
Abstract
Poly(m-phenylenediamine) (PmPDA) was synthesized via an oxidative polymerization using benzene-1,3-diaminium dodecyl sulfate (P2) as the monomer. P2 is a new concept of reactive surfactant because, unlike conventional polymerizable surfactants where the polymerizable group is a carbon–carbon double bond, therein, the polymer chain propagates through the diaminium group. Polymerization in aqueous micellar solution, chloroform/water interface, and xylene/water emulsion allowed successful synthesis of PmPDA. Infrared and UV–vis spectroscopy revealed that PmPDA share chemical structure, based on phenazine with open segments, rich in quinoid rings (pernigraniline-like), and partially doped, regardless of the method of synthesis. Conversely, electron microscopy exhibited important effect on morphology (nanofibers, pot-like, and globular nanoparticles) as a function of polymerization method and temperature. Additionally, cyclic voltammetry indicated electroactivity in all products, showing reduced/semi-oxidized and semi-oxidized/oxidized redox transitions with differences concerning synthesis method and temperature. A paraffin/PmPDA core–shell composite was obtained by emulsion polymerization method taking advantage of P2 amphiphilic properties. Electron microscopy evidenced microencapsulation, whereas thermal properties (melting temperature, melting enthalpy, thermal stability, and viscosity as function of temperature) suggested promising properties for the design of form-stable phase change materials.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyphenylenediamine (PPDA) belongs to the large family of polyanilines, sharing a structure based on quinoid and benzenoid groups. PPDA is derived either from ortho- (o-), meta- (m-), or para- (p-) phenylenediamine (PDA). Literature suggests that PPDA consists of a ladder-like structure of aromatic nitrogenous heterocycles (phenazine) irrespective of the monomer [1–3]; however, differences in the polymer ladder depending on the monomer used have been reported [3]. Likewise, polyaniline (PAni), depending on polymerization conditions, PPDA presents oxidation isomers [4]. Modifications to polymerization conditions, dopant, oxidizing agent, or pH allows improving PPDA properties, which has permitted to develop applications in, for example, biosensing or metal ion adsorption [3, 5–10].
Several groups have developed synthetic strategies that have enabled them to design and control PPDA microstructure. Thanks to this, they have developed materials with specific morphologies that have exhibited varied properties. For example, Wang et al. developed a method for synthesizing PpPDA in a controlled manner. Synthesis was achieved in aqueous medium with chloroauric acid as the oxidizing and polyvinilpirrolidona (PVP) as the surfactant. They found that the presence of PVP is an important condition in microstructure control (spindle-like, diamond-like, centrosymmetric leaf-like, and parallelogram-like) [11]. The group of Sun et al. synthesized core–shell composites using silver nanoparticles as the shell. The combination of PmPDA with silver nanoparticles, obtained by chemical polymerization in aqueous phase, allowed designing sensors for peroxide and nucleic acid sequences thanks, respectively, to the catalytic activity and fluorescence [6, 12]. The same group of Sun et al. reported the use of PmPDA with rod and belt microstructures obtained by chemical polymerization in aqueous phase and organic solvents, using ammonium persulfate (APS) as oxidant. In both cases, the microstructure was used as a platform for sensing nucleic acid sequences [13, 14]. Sang et al. reported the absorption of sulfate ions by microparticles of PmPDA synthesized in aqueous medium using APS as oxidant at 25 °C. They found that the concentration of the oxidizing agent and the pH are critical conditions in yield and ion adsorption efficiencies [15]. Wang et al. reported the design of a sensor for detecting metalloproteinase 2. In this investigation, spherical nanoparticles of PmPDA, synthesized in a mixture of water and methylpyrrolidone and catalyzed by APS, were used as sensor platform due to high electron density [16]. Furthermore, Jiang et al. reported a method to obtain different structures of PoPDA in aqueous media induced by the incorporation of NaCl (sodium chloride). They found that by modifying the concentration of NaCl, it is possible to modify the microstructure (belts, shaped bun, agglomerates, or long belts) [17].
Recently, we reported on the synthesis of polystyrene/PAni core–shell nanocomposites and nanographite/PAni composites using anilinium dodecyl sulfate as the monomer precursor of PAni [18–20]. In these works, the anilinium salt allowed, respectively, stabilizing styrene emulsion polymerizations and assisting graphite exfoliation. Subsequently, the PAni shell was produced by an oxidative polymerization catalyzed by ammonium persulfate, that is, the salt is, simultaneously, surfactant and monomer of PAni. Based on these properties, we considered it as a new concept of reactive surfactant, with the difference that the anilinium group is the reactive site and not a carbon–carbon double bond as in conventional reactive surfactants.
Herein, we report on the synthesis of poly(m-phenylenediamine) (PmPDA) using benzene-1,3-diaminium dodecyl sulfate (P2) as the monomer (Fig. 1a). In this two-tailed amphiphile, the diaminium group is the reactive site. Through this group, the ladder-like structure propagates via an oxidative polymerization. Additionally, the dodecyl sulfate counterions provide P2 with surface-active properties, that is, P2 is a surfactant. In this work, we studied polymerization method effect on PmPDA morphology, chemical structure, and electroactive properties. PDA chemical polymerization in the aqueous phase has been widely studied; however, heterophase systems have not been so popular. Emulsified and micellar systems were employed to synthesize nanocomposites and to control nanostructure [21–23]. Concerning interfacial polymerization, widely used method for the synthesis of polyaniline nanofibers, only one report was found [24]. Consequently, many aspects on PPDA synthesis related to monomer modification and synthesis method offer the opportunity to contribute with new knowledge in this field. Motivating results inspired the synthesis of paraffin/PmPDA core–shell composite, via emulsion polymerization method for stable-form phase change material application. With P2, we wish to contribute to the development of conjugated polymers and with the design of reactive surfactants with functional groups other than the conventional carbon–carbon double bonds, but keeping surfactant–monomer concept concurrently. Considering the significant difference in reactivity between P2 and the conventional reactive surfactants (Fig. 1b), no details on this issue are given. Relevant literature addressing this topic can be consulted [25, 26].

a Chemical structure of reactive amphiphile, benzene-1,3-diaminium dodecylsulfate (P2). b General structure of a conventional reactive surfactant which includes hydrophilic head-group, counterion, hydrophobic tail, and free radical polymerizable C–C double bond
Experimental
Reagents
m-Phenylenediamine (WAKO), ammonium persulfate (recrystallized from a saturated aqueous solution at 45 °C), sodium dodecyl sulfate (SDS), hydrochloric acid, ethanol, xylene, acetone, and paraffin (m.p. 45 °C) were used as delivered (Nacalai Tesque) unless something else was indicated. Tri-distilled quality water was used in all experiments.
Synthesis
P2 was synthesized by reacting m-phenylenediamine (mPDA) with protonated SDS at 60 °C. P2 purification was carried out by recrystallization from water; first, the product was dissolved in water at 60 °C and left to cool at room temperature. The crystals were recovered by filtration and evaporated at room temperature (purification procedure is repeated two times). P2FTIR 2940 cm−1 (C–H,νs,CH3); 2859 cm−1 (C–H,νs,CH2), 2920 cm−1 (C–H,νas,CH2); 1210 cm−1 (S=O2,νas,SO4), 1066 cm−1 (S=O2,νs,SO4), 805 cm−1 (S–O,νs,SO4); 2620 cm−1 (NH4 +,νovertone), 1520 cm−1 (N–H,νas, NH4 +), 1600 cm−1 (N–H,νs,NH4 +); 1495 cm−1 (C–H,ω,anilinium) 950 cm−1 (C–H,ρ,anilinium). P2UV–vis 360 nm (π-π*phenazin fragments).
Krafft temperature (KT, 50 °C) and critical micellar concentration (cmc, 1.5 mmol L−1) were determined by electrical conductivity measurements. Electrical conductometry is a classical method to determine KT and cmc [27–29]. This method is particularly suitable for ionic surfactants. It is based on the abrupt change of slope in the curve of electrical conductivity versus concentration of the surfactant in aqueous solution. At the cmc, the spontaneous self-assembly of the surfactant molecules occurs, because the micelles are bulkier than the surfactant molecules in solution, the conductivity decreases causing slop reduction. The procedure was as follows: an aqueous solution of P2 was prepared at 55 °C. In a glass vessel, demineralized water (conductivity 1.8 μS cm−1) was loaded and tempered at 55 °C. Progressive additions of 1 mL each time of the solution were achieved. The evaluation consisted in measuring the change in electrical conductivity as a function of salt concentration; as mentioned, the cmc was evidenced by the slope change.
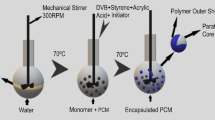
P2 (0.1 g, 0.156 mmol) was polymerized via aqueous micellar solution (50 mLwater) using mechanical stirring (100 rpm) for 24 h. Emulsion polymerization of P2 was achieved using xylene as the dispersed phase (10 mLxylene/50 mLwater); in this process, mechanical stirring (300 rpm) was applied for 24 h. Concerning interface polymerization, it was performed using a water/chloroform system (50 mLwater/50 mLcholoform). P2 contained in chloroform was the lower phase, whereas an APS aqueous solution was the upper phase; these polymerizations were left for 24 h without any stirring. In all polymerizations, APS (0.312 mmol) was used as the unique catalyst. Temperature effect (0, 25, or 50 °C) was evaluated. To purify the product, first, acetone was added to precipitate the polymer; subsequently, progressive additions of the solvent were made until no color was observed in the eluent. Experimentation details can be consulted in Supplementary Material.
Measurements
PmPDA was characterized using an infrared spectrometer (FTIR-4100, JASCO); the samples were dispersed by sonication in acetone and then deposited on potassium bromide compressed pellets with the aid of a capillary. UV–vis spectra were recorded using a spectrophotometer (V-370, JASCO); samples were prepared in N,N-dimethylacetamide. Melting-recrystallization cycling of the core–shell composite and paraffin were run in a differential scanning calorimeter (Thermal Analyst 2100, TA Instruments); sample were run under air atmosphere and heating rate of 10 °C min−1. Morphology was characterized using a field emission scanning electron microscope (JSM-7401F, JEOL Ltd.) and a field emission transmission electron microscope (JEM 2200FS, JEOL Ltd.); samples were prepared placing a droplet of polymerization product on a holey–carbon–copper grid and then left to dry at laboratory conditions. Additionally, PmPDAs were characterized by cyclic voltammetry (CV) using a potentiostate analyzer (model 1260 plus 1287, Solartron). Electrochemical measurements were performed in a standard three-electrode cell at room temperature using Pt square foil (area = 0.75 cm2) as the counter electrode and Ag/AgCl/saturated KCl as the reference electrode. The electrolyte was a sulfuric acid (H2SO4) solution 1 M. All analyses were performed at a scan rate of 25 mV s−1 by sweeping the potential between −0.5 and +1.0 V against Ag/AgCl reference electrode. Working electrodes were made with carbon paste; 5 mg of sample was deposited for measurement. Core–shell dispersion viscosity dependency with respect to temperature was evaluated using a rotational rheometer (MCR 501, Anton Paar Physica); samples were run using concentric cylinder geometry, swiping temperature from 25 to 60 °C, heating rate of 2 °C min−1, and shear constant of 0.001 s−1.
Results and discussion
Critical micellar concentration
The curves of specific conductivity versus temperature and specific conductivity versus P2 concentration are shown in Fig. S1 (Supplementary Material). P2 presented a Krafft temperature (KT) of 50 °C (Fig. S1a). This property indicates the critical temperature for micelle formation. Based on this result, cmc was determined at 55 °C (Fig. S1b), giving a value of 1.5 mmol L−1 which was, respectively, two and five times lower than those reported for the anilinium salt (2.99 mmol L−1) and SDS (7.9 mmol L−1) [18]. The substitution of the small hydrophilic counterion by a voluminous hydrophobic organic counterion results in changes in surfactant micellar behavior. When aromatic counterions are present, transitions from spherical micelles to rodlike, elongated, or wormlike micelles have been reported [30–33]. Cmc reduction, as a function of organic counterion hydrophobicity, has also been observed [34–36]. These changes were attributed to organic counterion fitting into the chain palisade [37]. In the present case, diaminium group location within the hydrocarbon micellar palisade seems restricted considering the sulfate anions pull the diaminium group to the micellar surface. Figure S2 shows the molecular models for P2, the anilinium salt, and SDS. As observed, the molecule of P2 occupies a larger area compared with the others, causing water–air interface saturation at lower concentration and the reduction of the cmc as a consequence (FTIR characterization of P2 is reported in Supplementary Material, Fig. S3).
Chemical structure of PmPDA
Figure 2 shows FTIR spectra of PmPDA. Characteristic peaks at 1620 and 1500 cm−1 were ascribed, respectively, to quinoid and benzenoid rings stretching. Intensity difference of these bands was related to the abundance of these rings in the polymer ladder [38]. This suggests that PmPDA contains a higher proportion of quinoid to benzenoid rings in all cases; consequently, in analogy with polyaniline, pernigraniline-like arrangements seem the preferred. The bands at 1400 and 1250 cm−1 were associated, respectively, to stretching of N–C of imine of phenazine structure and to N–C of benzenoid rings. The bands at 1110, 1050, and 620 cm−1 were ascribed to S–O stretching of sulfate group. Sang et al. reported the absorption sulfate ions by PmPDA at 1110 cm−1 [15], suggesting, in the present case, that the ammonium groups are counterions of dodecyl sulfate anions. Literature remarks the presence of the doping agent in this region [2, 39]. At high field, the bands at 3340 and 3160 cm−1 were attributed, respectively, to N–H stretching of –NH2 (amino) and –NH– (imino) groups. The proportion of these bands has also been related to the relative abundance of benzenoid to quinoid rings in the ladder structure [3], suggesting also higher content of quinoid rings. Finally, the small teeth observed at 2920 and 2850 cm−1 were ascribed, respectively, to C–H asymmetric and symmetric stretching of –CH2– groups of dodecyl sulfate anion. Literature has pointed equivalent bands for PAni doped by SDS [40]. According to Sulimenko et al. [41], PmPDA does not protonate even in acid medium, which seems the case of the PmPDA produced by emulsion polymerization; however, the PmPDA produced by micellar solution or interface presents, apparently, certain degree of doping.

Infrared spectra of PmPDA obtained by different synthetic methods. Aqueous micellar solution at 50 °C (W50), emulsion polymerization at 25 °C (XR) and 50 °C (X50), and interface polymerization at 25 °C (CR) and 50 °C (C50)
UV–vis spectrometry was performed as a complementary technique to FTIR. PmPDA spectra presented similar wide absorption pattern despite of the synthetic method, Fig. 3a. A maximum absorption band in the region close to 360 nm was attributed to π-π* transitions of phenazin fragments. Sulimenko et al. [41] reported similar spectra for PmPDA, suggesting chemical structure complexity, related to the variety of C–N possible links, as the possible reason. Sestrem et al. characterized PmPDA oligomers; in this study, phenazine absorption at 365 nm was reported [42]. Concerning chemical structure, Huang et al. [3] pointed differences in the PPDA obtained by the three PDA isomers, suggesting phenazine open structure for PmPDA.

a UV–vis spectra of PmPDA obtained by different synthetic methods. b PmPDA proposed chemical structure (model based on literature [3])
It is worth mentioning that the group of Li et al. has been one of the most active groups in this topic. They reported NMR studies of PoPDA and copolymers. 1H NMR spectra gave evidence of open rings in the ladder structure due to the presence of –NH2 groups on phenazine unit [43, 44]. They also reported that the 1H NMR spectrum of PmPDA had not been obtained because of the limited solubility of this compound [45]. However, later they reported the characterization of this polymer by 13C NMR of solids giving also evidence of open rings [46].
Consequently, we can say that the PmPDA produced by the three methods of polymerization, share a chemical structure based on phenazine with open segments, rich in quinoid rings (pernigraniline-like), and partially doped, Fig. 3b.
Morphology
Electron microscopy allowed studying synthesis method effect on the PmPDA morphology, Fig. 4. As for the aqueous micellar polymerization, a great deal of 2D nanostructures (nanomats) was observed, some of them apparently rigid (Fig. 4a) and others even rolled (Fig. 4b). Such 2D nanostructures are formed by the lateral self-assembling of fibers, as observed in the inset. Wideness of some 2D nanostructures coincided with fiber diameter (about 25 nm), suggesting that some of these structures are monolayers of finely interconnected fibers. It is noteworthy that only particle dispersions (no fibers or other structured arrangement) were obtained by the micellar polymerization of the anilinium salt [18]. Concerning emulsion polymerization, morphology indicated P2 polymerization at the water–xylene interface, as a great deal of pot-like nanostructures was observed, especially at 25 °C. Pot-like morphology suggested that during xylene evaporation, the fibers retracted giving the appearance as if they were sliced in half. The tendency to form 2D nanostructures at 50 °C was notable (Fig. 4d); however, fiber self-assembling was not as ordered as in the micellar polymerization. At 25 °C, the fibers look entangled around the pot-like structures which apparently restricted self-assembly (see the inset). Finally, interfacial polymerization (Fig. 4e, f) showed no tendency to form 2D structures or fibers; instead, interconnected spherical particles are observed (Fig. 4c). PPDA with microstructure, for example, nanorods, nanospheres, leaf-like, petal-like, diamond-like microparticles, multi-angula microrods, or dendalion-like microparticles, have been reported. In these, pH, oxidizing agent concentration, oxidizing agent to monomer molar ratio, or surface additives (surfactants or water-soluble polymers) were the shape driving conditions [5, 11, 24, 47, 48]. This indicates that diverse variables determine microstructure, most of them not related with the monomer itself.

Micrographs of PmPDA obtained by different synthetic methods: a, b aqueous micellar solution at 50 °C; c, d emulsion polymerization, respectively, at 25 and 50 °C; e, f interface polymerization, respectively, at 25 and 50 °C
Definitely, the conditions that give rise to the diverse microstructures in the PPDAs, and in the polyanilines, in general, are intriguing aspects that have not yet been resolved. In literature, it is possible to find reports that have addressed this issue, explaining the origins from different points of view. In some cases, the geometry is attributed to the coexistence of immiscible phases (oil/water), which allows the controlled generation of fibers [49, 50]; however, other reports have demonstrated that fibers can be obtained in the aqueous phase [51]. The presence of surfactants (ionic or nonionic), which form micellar soft-templates is another mechanism which has also been used to explain fibrous geometries [52–54]. The synthesis of the MCM-41 structure is a clear example of this quality of surfactants. Homogeneous nucleation (at the initial stage of the polymerization) and the use or not of agitation have been reported as other determining factors in the formation of ordered fibrous structures [55–57]. Therefore, a fast polymerization in the absence of agitation can be highly favorable for homogeneous fibrous structures. Other authors have proposed models based on the growth process of the polymer chain, where the primary oligomeric stages (nucleates) are essential in defining the geometry itself, being pH a fundamental variable [58]. Furthermore, the multilayer model sustains polyaniline nanostructures suffer from a variety of conformational changes along polymerization (from fibers, self-assembling of fibers and particles), where the π–π stacking and cross-linking play an important role [59]. As seen, it can be mentioned that, currently, there is no model describing the origin of the different morphologies reported that meets the different viewpoints. However, in the literature, there is a significant amount of work aimed at structural aspects of polyanilines and can be consulted [59–64].
In this study, we consider that the amphiphilic character of P2 and its interaction with the medium strongly influenced the final nanostructure. As mentioned, organic counterions favor formation of elongated, worm-like, or rod-like micelles [31–33]. Accordingly, it is possible to deduce that the fibers produced in the micellar system are the product of the polymerization of micelles with an elongated arrangement, as proposed by the soft-template model. Similar mechanisms were proposed by Wei et al. for the system β-naphthalene sulfonic acid/aniline [65] and by Zhang et al. for the system inorganic acid/aniline [66]; in these, the anilinium salts (product of aniline quaternization by the strong acids) micellar solutions played a template-like role (rod-like) in the synthesis of polyaniline nanotubes. It is worth mentioning that particles other than fibers were not observed during microscopy analysis, suggesting that polymerization of P2 was conducted, apparently, based on a unique elongated structure. Concerning the emulsified systems, partitioning of P2 at the aqueous (micellar form) and emulsified phases seems to occur. According to this, P2 polymerized at the xylene–water interface producing the pot-like structures, where the xylene microdroplets played a template role, while elongated micelle polymerization generated fibers as in the pure micellar polymerization. Interface polymerizations behaved differently, as fibers were not produced, suggesting that micelles were not formed in the aqueous phase. In interface polymerization, P2 was initially in the organic phase and progressively diffused to the chloroform–water (rich in APS) interface where polymerization was performed. Under this scenario, P2 polymerization at the interface generated the interconnected networks due to the high concentration of P2 in a restricted volume. A schematic description is shown in Fig. 5.

Model relating PmPDA morphology and P2 micellar structure and adsorption at the water–organic interface. a Micellar polymerization (fibers), b emulsion polymerization (pot-like and fibers), and c interface polymerization (globular-interconnected)
Electroactivity
Figure 6 shows voltammograms at sweep rate of 25 mV s−1 in 1.0 M sulfuric acid electrolyte. All samples exhibited electroactivity, with one anodic peak and one or two cathodic peaks corresponding, respectively, to reduced/semi-oxidized and semi-oxidized/oxidized redox transitions. This assignation was made based on Wu et al. report for CV analysis of PoPDA synthesized by electrochemical polymerization [4]. They determined using cyclic voltammetry and Raman spectroscopy the occurrence of at least three structures of PoPDA (reduced, semi-oxidized, and oxidized). From these, the semi-reduced structure is the most chemically stable. Concerning PmPDA, it is worth noting that in the present case, the method of synthesis produced an important effect, as the anodic peaks and the hysteresis cycles are much larger for the emulsified systems regarding the others. This feature indicates higher electroactivity which could be useful for sensor design. Zhou et al. reported the design of glucose biosensors based on PPDAs on platinized glassy graphite. Oxidation irreversibility during the electrochemical synthesis of the three PDAs, under this particular synthesis conditions, was found. The PmPDA-based biosensor exhibited, particularly, high sensitivity to glucose and low permeability to interfering species [67]. Furthermore, Li et al. developed an amperometric PmPDA-based biosensor and gold nanoparticles with horseradish peroxidase immobilized for peroxide detection. The presence of the gold particles, among other things, enhances the electrical conductivity of the PmPDA [68]. These studies demonstrate the great potential of the PmPDA as material for the design of electrochemical sensors.

Cyclic voltammetry plots of PmPDA obtained by different synthetic methods. Counter electrode of Pt, reference electrode Ag/AgCl/saturated KCl, 1 M sulfuric acid electrolyte, scan rate of 25 mV s−1, sweeping the potential between −0.5 and +1.0 V against Ag/AgCl reference electrode. Working electrodes were made with carbon paste. W50 corresponds to polymerization in aqueous micellar solution at 50 °C; X50 and XR correspond, respectively, to emulsion polymerization at 50 and 25 °C; and C50 and CR stand for interface polymerization, respectively, at 50 and 25 °C
Paraffin microencapsulation and characterization
As seen, P2 meets surfactant and monomer (of PmPDA) functions; therefore, application for practical purposes is quite challenging. Taking advantage of P2 adsorption at organic water interfaces, core–shell composite design, and microencapsulation seem primary lines to follow. Recently, phase change materials (PCM) development has received intense attention obeying worldwide necessities to save energy and to protect the atmosphere [69]. In this application, microencapsulation allows the integration of PCM and construction materials with excellent results [70]. Few reports concerning polyaniline-based PCM were found in literature, all of them achieved by the emulsion technique using in most cases a surfactant as stabilizer which was an important variable [71–75]. These systems presented both high thermal stability (TGA) and high phase change enthalpy (DSC) which makes these composites very attractive. It is noteworthy that any report using PPDA for an equivalent function was found.
Emulsion polymerization method allowed successful microencapsulation of paraffin (m.p. 45 °C) using a ration of P2 to paraffin of 1 to 4 (w/w) at 60 °C. Figure 7 portrays the as-prepared dispersion and dilution of microencapsulated paraffin (form-stable PCM). Irregularly shaped particles close to 20-μm diameter are mostly observed; irregular shape was associated to paraffin recrystallization. While well-defined particle surface, on the other hand, evidenced paraffin encapsulation, suggesting P2 polymerization on the surface of the emulsified droplets of melted paraffin, just as in the xylene–water emulsified systems. FTIR spectroscopy was performed in ATR mode to characterize the core–shell composite surface, Fig. 7a (inset). As references, spectra of pure paraffin and PmPDA obtained by emulsion polymerization were included. Characteristic signals of the methylene groups of paraffin at 2850 and 2920 cm−1 corresponding, respectively, to the symmetric and asymmetric vibration of –CH2– groups are observed in the core–shell. However, the bands at 3300, 1620, and 1110 cm−1 corresponding, respectively, to N–H stretching of –NH2 (amino), quinoid, and S–O stretching of sulfate group are also observed, which indicates the presence of PmPDA on the composite surface. The high intensity of the C–H bonds was associated with the highest concentration of paraffin in the composite (4:1). These results suggested that the PmPDA is on the surface as the composite shell stabilizing the paraffin microparticles.

Micrographs of microencapsulated paraffin by emulsion polymerization method at 60 °C. a As-prepared dispersion and b diluted dispersion. Inset: FTIR spectra of paraffin, form-stable PCM (core–shell) and PmPDA
To evaluate compartmentalization effect, the core–shell composite (form-stable PCM) was submitted to melting–crystallization cycling by differential scanning calorimetry (Fig. 8a). For comparison purposes, neat paraffin and PmPDA, obtained by emulsion polymerization, were also evaluated. The last cycle of a series of 20 heating–cooling cycles is included. As PmPDA shows no transition within the range of temperature (no thermal storage), the endothermic peaks are only attributable to paraffin. PCM and paraffin melting and crystallization temperatures were, respectively, 47 and 38 °C, and 48 and 36 °C. These results indicate slight melting point reduction (1 °C) and crystallization increment (2 °C). Endothermic peak sharpening in the core–shell thermogram is another aspect to note. Zhu et al. [73] observed similar behavior in palmitic acid/polyaniline/copper nanowire composites. Thermal resistance reduction, caused by copper nanowires, was suggested to explain temperature changes and peak sharpening. We consider that compartmentalization (confinement of paraffin in discrete volume) improved heat transference causing, as a consequence, shifting of melting and crystallization temperatures.

a DSC thermograms of form-stable PCM, neat paraffin, and PmPDA (last cycle of 20 cycles). b TGA traces of form-stable PCM, neat paraffin, and PmPDA obtained by emulsion polymerization method. c Viscosity dependency with respect to temperature of form-stable PCM obtained by emulsion polymerization at 60 °C
Melting enthalpy (∆H m ) showed 100 and140 J g−1, respectively, for the PCM and paraffin, indicating a difference of 40 J g−1. Organic acid/polyaniline composites (charged or not with certain nanomaterials) showed melting enthalpy variation as a function of organic acid loading, with a general reduction of PCM ∆H m . Absorption of the liquefied organic acid by PAni, above melting temperature, or by the nanomaterial was attributed to cause melting enthalpy reduction in the PCM regarding the pure organic acid in terms of liquefaction restriction [72, 74]. Surface interaction of other organic acids with carbon materials showed also important effects on ∆H m , melting and crystallization temperature of PCM confined in porous materials depending on the porosity scale [76]. In the present study, paraffin loading ((∆H m-PCM /∆H m-Paraffin ) × 100) of 71.4 wt% is in the upper middle range reported for PCM systems. We can say, consequently, that the method followed here is feasible for PCM applications, with the advantage that P2 permits paraffin emulsification and microencapsulation on its own, without needing any other additive, avoiding possible affections on paraffin physical properties, or high shear rate, making scaling feasible.
Figure 8b (inset) shows the traces of thermal degradation for PCM, paraffin, and PmPDA obtained by emulsion polymerization. Form-stable PCM presented three-step degradation process: first, at 115 °C, which was attributed to doping agent and oligomer evaporation; the second transition at 215 °C evidently corresponds to paraffin thermal degradation, as compared with the blank of paraffin (219 °C), and the third transition, at 452 °C, that fits with PmPDA backbone degradation (460 °C) [77]. The second degradation step suggests the limit of PCM thermal stability. By considering phase transition at 47 °C, we can say that the PCM is highly thermally stable.
Figure 8c shows dispersion viscosity dependency regarding temperature. Initially, aqueous suspension viscosity remained with little variation from 25 to 30 °C, followed by a progressive decrease until 35 °C. This viscosity reduction was attributed to the breaking of particle suspension initial structure. From 35 to 44 °C, a slight increase in viscosity, considered as a signal of suspension stability, is present. Close to 45 °C, an abrupt reduction of viscosity is observed; this temperature coincides with paraffin melting temperature, which suggests unencapsulated paraffin fusion. The last transition, from 50 to 60 °C, was interpreted as the re-emulsification of the melted unencapsulated paraffin in the aqueous phase. It is worth saying that the evaluation was run only until 60 °C considering temperature ranges of application of these systems. Stable melting–crystalization cycling, appropriate ∆H m , and thermal stability, indicate promising performance for the form-stable PCM. Additionally, the aqueous dispersion provides the possibility to produce formulation with, for example, polymer latexes without affecting stability in the range temperature bellow paraffin melting point. Based on these motivating results, a complete study on this topic is under development and will be published later.
Conclusion
In this work, new two-tailed reactive amphiphile, benzene-1,3-diaminium dodecylsulfate (P2), was successfully applied for the synthesis of PmPDA. Experimentation showed that products shared chemical structure based on phenazine openquinoid-rich rings, as FTIR and UV–vis indicated. Electron microscopy exhibited, moreover, that the synthetic method is a determining condition in PmPDA morphology, deeply related to the surface interaction of P2 with the medium. Emulsion polymerization method allowed P2 successful application in paraffin microencapsulation for the synthesis of stable-form PCM. Electron microscopy allowed suggesting the direct polymerization of P2 on the emulsified paraffin microdroplet surface. Thermal properties indicated that the form-stable PCM presents a great potential for heat storage application. Based on these results, P2 was classified as a reactive surfactant, as it is monomer and surfactant concurrently. With the difference that P2 can be used in applications that conventional reactive surfactants cannot accomplish by themselves. This work provided the foundations for further research based on P2 amphiphilic properties. P2 adsorption at interfaces and posterior polymerization stimulate the application of P2 and its polymer for a wide range of applications. Such applications range from sensor design and other devices, to heavy metal ion capture, or form-stable PCM design as reported here.
References
Chan HSO, Ng SC, Hor TSA, Sun J, Tan KL, Tan BTG (1991) Poly(m-phenylenediamine): synthesis and characterization by x-ray photoelectron spectroscopy. Eur Polym J 27:1303–1308
Lin X, Zhang H (1996) In situ external reflection FTIR spectroelectrochemical investigation of poly(o-phenylenediamine) film coated on a platinum electrode. Electrochim Acta 41:2019–2024
Huang M-R, Peng Q-Y, Li X-G (2006) Rapid and effective adsorption of lead ions on fine poly(phenylenediamine) microparticles. Chemistry 12:4341–4350
Wu L-L, Luo J, Lin Z-H (1996) Spectroelectrochemical studies of poly-o-phenylenediamine. Part 1. In situ resonance Raman spectroscopy. J Electroanal Chem 417:53–58
Zhang Y, Li H, Luo Y, Shi X, Tian J, Sun X (2011) Poly(m-phenylenediamine) nanospheres and nanorods: selective synthesis and their application for multiplex nucleic acid detection. PLoS One 6, e20569
Zhang Y, Wang L, Tian J, Li H, Luo Y, Sun X (2011) Ag@poly(m-phenylenediamine) core-shell nanoparticles for highly selective, multiplex nucleic acid detection. Langmuir 27:2170–2175
Zhang L, Chai L, Liu J, Wang H, Yu W, Sang P (2011) pH Manipulation: a facile method for lowering oxidation state and keeping goodyield of poly(m-phenylenediamine) and its powerful Ag+ adsorption ability. Langmuir 27:13729–13738
Liao F, Yang S, Li X, Yang L, Xie Z, Hu C, Yan S, Ren T, Liu Z (2014) Preparation of heteroatom doped poly(o-phenylenediamine) fluorescent nanospheres: tunable fluorescent spectrum and sensing performance. Synth Met 189:126–134
Ariffin AA, Neill RDO, Yahya MZA, Zain ZM (2012) Electropolymerization of ortho-phenylenediamine and its use for detection on hydrogen peroxide and ascorbic acid by electrochemical impedance spectroscopy. Int J Electrochem Sci 7:10154–10163
Wang Z, Liao F (2012) Synthesis of poly(ortho-phenylenediamine) fluffy microspheres and application for the removal of Cr(VI). J Nanomater 2012, ID 682802
Wang J-J, Jiang J, Hu B, Yu S-H (2008) Uniformly shaped poly(p-phenylenediamine) microparticles: shape-controlled synthesis and their potential application for the removal of lead ions from water. Adv Funct Mater 18:1105–1111
Tian J, Luo Y, Li H, Lu W, Chang G, Qin X, Sun X (2011) Ag@poly(m-phenylenediamine)-Ag core–shell nanoparticles: one-step preparation, characterization, and their application for H2O2 detection. Catal Sci Technol 1:1393–1398
Wang L, Zhang Y, Tian J, Li H, Sun X (2011) Conjugation polymer nanobelts: a novel fluorescent sensing platform for nucleic acid detection. Nucleic Acids Res 39:e37. doi:10.1093/nar/gkq1294
Zhang Y, Sun X (2011) A novel fluorescent aptasensor for thrombin detection: using poly(m-phenylenediamine) rods as an effective sensing platform. Chem Commun 47:3927–3929
Sang P, Wang Y, Zhang L, Chai L, Wang H (2013) Effective adsorption of sulfate ions with poly(m-phenylenediamine) in aqueous solution and its adsorption mechanism. Trans Nonferrous Metals Soc China 23:243–252
Wang Z, Li X, Feng D, Li L, Shi W (2014) Poly(m-phenylenediamine)-based fluorescent nanoprobe for ultrasensitive detection of matrix metalloproteinase 2. Anal Chem 86:7719–7725
Jiang K, Ma S, Bi H, Chen D, Han X (2014) Morphology controllable fabrication of poly-o-phenylenediamine microstructures tuned by the ionic strength and their applications in pH sensors. J Mater Chem A 2:19208–19213
Zaragoza-Contreras EA, Stockton-Leal M, Hernández-Escobar CA, Hoshina Y, Guzmán-Lozano JF, Kobayashi T (2012) Synthesis of core–shell composites using an inverse surfmer. J Colloid Interface Sci 377:231–236
Vega-Rios A, Rentería-Baltiérrez FY, Hernández-Escobar CA, Zaragoza-Contreras EA (2013) A new route toward graphene nanosheet/polyaniline composites using a reactive surfactant as polyaniline precursor. Synth Met 184:52–60
Vega-Rios A, Hernández-Escobar CA, Zaragoza-Contreras EA, Kobayashi T (2013) Electrical and electrochemical properties of polystyrene/polyaniline core–shell materials prepared with the use of a reactive surfactant as the polyaniline shell precursor. Synth Met 167:64–71
Lakouraj MM, Zare EN, Moghadam PN (2014) Synthesis of novel conductive poly(p-phenylenediamine)/Fe3O4 nanocomposite via emulsion polymerization and investigation of antioxidant activity. Adv Polym Technol 33:21385
Yong Y, Lu-Yuan M, Liu-Zhu L, Xiao-Gang L, Jun S, Shao-kui C (2004) Polymerization of o-phenylenediamine catalyzed by hemeproteins encapsulated in reversed micelle. Chem Res Chin Univ 20:240–243
Ichinohe D, Saitoh N, Kise H (1998) Oxidative polymerization of phenylenediamines in reversed micelles. Macromol Chem Phys 199:1241–1245
Hao Q, Sun B, Yang X, Lu L, Wang X (2009) Synthesis and characterization of poly (o-phenylenediamine) hollow multi-angular microrods by interfacial method. Mater Lett 63:334–336
Guyot A, Tauer K (1994) Reactive surfactants in emulsion polymerization. Adv Polym Sci 111:43–65
Guyot A (2004) Advances in reactive surfactants. Adv Colloid Interf Sci 108-109:3–22
Wright KA, Abbott AD, Sivertz V, Tartar HV (1939) Studies of sulfonates. V. Electrical conductance of sodium decyl, dodecyl, and hexadecyl sulfonate solutions at 40, 60 and 80°-micelle formation. J Am Chem Soc 61:549–554
Domínguez A, Fernández A, González N, Iglesias E, Montenegro L (1997) Determination of critical micelle concentration of some surfactants by three techniques. J Chem Educ 74:1227–1231
Kunleda H, Shinoda K (1976) Krafft points, critical micelle concentrations, surface tension, and solubilizing power of aqueous solutions of fluorinated surfactants. J Phys Chem 80:2468–2470
Hassan PA, Raghavan SR, Kaler EW (2002) Microstructural changes in SDS micelles induced by hydrotropic salt. Langmuir 18:2543–2548
Hartmann PC, Dieudonné P, Sanderson RD (2005) Self-assembly and influence of the organic counterion in the ternary systems dodecylamine/acrylic acid/water and dodecylamine/methacrylic acid/water. J Colloid Interface Sci 284:289–297
Bijma K, Rank E, Engberts J (1998) Effect of counterion structure on micellar growth of alkylpyridinium surfactants in aqueous solution. J Colloid Interface Sci 205:245–256
Buwalda RT, Stuart MCA, Engberts JBFN (2000) Wormlike micellar and vesicular phases in aqueous solutions of single-tailed surfactants with aromatic counterions. Langmuir 16:6780–6786
Anacker EW, Underwood AJ (1981) Organic counterions and micellar parameters. n-Alkyl carboxylates. J Phys Chem 85:2463–2466
Underwood A, Anacker E (1984) Organic counterions and micellar parameters: substituent effects in a series of benzoates. J Phys Chem 88:2390–2393
Gamboa C, Rios H, Sepulveda L (1989) Effect of the nature of counterions on the sphere-to-rod transition in cetyltrimethylammonium micelles. J Phys Chem 93:5540–5543
Bachofer S, Simonis U (1996) Determination of the ion exchange constants of four aromatic organic anions competing for a cationic micellar interface. Langmuir 12:1744–1754
Li X, Huang M, Chen R, Jin YI, Yang Y-L (2001) Preparation and characterization of poly (p-phenylenediamine-co-xylidine). J Appl Polym Sci 81:3107–3116
Parsa A, Ghani SA (2008) Electrocopolymerization of aniline and ortho-phenylenediamine via facile negative shift of polyaniline redox peaks. Polymer 49:3702–3708
Zhou C, Han J, Guo R (2008) Dilute anionic surfactant solution route to polyaniline rectangular sub-microtubes as a novel nanostructure. J Phys Chem B 112:5014–5019
Sulimenko T, Stejskal J, Prokes J (2001) Poly(phenylenediamine) dispersions. J Colloid Interface Sci 236:328–334
Sestrem RH, Ferreira DC, Landers R, Temperini MLA, do Nascimento GM (2010) Synthesis and spectroscopic characterization of polymer and oligomers of ortho-phenylenediamine. Eur Polym J 46:484–493
Huang M-R, Li X-G, Yang Y (2001) Oxidative polymerization of o-phenylenediamine and pyrimidylamine. Polym Degrad Stab 71:31–38
Huang M-R, Li X-G, Duan W (2005) Effect of polymerization conditions on o-phenylenediamine and o-phenetidine oxidative copolymers. Polym Int 54:70–82
Li X-G, Duan W, Huang M-R, Yang Y-L, Zhao D-Y, Dong Q-Z (2003) A soluble ladder copolymer from m-phenylenediamine and ethoxyaniline. Polymer (Guildf) 44:5579–5595
Li X-G, Duan W, Huang M-R, Yang Y-L (2001) Preparation and characterization of soluble terpolymers from m-phenylenediamine, o-anisidine, and 2,3-xylidine. J Polym Sci A Polym Chem 39:3989–4000
Guo T, Liao F, Wang Z, Yang S (2012) Shape-controlled synthesis of dandelion-like poly(m-phenylenediamine). J Mater Sci Res 1:25–30
Wang M, Zhang H, Wang C, Hu X, Wang G (2013) Direct electrosynthesis of poly-o-phenylenediamine bulk materials for supercapacitor application. Electrochim Acta 91:144–151
Abdolahi A, Hamzah E, Ibrahim Z, Hashim S (2012) Synthesis of uniform polyaniline nanofibers through interfacial polymerization. Materials (Basel) 5:1487–1494
Huang J, Virji S, Weiller BH, Kaner RB (2003) Polyaniline nanofibers: facile synthesis and chemical sensors. J Am Chem Soc 125:314–315
Tran HD, Kaner RB (2006) A general synthetic route to nanofibers of polyaniline derivatives. Chem Commun 37:3915–3917
Permpool T, Sirivat A, Aussawasathien D (2014) Synthesis of polydiphenylamine with tunable size and shape via emulsion polymerization. Polym Int 63:2076–2083
Jeevananda T, Lee J (2008) Preparation of polyaniline nanostructures using sodium dodecylsulphate. Mater Lett 62:3995–3998
Li J, Jia Q, Zhu J, Zheng M (2008) Interfaial polymerization of morphologically modified polyaniline: from hollow microspheres to nonowires. Polym Int 57:337–341
Li D, Kaner RB (2007) How nucleation affects the aggregation of nanoparticles. J Mater Chem 17:2279
Li D, Kaner RB (2006) Shape and aggregation control of nanoparticles: not shaken, not stirred. J Am Chem Soc 128:968–975
Dan LI, Huang J, Kaner RB (2009) Polyaniline nanofibers: a unique polymer nanostructure for versatile applications. Acc Chem Res 42:135–145
Stejskal J, Sapurina I, Trchová M (2010) Polyaniline nanostructures and the role of aniline oligomers in their formation. Prog Polym Sci 35:1420–1481
Laslau C, Zujovic Z, Travas-Sejdic J (2010) Theories of polyaniline nanostructure self-assembly: towards an expanded, comprehensive Multi-Layer Theory (MLT). Prog Polym Sci 35:1403–1419
Stejskal J, Sapurina I, Trchova M, Konyushenko EN, Holler P (2006) The genesis of polyaniline nanotubes. Polymer (Guildf) 47:8253–8262
Shishov MA, Moshnikov VA, Sapurina IY (2011) Nanostructures of oligoaniline and polyaniline and their properties. Glas Phys Chem 37:106–110
Konyushenko EN, Stejskal J, Šeděnková I, Trchová M, Sapurina I, Cieslar M, Prokeš J (2006) Polyaniline nanotubes: conditions of formation. Polym Int 55:31–39
Stejskal J, Sapurina I, Trchová M, Konyushenko EN (2008) Oxidation of aniline: polyaniline granules, nanotubes, and oligoaniline microspheres. Macromolecules 41:3530–3536
Liu P, Zhang L (2009) Hollow nanostructured polyaniline: preparation, properties and applications. Crit Rev Solid State Mater Sci 34:75–87
Wei Z, Zhang Z, Wan M (2002) Formation mechanism of self-assembled polyaniline micro/nanotubes. Langmuir 18:917–921
Zhang Z, Wei Z, Wan M (2002) Nanostructures of polyaniline doped with inorganic acids. Macromolecules 35:5937–5942
Zhou DM, Dai YQ, Shiu KK (2010) Poly(phenylenediamine) film for the construction of glucose biosensors based on platinized glassy carbon electrode. J Appl Electrochem 40:1997–2003
Li J, Xiao LT, Liu XM, Zeng GM, Huang GH, Shen GL, Yu RQ (2003) Amperometric biosensor with HRP immobilized on a sandwiched nano-Au/polymerized m-phenylenediamine film and ferrocene mediator. Anal Bioanal Chem 376:902–907
Kenisarin MM, Kenisarina KM (2012) Form-stable phase change materials for thermal energy storage. Renew Sustain Energy Rev 16:1999–2040
Tyagi VV, Kaushik SC, Tyagi SK, Akiyama T (2011) Development of phase change materials based microencapsulated technology for buildings: a review. Renew Sustain Energy Rev 15:1373–1391
Zeng JL, Zhang J, Liu YY, Cao ZX, Zhang ZH, Xu F, Sun LX (2008) Polyaniline/1-tetradecanol composites-Form-stable PCMS and electrical conductive materials. J Therm Anal Calorim 91:455–461
Zeng J-L, Zhu F-R, Yu S-B, Xiao Z-L, Yan W-P, Zheng S-H, Zhang L, Sun L-X, Cao Z (2013) Myristic acid/polyaniline composites as form stable phase change materials for thermal energy storage. Sol Energy Mater Sol Cells 114:136–140
Zhu F-R, Zhang L, Zeng J-L, Zhu L, Zhu Z, Zhu X-Y, Li R-H, Xiao Z-L, Cao Z (2013) Preparation and thermal properties of palmitic acid/polyaniline/copper nanowires form-stable phase change materials. J Therm Anal Calorim 115:1133–1141
Zeng J-L, Zheng S-H, Yu S-B, Zhu F-R, Gan J, Zhu L, Xiao Z-L, Zhu X-Y, Zhu Z, Sun L-X, Cao Z (2014) Preparation and thermal properties of palmitic acid/polyaniline/exfoliated graphite nanoplatelets form-stable phase change materials. Appl Energy 115:603–609
Zeng JL, Liu YY, Cao ZX, Zhang J, Zhang ZH, Sun LX, Shu F (2008) Thermal conductivity enhancement of MWNTs on the PANI/tetradecanol form-stable PCM. J Therm Anal Calorim 91:443–446
Li B, Liu T, Hu L, Wang Y, Nie S (2013) Facile preparation and adjustable thermal property of stearic acid–graphene oxide composite as shape-stabilized phase change material. Chem Eng J 215-216:819–826
Archana S, Jaya SR (2014) Synthesis and characterization of poly (p-phenylenediamine) in the presence of sodium dodecyl sulfate. Res J Chem Sci 4:60–67
Acknowledgments
Authors wish to thank the Nagaoka University of Technology through Pacific RIM Project. We also wish to thank W. Antunez, C. Ornelas, K. Campos, D. Lardizabal, E.I. López, L. de la Torre, and S. Noguchi for their enthusiastic support during this research.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 113 kb)
Rights and permissions
About this article

Cite this article
Zaragoza-Contreras, E.A., Hernández-Escobar, C.A., Vega-Rios, A. et al. Diaminium salt as reactive amphiphile for the synthesis of poly(m-phenylenediamine) and paraffin microencapsulation. Colloid Polym Sci 293, 2635–2645 (2015). https://doi.org/10.1007/s00396-015-3667-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-015-3667-8