymer 48:7241–7248. doi:10.1016/j. polymer .2007.10.019
Xu G, Deng L, Wen X, Pi P, Zheng D, Cheng J, Yang Z (2011) Synthesis and characterization of fluorine-containing poly-styrene-acrylate latex with core–shell structure using a reactive surfactant. J Coat Technol Res 8:401–407. doi:10.1007/s11998-010-9308-8
Briggs D (1989) Characterization of surfaces. In: Booth C, Price C (eds) Comprehensive polymer science. The synthesis, characterization, reactions & applications of polymers, 1st edn. Pergamon Press, Oxford, pp 543–559
Brindley A, Davis SS, Davies MC, Watts JF (1995) Polystyrene colloids with surface-grafted polyethylene oxide as model systems for site-specific drug delivery: I. Preparation and Surface Chemical Characterization Using SIMS and XPS J Colloid Interf Sci 171:150–161. doi:10.1006/jcis.1995.1161
Synthesis and properties of core-shell fluorosilicone-containing acrylate latexes.
Basinska T, Slomkowski S, Kazmierski S, Dworak A, Chehimi MM (2004) Studies of the surface layer structure and properties of poly (styrene/alpha-t-butoxy-omega-polyglycidol) microspheres by carbon nuclear magnetic resonance, X-ray photoelectron spectroscopy, and the adsorption of human serum albumin and gamma-globulins. J Polym Sci A Pol Chem 42:615–628. doi:10.1002/pola.10863
Rabanel J-M, Hildgen P, Banquy X (2014) Assessment of PEG on polymeric particles surface, a key step in drug carrier translation. J Control Release 185:71–87. doi:10.1016/j.jconrel.2014.04.017
Gam-Derouich S, Gosecka M, Lepinay S, Turmine M, Carbonnier B, Basinska T, Slomkowski S, Millot M-C, Othmane A, Hassen-Chehimi DB, Chehimi MM (2011) Highly hydrophilic surfaces from polyglycidol grafts with dual antifouling and specific protein recognition properties. Langmuir 27:9285–9294. doi:10.1021/la200290k
Meng FH, Engbers GHM, Gessner A, Müller RH, Feijen J (2004) Pegylated polystyrene particles as a model system for artificial cells. J Biomed Mater Res A 70:97–106. doi:10.1002/jbm.a.30068
Guyot A, McKenna T, Schneider M (2002) Functional polymer colloids from functional monomers and macromers. In: Arshady R, Guyot A (eds) Functional polymer colloids & microparticles. Citus Books, London, pp 39–76
Bai R, Qiu T, Duan M, Ma G, He L, Li X (2012) Synthesis and characterization of core–shell polysilsesquioxane-poly(styrene-butyl acrylate-fluorinated acrylate) hybrid latex particles. Colloids Surf A 396:251–257. doi:10.1016/j.colsurfa.2012.01.003
Griffete N, Dybkowska M, Glebocki B, Basinska T, Connan C, Maître A, Chehimi MM, Slomkowski S, Mangeney C (2010) Thermoresponsive colloidal crystals built from core–shell poly(styrene/α-tert-butoxy-ω-vinylbenzylpolyglycidol) microspheres. Langmuir 26:11550–11557. doi:10.1021/la100537v
Jönsson J-E, Karlsson OJ, Hassander H, Törnell B (2007) Semi-continuous emulsion polymerization of styrene in the presence of poly(methyl methacrylate) seed particles. Polymerization conditions giving core–shell particles. Eur Polym J 43:1322–1332. doi:10.1016/j.eurpolymj.2007.01.027
Kirsch S, Doerk A, Bartsch E, Sillescu H, Landfester K, Spiess HW, Maechtle W (1999) Synthesis and characterization of highly cross-linked, monodisperse core-shell and inverted core-shell colloidal particles. Polystyrene/poly(tert-butyl acrylate) core-shell and inverse core-shell particles. Macromolecules 32:4508–4518. doi:10.1021/ma980916e
Gaillard C, Fuchs G, Plummer CJG, Stadelmann PA (2007) The morphology of submicronsized core-shell latex particles: an electron microscopy study. Micron 38:522–535. doi:10.1016/j.micron.2006.07.023
Trent JS, Scheinbeim JI, Couchman PR (1983) Ruthenium tetraoxide staining of polymers for electron microscopy. Macromolecules 16:589–598. doi:10.1021/ma00238a021
Janik H, Walch E, Gaymans RJ (1992) Ruthenium tetraoxide staining of polybutylene terephatalate and polyisobutylene-b-PBT segmented block copolymers. Polymer 33:3522–3524. doi:10.1016/0032-3861(92)91114-H
Sawyer LC, Grubb DT (1996) Polymer microscopy. Chapman & Hall, Oxford
Serizawa T, Takehara S, Akashi M (2000) Transmission electron microscopic study of cross-sectional morphologies of core–corona polymeric nanospheres. Macromolecules 33:1759–1764. doi:10.1021/ma9907486
Ito K, Cao J, Kawaguchi S (2002) Functional polymer colloids from functional monomers and macromers. In: Arshady R, Guyot A (eds) Functional polymer colloids & microparticles. Citus Books, London, pp 109–135
De Jaeger N, Demeyere H, Finsy R, Sneyer R, Vanderdeelen J, van der Meeren P, van Laethem M (1991) Particle sizing by photon correlation spectroscopy part I: monodisperse latices: influence of scattering angle and concentration of dispersed material. Part Part Syst Charact 8:179–186. doi:10.1002/ppsc.19910080134
Kirsch S, Landfester K, Shaffer O, El-Aasser MS (1999) Particle morphology of carboxylated poly-(n-butyl acrylate)/poly(methyl methacrylate) composite latex particles investigated by TEM and NMR. Acta Polym 50:347–362. doi:10.1002/(SICI)1521-4044(19991001)50:10<347::AID-APOL347>3.0.CO;2-C
Li P, Zhu J, Sunintaboon P, Harris FW (2002) New route to amphiphilic core − shell polymer nanospheres: graft copolymerization of methyl methacrylate from water-soluble polymer chains containing amino groups. Langmuir 18:8641–8646. doi:10.1021/la0261343
Li W, Li P (2007) Synthesis of well-defined amphiphilic core–shell particles containing amine-rich shells. Macromol Rapid Commun 28:2267–2271. doi:10.1002/marc.200700449
You B, Zhou D, Yang F, Ren X (2011) Synthesis and characterization of core–shell polyacrylate particles containing hindered amine light stabilizers. Colloids Surf A 392:365–370. doi:10.1016/j.colsurfa.2011.10.019
Liu BL, Zhang BT, Cao SS, Deng XB, Hou X, Chen H (2008) Preparation of the stable core–shell latex particles containing organic-siloxane in the shell. Prog Org Coat 61:21–27. doi:10.1016/j.porgcoat.2007.08.008
Stubbs JM, Sundberg DC (2005) A round robin study for the characterization of latex particle morphology—multiple analytical techniques to probe specific structural features. Polymer 46:1125–1138. doi:10.1016/j. polymer .2004.11.079
Jang J, Oh JH (2005) Fabrication of a highly transparent conductive thin film from polypyrrole/poly(methyl methacrylate) core/shell nanospheres. Adv Funct Mater 15:494–502. doi:10.1002/adfm.200400095
Polzer F, Heigl J, Schneider C, Ballauff M, Borisov OV (2011) Synthesis and analysis of zwitterionic spherical polyelectrolyte brushes in aqueous solution. Macromolecules 44:1654–1660. doi:10.1021/ma102927c
Almgren M, Edwards K, Karlsson G (2000) Cryo transmission electron microscopy of liposomes and related structures. Colloids Surf A 174:3–21. doi:10.1016/S0927-7757(00)00516-1
Crassous JJ, Wittemann A, Siebenburger M, Schrinner M, Drechsler M, Ballauff M (2008) Direct imaging of temperature-sensitive core-shell latexes by cryogenic transmission electron microscopy. Colloid Polym Sci 286:805–812. doi:10.1007/s00396-008-1873-3
Dingenouts N, Norhausen C, Ballauff M (1998) Observation of the volume transition in thermosensitive core-shell latex particles by small-angle X-ray scattering. Macromolecules 31:8912–8917. doi:10.1021/ma980985t
Crassous JJ, Ballauff M, Drechsler M, Schmidt J, Talmon Y (2006) Imaging the volume transition in thermosensitive core–shell particles by cryo-transmission electron microscopy. Langmuir 22:2403–2406. doi:10.1021/la053198f
Samokhina L, Schrinner M, Ballauff M, Drechsler M (2007) Binding of oppositely charged surfactants to spherical polyelectrolyte brushes: a study by cryogenic transmission electron microscopy. Langmuir 23:3615–3619. doi:10.1021/la063178t
Wang S, Zhao W, Song J, Cheng S, Fan L-J (2013) A platform for preparation of monodispersed fluorescent conjugated polymer microspheres with core-shell structures. Macromol Rapid Commun 34:102–108. doi:10.1002/marc.201200560
Fraga I, Montserrat S, Hutchinson M (2007) TOPEM, a new temperature modulated dsc technique—application to the glass transition of polymers. J Therm Anal Calorim 87:119–124. doi:10.1007/s10973-006-7969-4
Song M, Hourston DJ, Schafer F-U, Pollock HM, Hammiche A (1998) Modulated differential scanning calorimetry: XVI. Degree of mixing in interpenetrating polymer networks. Thermochim Acta 315:25–32. doi:10.1016/S0040-6031(98)00273-1
Hourston DJ, Song M, Schafer F-U, Pollock HM, Hammiche A (1998) Modulated differential scanning calorimetry: 13. Analysis of morphology of poly(ethyl methacrylate)/polyurethane interpenetrating polymer networks. Thermochim Acta 324:109–121. doi:10.1016/S0040-6031(98)00528-0
Duan M, Qiu T, Huang C, Xu G, Guo L, Li X (2013) Synthesis of poly(acrylate-styrene)/poly(acrylate-styrene) core/shell latex and TOPEM-DSC characterization. Progress in Organic Coatings 76:216–223. doi:10.1016/j.porgcoat.2012.09.019
Song M, Liao B (2004) A modulated DSC characterization of morphology of composite latex particles. Thermochim Acta 423:57–61. doi:10.1016/j.tca.2004.04.025
Hourston DJ, Song M, Pang Y (2001) A modulated differential scanning calorimetry and small-angle X-ray scattering study of the interfacial region in structured latices. J Braz Chem Soc 12:87–92. doi:10.1590/S0103-50532001000100012
Hourston D, Song M, Hammiche A, Pollock HM, Reading M (1997) Modulated differential scanning calorimetry: 6. Thermal characterization of multicomponent polymers and interfaces. Polymer 38:1–7. doi:10.1016/S0032-3861(96)00466-1
Mu Y, Qiu T, Li X, Guan Y, Zhang S, Li X (2011) Layer-by-layer synthesis of multilayer core-shell latex and the film formation properties. Langmuir 27:4968–4978. doi:10.1021/la104862h
Ballauff M (2001) SAXS and SANS studies of polymer colloids. Curr Opin Colloid Interface Sci 6:132–139. doi:10.1016/S1359-0294(01)00072-3
Dingenouts N, Seelenmeyer S, Deike I, Rosenfeldt BM, Lindner P, Narayanan T (2001) Analysis of thermosensitive core-shell colloids by small-angle neutron scattering including contrast variation. Phys Chem Chem Phys 3:1169–1174. doi:10.1039/b009104i
Bolze J, Hörner KD, Ballauff M (1996) Adsorption of the nonionic surfactant triton X-405 on polystyrene latex particles as monitored by small-angle X-ray scattering. Langmuir 12:2906–2912. doi:10.1021/la951073c
Bolze J, Hörner KD, Ballauff M (1996) Competitive adsorption of an anionic and a nonionic surfactant on polystyrene latex particles as monitored by small x-ray scattering. Colloid Polym Sci 274:1099–1108. doi:10.1007/BF00655680
Seelenmeyer S, Ballauff M (2000) Analysis of surfactants adsorbed onto the surface of latex particles by small-angle X-ray scattering. Langmuir 16:4094–4099. doi:10.1021/la990998f
Dingenouts N, Kim YS, Ballauff M (1994) The interface between immiscible polymers in composite latexes: a small-angle x-ray scattering study employing contrast variation. Colloid Polym Sci 272:1380–1387. doi:10.1007/BF00654168
Grunder R, Urban G, Ballauff M (1993) Small-angle x-ray analysis of latex particles with core-shell morphology. Colloid Polym Sci 271:563–572. doi:10.1007/BF00657987
Chen X, Zhang J, Yi Z, Wang Q, Li X, Bian F, Wang J, Men Y (2011) Radial structure of commercial styrene-co-butyl acrylate latex particles by means of synchrotron small-angle X-ray scattering under contrast-variation conditions. J Coat Technol Res 8:489–496. doi:10.1007/s11998-010-9314-x
Ballauff M (2003) Nanoscopic polymer particles with a well-defined surface: synthesis, characterization, and properties. Macromol Chem Phys 204:220–234. doi:10.1002/macp.200290072
Goldman M, Shen L (1961) Spin-spin relaxation in LaF3. Phys Rev 144:321–331. doi:10.1103/PhysRev.144.321
Landefester K, Spiess HW (1996) Optimum measurement temperature for elucidating incomplete phase separation in core-shell latexes by solid state NMR. Macromol Rapid Commun 17:875–883. doi:10.1002/marc.1996.030171206
Ishida M, Oshima J, Yoshinaga K, Horii F (1999) Structural analysis of core-shell type polymer particles composed of poly(butyl acrylate) and poly(methyl methacrylate) by high-resolution solid-state 13C n.m.r. spectroscopy. Polymer 40:3323–3329. doi:10.1016/S0032-3861(98)00541-2
Acknowledgments
The authors gratefully acknowledge Prof. Stanislaw Slomkowski for his helpful discussions in reviewing this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gosecka, M., Gosecki, M. Characterization methods of polymer core–shell particles. Colloid Polym Sci 293, 2719–2740 (2015). https://doi.org/10.1007/s00396-015-3728-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-015-3728-z