
Abstract
Although infiltration of CD8+ T cells in human atherosclerotic lesions has been described 30 years ago, the role of these cells in lesion development has long remained enigmatic. While experimental models hinted at their pro-atherogenic role based on circumstantial evidence, genetic mouse models of cytotoxic CD8+ T cell-specific immune deficiency suggested no crucial role of these cells in lesion development. However, in recent years, more refined models of adoptive cell transfer, disruption of specific immune regulatory pathways or monoclonal antibody-mediated cell depletion have proposed both atheroprotective and pro-atherogenic functions for CD8+ T cells in atherosclerosis. In particular, MHC class I-restricted CD8+ T cell responses may protect from atherosclerosis, and Qa-1 restricted regulatory CD8+ T cells have been defined. In addition, regulatory CD8+CD25+ T cells possess atheroprotective properties. However, CD8+ T cells can also promote monopoiesis in hyperlipidemia, and exert prototypical cytotoxic functions to promote vascular inflammation and macrophage accumulation leading to atherosclerotic lesion development. Here, we review these findings, mostly from experimental studies that reveal a previously unrecognized complexity and important role of CD8+ T cells in atherosclerosis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Atherosclerosis is characterized by a chronic inflammatory process in the vascular wall that is initially triggered by lipoprotein infiltration in the vascular intima [24]. Accumulation of macrophages, which turn into foam cells upon ingestion of modified lipoprotein, is a hallmark of atherosclerotic lesion formation [13, 28, 32, 53]. Macrophages accumulate either through recruitment of their circulating precursors, i.e., monocytes, or through local proliferation in advanced plaques [13, 42]. Recently, a population of self-renewing resident arterial macrophages, which populate vessels shortly after birth and originate from CX3CR1+ precursors and circulating monocytes has been described, but whether it participates in macrophage accumulation during plaque formation remains to be determined [16]. Even though macrophages constitute the vast majority of immune cells in atherosclerotic lesions, it is now well recognized that adaptive immunity can modulate lesion growth [1]. Infiltration of T cells in atherosclerotic lesions in humans and animals has been demonstrated in pioneering studies [31]. Several subsets of CD4+ T helper cells have subsequently been defined and their various contributions to atherosclerosis were uncovered in experimental models. Th1 polarized T cells, which produce the pro-atherogenic cytokine IFNγ, promote atherosclerosis, while immunosuppressive Foxp3+ regulatory T cells limit lesion progression [1]. Th2 polarized T cells are thought to be mostly anti-atherogenic, although results are less clear-cut [1]. The role of IL-17 producing Th17 cells is still debated as various studies have produced contradictory results [1]. Although the presence of CD8+ T cells in atherosclerotic lesions alongside CD4+ T cells has long been documented [31], evidence pointing towards a role of these cells in atherosclerosis has long remained circumstantial, or was regarded to not influence lesion formation. In the past few years, however, a number of studies have focused on CD8+ T cells in atherosclerosis, and suggested that, similar to CD4+ T cells, the role of CD8+ T cells in atherogenesis is complex and subset-dependent.
CD8+ T cells
Cytotoxic CD8+ T cells are known as critical players in host defense. To become effector cytotoxic T lymphocytes (CTLs), naive antigen-specific CD8+ T cells need to be activated by antigen-presenting cells (APCs) that present antigen on MHC class I molecules on the cell surface. Antigens are either derived from endogenous peptides (from modified self or foreign proteins), or exogenous antigens acquired by APCs (a mechanism known as cross-presentation) [30]. Killing of transformed or infected cells that present such antigens via MHCI is thought to occur through granule exocytosis and delivery of key effector molecule, the pore-forming protein perforin as well as other serine proteinases known as granzymes that then induce apoptosis of target cells. In addition, CD8+ T cells produce several cytokines, such as tumour-necrosis factor (TNF)-α and interferon (IFN)-γ, that can have cytotoxic action when secreted in the vicinity of target cells [3]. Besides these classical, well established effector functions of CD8+ T cells, CD8+ T cells with immune suppressive functions have been described. Namely, a small subset of CD25+CD8+ T cells with immune suppressive activity has been uncovered in humans and mice [8], and a population of immune regulatory CD8+ T cells that expresses a T cell receptor exclusively interacting with the MHC class Ib molecule Qa-1 has been identified in mice, with HLA-E restricted CD8+ T cells being the potential equivalent in humans [25, 29].
CD8+ T cells in atherosclerosis in humans
In a seminal study by Jonasson and colleagues, immunohistochemical analysis of human carotid artery plaques revealed CD8+ T cell infiltration in atherosclerotic lesions [31]. We could recently corroborate abundant CD8+ T cell infiltrates at various locations in atherosclerotic lesions in the carotid artery, including the plaque shoulder and the vicinity of the necrotic core [12]. Other studies have furthermore shown the presence of CD8+ T cells in the vascular wall in occlusive aortic atherosclerosis [36] and in atherosclerotic lesions at various stages [55]. Notably, CD8+ T cells isolated from human plaque atherectomy specimens are highly activated, much more so than plaque CD4+ T cells, or T cells isolated from the blood of the same patients [22]. Abundance of CD8+ T cells in the vascular intima was found to increase with atherosclerotic disease severity [21]. In vitro, CD8+ T cells were unable to infiltrate early atherosclerotic lesions, whereas they efficiently infiltrated advanced human lesion tissue [21], suggesting that vascular inflammation in advanced plaques may support recruitment of circulating CD8+ T cells to the vascular wall.
Associations between blood levels of specific CD8+ T cell subsets and cardiovascular disease was further proposed in a study analyzing CD8+ T cell proportions in frozen blood samples obtained during baseline investigations in 700 subjects in the cardiovascular arm of the Malmö Diet and Cancer Study [37]. Here, a negative correlation of the frequency of IFNγ+ CD8+ T cells among total blood CD3+ T cells with the degree of carotid stenosis was found that remained significant after adjustment for major cardiovascular risk factors [37]. However, subjects in the two highest tertiles of CD8+ T cells among CD3+ T cells showed a trend towards an increased incidence of coronary events during the 15 years of follow-up [37]. In other studies, an expansion of CD8+ T cells expressing the IL-6 receptor α chain [27] or co-expressing the inhibitory receptors PD-1 and Tim-3 [49] has been described in patients with clinical manifestations of atherosclerosis. The relevance of these findings remains to be elucidated. In particular, it is still unknown if numbers of CD8+ T cells correlate with their infiltration into the vessel wall (or other organs, shown to be important for their functional contribution to atherosclerosis, as outlined below), or whether a certain cytokine or surface marker expression profile defines or contributes to the role of CD8+ T cells in lesion formation.
CD8+ T cells in experimental models of atherosclerosis
In murine models of atherosclerosis, CD8+ T cells have been observed in atherosclerotic lesions by immunohistochemistry in Apoe −/− mice [59] or by flow cytometric analyses of vascular tissue in Ldlr −/− animals [12]. CD8+ T cells can also be found in adventitial tertiary lymphoid organs in advanced atherosclerotic vessels [10, 46].
A principal role of antigen-driven T cell responses was examined in Apoe −/− mice expressing β-Galactosidase as an artificial antigen in vascular smooth muscle cells in the aorta and lung arteries. Immunized with dendritic cells presenting a β-galactosidase-derived immunogenic peptide, these mice developed strong CD8+ T cell responses against cells expressing β-galactosidase, leading to non-resolving inflammation and a massive infiltration of the vascular wall with CD8+ T cells and F4/80+ macrophages, resulting in larger atherosclerotic lesions than in Apoe −/− control mice [44]. These data suggested that CD8+ T cells that antigen-specifically react against vascular cells promote arterial inflammation and lesion formation.
In more standard murine models of atherosclerosis, high fat diet feeding over 4 weeks in Apoe −/− mice preferentially induced CD8+ rather than CD4+ T cell activation in the spleen [38], and these splenic CD8+ T cells produced higher levels of both IL-10 and IFNγ, suggesting that hypercholesterolemia can trigger both anti- and pro-inflammatory programs in CD8+ T cells [38]. Indirect lines of evidence further suggested pro-atherogenic functions of CD8+T cells: increased CD8+ T cell infiltration and/or activation in inflamed vessels coincided with increased lesion development in some murine models. In mice treated with an agonist of the T cell activating co-stimulatory receptor CD137, increased CD8+ T cell infiltrates in plaques were associated with enhanced lesion development [48]. Deficiency in the co-inhibitory receptor PD-1 (programmed cell death-1), which acts as an inhibitor of T cell activation, led to an infiltration of activated CD8+ T cells and increased lesion formation in Ldlr −/− mice [11]. In mice with a dendritic cell-specific deficiency of TGFβRII, resulting in blunted anti-inflammatory TGFβ signaling in dendritic cells, increased CD8+ T cell infiltration in plaques and augmented lesion formation were observed [41]. These data, however, present only circumstantial evidence that link CD8+ T cells with atherosclerotic lesion progression. Studies more directly assessing the contribution of these cells to atherosclerosis have provided contradictory results.
Apoe −/− Cd8 −/− mice deficient in CD8+ T cells display unaltered atherosclerosis
To explore the role of CD8+ T cells in atherosclerosis, Elhage and colleagues analyzed atherosclerotic lesion development in CD8+ T cell-deficient Apoe −/− Cd8 −/− mice [15]. In these mice, cytotoxic T cell responses were dramatically decreased, whereas B- as well as CD4+ T lymphocyte populations and function were described to be unaltered. Apoe −/− mice lacking CD8+ T cells and fed a normal chow for 18 weeks or 1 year did not show any significant changes in atherosclerotic lesion size in the aortic root and descending aorta, suggesting that CD8+ T cells do not play a major role in atherosclerosis. It should be noted, however, that in this study, lesions were analyzed at two time points that represent very early lesion formation, as reflected by minimal lesions in 18 week old Apoe −/−mice, and very large, advanced lesions in 1 year old mice [15]. It thus remains possible that an effect of CD8+ T cells at intermediate stages of lesion development may have gone unnoticed. A possible compensation of CD8+ T cell loss by other T cell subsets in CD8+ T cell deficient mice was also not addressed.
MHC class I-restricted CD8+ T cell responses in atherosclerosis
Endogenous peptides are displayed on the cell surface via MHC class I molecules that are recognized by cytotoxic CD8+ T cells, activated by APCs [30]. Several studies have explored MHC class I-restricted CD8+ T cell responses in atherosclerosis. In an early study aiming at the evaluation of the role of adaptive immunity in atherosclerosis, C57BL/6 mice deficient for surface expression of MHC Class I were employed, created by gene targeting of the β2 microglobulin locus, which are unable to mount MHC-Class I specific responses and lack cytotoxic T cells. Compared to control C57BL/6 mice, these MHCI-deficient C57BL/6 mice displayed an increased fatty streak development when fed a high fat, high cholesterol diet [18]. Although this finding suggested that MHCI-dependent antigen-presentation and the induction of CD8+ T cells is anti-atherogenic, increased atherosclerosis in β2-microglobulin deficient mice may have originated from processes unrelated to CD8+ T cells. For example, β2-microglobulin deficiency in mice is associated with drastically altered iron homeostasis [50] and exacerbated Toll-like receptor-dependent inflammatory responses [56].
In another study, Apoe −/− mice deficient in TAP-1 (transporter associated with antigen-processing 1), which is required for MHC class I antigen presentation, showed no alterations in plaque formation after 8 or 20 weeks of high fat diet feeding despite drastically reduced CD8+ T cell levels compared to Apoe −/− controls [38]. However, in these mice, total T cell infiltration into plaques was similar in both groups, and CD4+ T cell numbers were expanded in spleen, vessel draining lymph nodes and blood of Apoe −/− Tap1 −/−mice. Moreover, splenic CD4+ T cells from Apoe −/− Tap1 −/− mice isolated after 8 weeks of high fat diet feeding showed an increased proliferation when compared to controls [38]. This suggests that the loss of CD8+ T cells was compensated by CD4+ T cell expansion and activation in TAP-1 deficient mice.
That cross-priming of CD8+ T cells may be of subordinate importance is supported by a study using Ldlr −/− mice that displayed a robust reduction in the APC-mediated cross-priming capacity due to hematopoietic deficiency in Batf3. Despite a severe reduction in OT-I CD8+ T cell proliferation in response to OVA as an artificial antigen Batf3 −/− Ldlr −/− mice did not show any alterations in atherosclerotic lesion burden in the aortic arch and root after Wester type diet-feeding [40].
Although these studies suggest non-essential or even protective roles of CD8+ T cells in atherosclerosis, the experimental designs and potential biases induced by compensation or alterations in other cell types complicates their interpretation.
Regulatory CD8+CD25+ T cells with anti-atherogenic functions
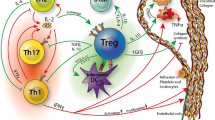
The first description of a putative anti-atherogenic CD8+ T cell subset came from a study were atherosclerosis-prone Apoe −/− mice were immunized with the ApoB-100 p210-derived peptide [9]. ApoB-100 derived peptides are considered as atherosclerosis-related antigens [34] and immunization of Apoe −/− mice with the ApoeB-100 p210 peptide has already been associated with decreased atherosclerosis [35]. Reduced atherosclerosis in mice immunized with the ApoB-100 p210 peptide was associated with an expansion of a CD8+CD25+IL10+ T cell population in the spleen and reduced dendritic cell (DC) levels in atherosclerotic lesions. Mechanistically, CD8+ T cells from p210 immunized mice had increased cytolytic activity towards DCs in vitro over CD8+ T cells from the control group, an effect that was lost when CD25+ cells were depleted from the CD8+ T cell population. Moreover, adoptive transfer of CD8+ T cells from p210-immunized mice, but not from control mice, reduced lesion development in recipient Apoe −/− mice, and this effect was dependent on the presence of CD25+ cells in the transferred CD8+ T cell population [9]. Recently, the same authors furthermore demonstrated that immunization with the ApoB-100 p210 peptide reduces angiotensin II-induced aortic aneurysm formation and rupture in Apoe −/− mice, an effect that was lost when CD8+ T cells were depleted using an anti-CD8 monoclonal antibody [23]. In this study, CD8+ T cells from p210-immunized mice had increased cytolytic activity towards angiotensin-II stimulated macrophages, and inhibited CD4+ T cell differentiation into Th17 cells [23]. Altogether, these studies suggest that a population of CD8+ T cells protects from vascular inflammation in the context of ApoB-100 p210 peptide immunization, possibly by reducing levels of DCs or macrophages through cytotoxic effects, by modulating CD4+ T cell responses, and anti-inflammatory effects related to IL-10 expression (Fig. 1). In another study, transfer of CD8+CD25+ but not CD8+CD25− cells sorted from non-immunized Apoe −/− mice similarly reduced lesion development in recipient mice [58]. A subset of CD8+CD25+ T cells may thus endogenously possess atheroprotective properties, in line with the description of these cells as anti-inflammatory, regulatory CD8+ T cells [5] (Fig. 1). The exact mechanisms mediating the anti-atherogenic properties of this CD8+CD25+ T cell subset and the effector molecules involved remain to be determined. Interestingly, CD8+CD25+ Tregs have also been described in humans, were they represent 0.1-1% of circulating CD8+ T cells [8], raising the possibility that CD8+CD25+ T cell-mediated atheroprotection may also occur in humans.

Anti-atherogenic functions of CD8+CD25+ T cells. CD8+CD25+ T cells have been shown to limit vascular inflammation, atherosclerotic plaque and aortic aneurysm formation through secretion of anti-inflammatory IL-10 (1), cytotoxic activity towards pro-inflammatory Angiotensin-II stimulated macrophages and immunostimulatory dendritic cells (2), and inhibition of naive CD4+ T cell differentiation into Th17 cells (3)
Atheroprotective Qa-1 restricted regulatory CD8+ T cells and the Tfh-GC B cell axis
Although the idea of CD8+ immune-suppressive T cells (CD8+ Tregs) had long been met with skepticism [7], a subset of immune regulatory CD8+ T cells that exclusively interact with the MHC class Ib molecule Qa-1 on target cells has been uncovered in 2004 in mice deficient for Qa-1 [25]. Subsequently, Qa-1 restricted CD8+ Tregs were described to inhibit follicular helper T cells (Tfh), which express high levels of Qa-1 [33]. This process was crucial for the maintenance of self-tolerance, as genetically modified mice with a D227 K point mutation in Qa-1 (Qa-1-D227 K mice) disrupting its binding to the T cell receptor/CD8-coreceptor, and thus interfering with CD8+ Treg function, developed autoimmune disease [33].
Recently, Clement et al. could first demonstrate that proportions of Qa-1-restricted CD8+ Tregs were reduced in the spleen of aged Apoe −/− mice compared to young controls, while numbers of Tfh and germinal center B cells (GC B cells) were increased. Transfer of young CD8+ Tregs into old Apoe −/− mice reduced splenic Tfh cell levels [10], and aged Apoe −/− mice crossed to Qa-1-D227K mice showed increased atherosclerotic lesion formation and vascular inflammation, associated with increased Tfh and GC B cell levels [10]. In these mice, depositions of IgG in atherosclerotic lesions were also increased [10].
To limit Tfh expansion in Apoe −/−-Qa-1-D227 K mice, the authors targeted the co-stimulatory molecule ICOS (inducible T cell costimulator), which has a critical role in Tfh cell generation [2] by treating mice with neutralizing anti-ICOS antibodies. This rescued the pro-atherogenic phenotype observed in Apoe −/−-Qa-1-D227K mice and IgG deposition in lesions, suggesting that Qa-1 restricted regulatory T cell can limit atherosclerotic lesion development through inhibition of Tfh cell-mediated activation of GC B cells [10] (Fig. 2 ).

Atheroprotective Qa-1-restricted CD8+regulatory T cells. In lymphoid organs of hypercholesterolemic Apoe −/− mice (left), Qa-1 restricted CD8+ regulatory T cells (Tregs) interact with Qa-1 expressing follicular helper T cell (Tfh) to inhibit their activation. This limits Tfh-mediated activation of germinal center B cells (GC B cells). When introducing a D227 K point mutation in Qa-1 that prevents its interaction with the T cell receptor (TCR) of Qa-1-restricted CD8+ Tregs (right, Apoe −/−Qa-1D227K mice), CD8+ Treg-dependent Tfh inhibition is abrogated, thus leading to the accumulation and activation of Tfh, which in turn activate GC B cells, leading to increased atherosclerosis with increased IgG deposition in lesions
Interestingly, formation of adventitial tertiary lymphoid organs (ATLOs) was accelerated in Apoe −/−-Qa-1-D227 K mice, indicating that Qa-1 restricted CD8+ Tregs inhibit formation of ATLO in advanced atherosclerosis [10]. ATLOs have been described in advanced atherosclerosis in mice [46] and in human atherosclerotic aneurysmal arteries [10]. The link between Qa-1 restricted CD8+ regulatory T cells, ATLO formation and atherosclerotic lesion development, however, is difficult to pinpoint as ATLOs were recently proposed to be atheroprotective in advanced atherosclerosis in mice [26]. In humans, HLA-E restricted CD8+ T cells have been postulated to be functionally equivalent to murine Qa-1 restricted regulatory CD8+ T cells, and a defect in antigen recognition by these cells has been proposed to contribute to the development of autoimmune type 1 diabetes [29]. So far, however, no studies have investigated whether HLA-E restricted CD8+ T cells may influence atherosclerosis in humans.
Pro-atherogenic effects of CD8+ T cells
Recently, two studies by Kyaw et al. [39] and our own group [12] used a similar strategy of CD8+ T cell depletion in atherosclerosis-prone mice using anti-CD8α or anti-CD8β monoclonal antibodies. Although this model also comes with some limitations such as the generation of antibodies neutralizing the injected antibody, it nevertheless has been shown to efficiently allow depleting CD8+ T cells in adult animals with an otherwise intact immune system, thereby circumventing biases inherent to aforementioned genetic models. In line with these considerations, both studies failed to observe any alterations in CD4+ T cell levels, activation or polarization in Ldlr −/− or Apoe −/− mice after CD8+ T cell depletion [12, 39], ruling out a compensation of the CD8+ T cell loss by CD4+ T cells. In Apoe −/− mice fed a Western diet for 8 weeks, Kyaw et al. observed reduced atherosclerotic lesion formation in mice treated with anti-CD8α or anti-CD8β monoclonal antibodies, associated with reduced macrophage accumulation in plaques [39]. We observed a similar phenotype of reduced lesion formation and macrophage accumulation in Ldlr −/− mice after 6 weeks of high fat diet feeding and weekly injections of CD8-depleting antibodies [12]. Interestingly, in both studies, levels of the chemokine MCP-1/CCL-2, which promotes monocyte infiltration into lesions [54], were decreased in vascular tissue of CD8+ T cell-depleted mice, suggesting that CD8+ T cells promote recruitment of monocytes into lesions. However, in adoptive transfer experiments, we could demonstrate that CD8+ T cell depletion does not affect monocyte recruitment into atherosclerotic aortae.
CD8+ T cells promote monopoiesis in hyperlipidemia
The accumulation of monocytes in lesions has previously been demonstrated to directly correlate with circulating monocyte levels [14]. Notably, reduced numbers of blood Ly6Chi monocytes were observed in CD8+ T cell depleted Ldlr −/− mice [12]. Ly6Chi monocytes, also known as inflammatory or classical monocytes, are the murine counterparts of human CD14+CD16− monocytes [19], and are thought to be the main circulating precursors of plaque macrophages [52]. Hence, reduced Ly6Chi monocyte levels after CD8+ T cell depletion was suggested to secondarily cause the decrease in macrophage accumulation in lesions upon CD8+ T cell depletion. As depletion of CD8+ T cells was associated with reduced plasma levels of the monocyte-mobilizing chemokine CCL2, a reduced egress of medullar monocytes likely contributed to reduced circulating Ly6Chi monocyte levels. In addition, we could demonstrate that CD8+ T cell depletion affected monocyte generation in the bone marrow, with reduced medullar levels of monocyte and late-committed monocyte progenitors (Fig. 3).

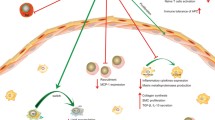
Pro-atherogenic roles of CD8+ T cells. In the bone marrow, CD8+ T cells promote monopoiesis (1), thereby increasing medullar monocyte levels. This subsequently entails increased circulating levels of Ly6Chigh monocytes (2), the precursors of plaque macrophages, hence indirectly leading to an increased accumulation of macrophages within plaques (3). Within lesions, CD8+ T cells secrete the cytotoxic effector molecules granzyme B and perforin, which promote death of vascular cells (4). CD8+ T cells also secrete pro-inflammatory cytokines, such as TNFα and IFNγ that may fuel vascular inflammation through local-pro-inflammatory effects on e.g. endothelial cells or macrophages (5)
Although the mechanisms underlying CD8+ T cell-mediated promotion of monocyte generation in the bone marrow during atherosclerosis remain to be elucidated, a similar property of CD8+ T cells has been described in the context of acute viral infection [51]. Schurch et al. proposed that effector CD8+ T cells indirectly induce proliferation and myeloid differentiation of hematopoietic progenitors during viral infection. In this model, IFNγ produced by CD8+ T cells triggerd the secretion of cytokines (e.g. IL-6) by bone marrow stromal cells that in turn promoted myelopoiesis, rather than directly affecting hematopoietic progenitors [51]. In hypercholesterolemic Ldlr −/− mice, IFNγ neutralization indeed partially reproduced the phenotype triggered by CD8+ T cell depletion and entailed a decrease in late-committed monocyte progenitors in the bone marrow. In addition, however, IFNγ blockade in mice also induced a retention of monocytes in the bone marrow, reflected by increased levels of medullar monocytes, while blood monocyte levels were decreased [12].
Cytotoxic CD8+ T cells induce plaque cell apoptosis and inflammation
In addition, a role of prototypical cytotoxic effector molecules for the pro-atherogenic functions of CD8+ T cells has been explored. In both studies of antibody-mediated CD8+ T cell depletion in Apoe −/− or Ldlr −/− mice, relative size of the necrotic core was reduced in CD8+ T cell depleted mice, indicating that cytotoxic properties of CD8+ T cells may contribute to their pro-atherogenic effects [12, 39]. This notion was corroborated in adoptive transfer experiment into T cell-deficient Apoe −/− Rag2 −/− mice, which demonstrated that CD8+ T cells deficient for the cytotoxic enzymes perforin or granzyme-B lost their pro-atherogenic potential. Vascular inflammation was shown to associate with increased numbers of apoptotic cells (identified as macrophages, endothelial and smooth muscle cells) in mice receiving perforin or granzyme-B-competent cells, and it was suggested that cytotoxic CD8+ T cells contribute to the genesis of apoptotic cells and necrotic cores in atherosclerotic lesions, and that macrophages are target cells for cytolyic CD8+ T cells in atherosclerosis (Fig. 3).
In these same experiments, CD8+ T cells lacking TNFα were furthermore discovered to not affect atherosclerotic lesion formation, whereas IFNγ-deficient CD8+ T cells promoted atherosclerosis similarly to controls, suggesting that also TNFα but not IFNγ, is instrumental to CD8+ T cell-induced atherosclerosis (Fig. 3). Although this study provides clear evidence that CD8+ T cells exert pro-atherogenic functions by cytotoxic mechanisms and TNFα production, there are some limitations to these experiments that may preclude reaching definitive conclusions about the respective role of CD8+ T cell effector molecules in atherosclerosis. CD8+ T cell reconstitution was documented in the spleen but not in vascular tissue. In addition, CD4+ T cell help critically influences several aspects of CD8+ T cell responses [4], so that reconstitution of CD8+ T cells in mice lacking CD4+ T cells may have prevented full deployment of CD8+ T cell functions.
CD8+ T cells and infection-induced atherosclerosis
A long standing hypothesis in the field of atherosclerosis is that chronic vascular inflammation may be sustained by microbial pathogens, either through direct infection of the vascular wall and vascular cells, or through systemic effects such as induction of pro-atherogenic cytokines [6], and several studies have proposed associations of infection with specific pathogens and an increased development of atherosclerotic lesions [6]. Among those pathogens, Chlamydia pneumoniae has been linked with lesion formation in several clinical and experimental studies [17]. Interestingly, a study examining atherosclerotic plaques from carotid arteries showed an increased infiltration of CD8+ T cells in plaques positive for Chlamydia pneumoniae [47]. In an experimental study, infection of C57BL/6 J mice with Chlamydia pneumoniae induced increased lesion formation in the aorta in response to hypercholesterolemia, an effect that was lost in CD8+ T cell-deficient C57BL/6 J mice [57], suggesting a causal role of CD8+ T cells in Chlamydia pneumoniae infection-induced acceleration of atherosclerosis. The mechanistic basis of these findings [57] remains to be explored. Given the very low level of lesion formation in these C57BL/6 J mice, experiments in atherosclerosis-prone mice should in addition be considered.
Conclusion and future perspectives
Although these recent studies begin to shed light on the complex role of CD8+ T cells in atherosclerosis, further research is necessary to completely apprehend their multifaceted functions in disease. Similar to other cell populations, current evidence suggests that both atheroprotective and pro-atherogenic CD8+ T cell subsets exist. CD8+ T cells seem to affect atherosclerosis locally, i.e., in the plaque itself, but also systemically, with e.g. effects observed in germinal centers of lymphoid organs, ATLOs [10] or in the bone marrow [12]. Hence, uncovering endogenous pro- and anti-atherogenic CD8+ T cell subsets and studying their dynamics at various sites during atherosclerosis progression would be of interest. In addition, surface markers and effector molecules, e.g., certain cytokines that define atheroprotective versus pro-atherogenic CD8+ T cell subsets relevant in atherosclerosis remain to be established. In particular, models of antibody-mediated CD8+ T cell depletion that lead to an undiscriminating elimination of all CD8 expressing T cells, do not allow determining whether particular T cells subsets (e.g., activated effector T cells, memory T cells) have different effects on lesion formation. Also the mechanisms of activation of CD8+ T cells in atherosclerosis are still unclear. So far, and in contrast to CD4+ T cells [34], no atherosclerosis relevant antigen that may activate CD8+ T cells has been described. Given the critical role of lipid metabolism in T cell function [43], hypercholesterolemia may also directly affect CD8+ T cell activation. Finally, the relevance of these recent studies performed in murine models of atherosclerosis for human disease is difficult to pinpoint. For example, although potential equivalents of murine CD8+CD25+ and Qa-1 restricted regulatory T cells have been described in humans, their association with atherosclerotic disease is still unknown. Likewise, whether pro-atherogenic effector functions of CD8+ T cells such as cytotoxicity towards vascular cells and cytokine-mediated pro-inflammatory effects are also relevant in human disease, remains to be determined. In addition, although models of high fat diet-induced atherosclerosis in Ldlr −/− or Apoe −/− mice recapitulate some major features of human atherosclerosis, these models suffer from numerous biases as reviewed in [20], in addition to the differences between the human and the murine immune system [45]. Findings obtained from studies using these models may thus not necessarily translate to the human situation, and additional research will be required in the future to fully address and understand the detailed mechanisms by which the different CD8+ T cell subpopulations affect atherosclerotic lesion formation.
References
Ait-Oufella H, Sage AP, Mallat Z, Tedgui A (2014) Adaptive (T and B cells) immunity and control by dendritic cells in atherosclerosis. Circ Res 114:1640–1660. doi:10.1161/CIRCRESAHA.114.302761
Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, Ma J, Tezuka K, Yagita H, Okumura K (2005) The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol 175:2340–2348
Barry M, Bleackley RC (2002) Cytotoxic T lymphocytes: all roads lead to death. Nat Rev Immunol 2:401–409. doi:10.1038/nri819
Bevan MJ (2004) Helping the CD8(+) T-cell response. Nat Rev Immunol 4:595–602. doi:10.1038/nri1413
Bisikirska B, Colgan J, Luban J, Bluestone JA, Herold KC (2005) TCR stimulation with modified anti-CD3 mAb expands CD8+ T cell population and induces CD8+ CD25+ Tregs. J Clin Invest 115:2904–2913. doi:10.1172/JCI23961
Campbell LA, Rosenfeld ME (2015) Infection and atherosclerosis development. Arch Med Res 46:339–350. doi:10.1016/j.arcmed.2015.05.006
Chess L, Jiang H (2004) Resurrecting CD8+ suppressor T cells. Nat Immunol 5:469–471. doi:10.1038/ni0504-469
Churlaud G, Pitoiset F, Jebbawi F, Lorenzon R, Bellier B, Rosenzwajg M, Klatzmann D (2015) Human and mouse CD8(+)CD25(+)FOXP3(+) regulatory T cells at steady state and during interleukin-2 therapy. Front Immunol 6:171. doi:10.3389/fimmu.2015.00171
Chyu KY, Zhao X, Dimayuga PC, Zhou J, Li X, Yano J, Lio WM, Chan LF, Kirzner J, Trinidad P, Cercek B, Shah PK (2012) CD8+ T cells mediate the athero-protective effect of immunization with an ApoB-100 peptide. PLoS One 7:e30780. doi:10.1371/journal.pone.0030780
Clement M, Guedj K, Andreata F, Morvan M, Bey L, Khallou-Laschet J, Gaston AT, Delbosc S, Alsac JM, Bruneval P, Deschildre C, Le Borgne M, Castier Y, Kim HJ, Cantor H, Michel JB, Caligiuri G, Nicoletti A (2015) Control of the T follicular helper-germinal center B-cell axis by CD8(+) regulatory T cells limits atherosclerosis and tertiary lymphoid organ development. Circulation 131:560–570. doi:10.1161/CIRCULATIONAHA.114.010988
Cochain C, Chaudhari SM, Koch M, Wiendl H, Eckstein HH, Zernecke A (2014) Programmed cell death-1 deficiency exacerbates T cell activation and atherogenesis despite expansion of regulatory T cells in atherosclerosis-prone mice. PLoS One 9:e93280. doi:10.1371/journal.pone.0093280
Cochain C, Koch M, Chaudhari SM, Busch M, Pelisek J, Boon L, Zernecke A (2015) CD8+ T cells regulate monopoiesis and circulating Ly6C-high monocyte levels in atherosclerosis in mice. Circ Res 117:244–253. doi:10.1161/CIRCRESAHA.117.304611
Cochain C, Zernecke A (2015) Macrophages and immune cells in atherosclerosis: recent advances and novel concepts. Basic Res Cardiol 110:34. doi:10.1007/s00395-015-0491-8
Combadiere C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedgui A, Mallat Z (2008) Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 117:1649–1657. doi:10.1161/CIRCULATIONAHA.107.745091
Elhage R, Gourdy P, Brouchet L, Jawien J, Fouque MJ, Fievet C, Huc X, Barreira Y, Couloumiers JC, Arnal JF, Bayard F (2004) Deleting TCR alpha beta+ or CD4+ T lymphocytes leads to opposite effects on site-specific atherosclerosis in female apolipoprotein E-deficient mice. Am J Pathol 165:2013–2018
Ensan S, Li A, Besla R, Degousee N, Cosme J, Roufaiel M, Shikatani EA, El-Maklizi M, Williams JW, Robins L, Li C, Lewis B, Yun TJ, Lee JS, Wieghofer P, Khattar R, Farrokhi K, Byrne J, Ouzounian M, Zavitz CC, Levy GA, Bauer CM, Libby P, Husain M, Swirski FK, Cheong C, Prinz M, Hilgendorf I, Randolph GJ, Epelman S, Gramolini AO, Cybulsky MI, Rubin BB, Robbins CS (2016) Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat Immunol 17:159–168. doi:10.1038/ni.3343
Filardo S, Di Pietro M, Farcomeni A, Schiavoni G, Sessa R (2015) Chlamydia pneumoniae-mediated inflammation in atherosclerosis: a meta-analysis. Mediators Inflamm 2015:378658. doi:10.1155/2015/378658
Fyfe AI, Qiao JH, Lusis AJ (1994) Immune-deficient mice develop typical atherosclerotic fatty streaks when fed an atherogenic diet. J Clin Invest 94:2516–2520. doi:10.1172/JCI117622
Geissmann F, Jung S, Littman DR (2003) Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19:71–82
Getz GS, Reardon CA (2012) Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol 32:1104–1115. doi:10.1161/ATVBAHA.111.237693
Gewaltig J, Kummer M, Koella C, Cathomas G, Biedermann BC (2008) Requirements for CD8 T-cell migration into the human arterial wall. Hum Pathol 39:1756–1762. doi:10.1016/j.humpath.2008.04.018
Grivel JC, Ivanova O, Pinegina N, Blank PS, Shpektor A, Margolis LB, Vasilieva E (2011) Activation of T lymphocytes in atherosclerotic plaques. Arterioscler Thromb Vasc Biol 31:2929–2937. doi:10.1161/ATVBAHA.111.237081
Honjo T, Chyu KY, Dimayuga PC, Yano J, Lio WM, Trinidad P, Zhao X, Zhou J, Chen S, Cercek B, Arditi M, Shah PK (2015) ApoB-100-related peptide vaccine protects against angiotensin II-induced aortic aneurysm formation and rupture. J Am Coll Cardiol 65:546–556. doi:10.1016/j.jacc.2014.11.054
Hopkins PN (2013) Molecular biology of atherosclerosis. Physiol Rev 93:1317–1542. doi:10.1152/physrev.00004.2012
Hu D, Ikizawa K, Lu L, Sanchirico ME, Shinohara ML, Cantor H (2004) Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat Immunol 5:516–523. doi:10.1038/ni1063
Hu D, Mohanta SK, Yin C, Peng L, Ma Z, Srikakulapu P, Grassia G, MacRitchie N, Dever G, Gordon P, Burton FL, Ialenti A, Sabir SR, McInnes IB, Brewer JM, Garside P, Weber C, Lehmann T, Teupser D, Habenicht L, Beer M, Grabner R, Maffia P, Weih F, Habenicht AJ (2015) Artery tertiary lymphoid organs control aorta immunity and protect against atherosclerosis via vascular smooth muscle cell lymphotoxin beta receptors. Immunity 42:1100–1115. doi:10.1016/j.immuni.2015.05.015
Hwang Y, Yu HT, Kim DH, Jang J, Kim HY, Kang I, Kim HC, Park S, Lee WW (2016) Expansion of CD8(+) T cells lacking the IL-6 receptor alpha chain in patients with coronary artery diseases (CAD). Atherosclerosis 249:44–51. doi:10.1016/j.atherosclerosis.2016.03.038
Jansen MF, Hollander MR, van Royen N, Horrevoets AJ, Lutgens E (2016) CD40 in coronary artery disease: a matter of macrophages? Basic Res Cardiol 111:38. doi:10.1007/s00395-016-0554-5
Jiang H, Canfield SM, Gallagher MP, Jiang HH, Jiang Y, Zheng Z, Chess L (2010) HLA-E-restricted regulatory CD8(+) T cells are involved in development and control of human autoimmune type 1 diabetes. J Clin Invest 120:3641–3650. doi:10.1172/JCI43522
Joffre OP, Segura E, Savina A, Amigorena S (2012) Cross-presentation by dendritic cells. Nat Rev Immunol 12:557–569. doi:10.1038/nri3254
Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK (1986) Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis 6:131–138
Keul P, Lucke S, von Wnuck Lipinski K, Bode C, Graler M, Heusch G, Levkau B (2011) Sphingosine-1-phosphate receptor 3 promotes recruitment of monocyte/macrophages in inflammation and atherosclerosis. Circ Res 108:314–323. doi:10.1161/CIRCRESAHA.110.235028
Kim HJ, Verbinnen B, Tang X, Lu L, Cantor H (2010) Inhibition of follicular T-helper cells by CD8(+) regulatory T cells is essential for self tolerance. Nature 467:328–332. doi:10.1038/nature09370
Kimura T, Tse K, Sette A, Ley K (2015) Vaccination to modulate atherosclerosis. Autoimmunity 48:152–160. doi:10.3109/08916934.2014.1003641
Klingenberg R, Lebens M, Hermansson A, Fredrikson GN, Strodthoff D, Rudling M, Ketelhuth DF, Gerdes N, Holmgren J, Nilsson J, Hansson GK (2010) Intranasal immunization with an apolipoprotein B-100 fusion protein induces antigen-specific regulatory T cells and reduces atherosclerosis. Arterioscler Thromb Vasc Biol 30:946–952. doi:10.1161/ATVBAHA.109.202671
Koch AE, Haines GK, Rizzo RJ, Radosevich JA, Pope RM, Robinson PG, Pearce WH (1990) Human abdominal aortic aneurysms. Immunophenotypic analysis suggesting an immune-mediated response. Am J Pathol 137:1199–1213
Kolbus D, Ljungcrantz I, Andersson L, Hedblad B, Fredrikson GN, Bjorkbacka H, Nilsson J (2013) Association between CD8+ T-cell subsets and cardiovascular disease. J Intern Med 274:41–51. doi:10.1111/joim.12038
Kolbus D, Ramos OH, Berg KE, Persson J, Wigren M, Bjorkbacka H, Fredrikson GN, Nilsson J (2010) CD8+ T cell activation predominate early immune responses to hypercholesterolemia in Apoe(−)(/)(−) mice. BMC Immunol 11:58. doi:10.1186/1471-2172-11-58
Kyaw T, Winship A, Tay C, Kanellakis P, Hosseini H, Cao A, Li P, Tipping P, Bobik A, Toh BH (2013) Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in apoE-deficient mice. Circulation 127:1028–1039. doi:10.1161/CIRCULATIONAHA.112.001347
Legein B, Janssen EM, Theelen TL, Gijbels MJ, Walraven J, Klarquist JS, Hennies CM, Wouters K, Seijkens TT, Wijnands E, Sluimer JC, Lutgens E, Zenke M, Hildner K, Biessen EA, Temmerman L (2015) Ablation of CD8alpha(+) dendritic cell mediated cross-presentation does not impact atherosclerosis in hyperlipidemic mice. Sci Rep 5:15414. doi:10.1038/srep15414
Lievens D, Habets KL, Robertson AK, Laouar Y, Winkels H, Rademakers T, Beckers L, Wijnands E, Boon L, Mosaheb M, Ait-Oufella H, Mallat Z, Flavell RA, Rudling M, Binder CJ, Gerdes N, Biessen EA, Weber C, Daemen MJ, Kuiper J, Lutgens E (2013) Abrogated transforming growth factor beta receptor II (TGFbetaRII) signalling in dendritic cells promotes immune reactivity of T cells resulting in enhanced atherosclerosis. Eur Heart J 34:3717–3727. doi:10.1093/eurheartj/ehs106
Lindau A, Hardtner C, Hergeth SP, Blanz KD, Dufner B, Hoppe N, Anto-Michel N, Kornemann J, Zou J, Gerhardt LM, Heidt T, Willecke F, Geis S, Stachon P, Wolf D, Libby P, Swirski FK, Robbins CS, McPheat W, Hawley S, Braddock M, Gilsbach R, Hein L, von zur Muhlen C, Bode C, Zirlik A, Hilgendorf I (2016) Atheroprotection through SYK inhibition fails in established disease when local macrophage proliferation dominates lesion progression. Basic Res Cardiol 111:20. doi:10.1007/s00395-016-0535-8
Lochner M, Berod L, Sparwasser T (2015) Fatty acid metabolism in the regulation of T cell function. Trends Immunol 36:81–91. doi:10.1016/j.it.2014.12.005
Ludewig B, Freigang S, Jaggi M, Kurrer MO, Pei YC, Vlk L, Odermatt B, Zinkernagel RM, Hengartner H (2000) Linking immune-mediated arterial inflammation and cholesterol-induced atherosclerosis in a transgenic mouse model. Proc Natl Acad Sci USA 97:12752–12757. doi:10.1073/pnas.220427097
Mestas J, Hughes CC (2004) Of mice and not men: differences between mouse and human immunology. J Immunol 172:2731–2738
Mohanta SK, Yin C, Peng L, Srikakulapu P, Bontha V, Hu D, Weih F, Weber C, Gerdes N, Habenicht AJ (2014) Artery tertiary lymphoid organs contribute to innate and adaptive immune responses in advanced mouse atherosclerosis. Circ Res 114:1772–1787. doi:10.1161/CIRCRESAHA.114.301137
Nadareishvili ZG, Koziol DE, Szekely B, Ruetzler C, LaBiche R, McCarron R, DeGraba TJ (2001) Increased CD8(+) T cells associated with Chlamydia pneumoniae in symptomatic carotid plaque. Stroke 32:1966–1972
Olofsson PS, Soderstrom LA, Wagsater D, Sheikine Y, Ocaya P, Lang F, Rabu C, Chen L, Rudling M, Aukrust P, Hedin U, Paulsson-Berne G, Sirsjo A, Hansson GK (2008) CD137 is expressed in human atherosclerosis and promotes development of plaque inflammation in hypercholesterolemic mice. Circulation 117:1292–1301. doi:10.1161/CIRCULATIONAHA.107.699173
Qiu MK, Wang SC, Dai YX, Wang SQ, Ou JM, Quan ZW (2015) PD-1 and Tim-3 Pathways Regulate CD8+ T Cells Function in Atherosclerosis. PLoS One 10:e0128523. doi:10.1371/journal.pone.0128523
Santos M, Schilham MW, Rademakers LH, Marx JJ, de Sousa M, Clevers H (1996) Defective iron homeostasis in beta 2-microglobulin knockout mice recapitulates hereditary hemochromatosis in man. J Exp Med 184:1975–1985
Schurch CM, Riether C, Ochsenbein AF (2014) Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell 14:460–472. doi:10.1016/j.stem.2014.01.002
Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ (2007) Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest 117:195–205. doi:10.1172/JCI29950
Tabas I, Bornfeldt KE (2016) Macrophage phenotype and function in different stages of atherosclerosis. Circ Res 118:653–667. doi:10.1161/CIRCRESAHA.115.306256
Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ (2007) Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest 117:185–194. doi:10.1172/JCI28549
Xu QB, Oberhuber G, Gruschwitz M, Wick G (1990) Immunology of atherosclerosis: cellular composition and major histocompatibility complex class II antigen expression in aortic intima, fatty streaks, and atherosclerotic plaques in young and aged human specimens. Clin Immunol Immunopathol 56:344–359
Xu S, Liu X, Bao Y, Zhu X, Han C, Zhang P, Zhang X, Li W, Cao X (2012) Constitutive MHC class I molecules negatively regulate TLR-triggered inflammatory responses via the Fps-SHP-2 pathway. Nat Immunol 13:551–559. doi:10.1038/ni.2283
Zafiratos MT, Manam S, Henderson KK, Ramsey KH, Murthy AK (2015) CD8+ T cells mediate Chlamydia pneumoniae-induced atherosclerosis in mice. Pathog Dis. doi:10.1093/femspd/ftv052
Zhou J, Dimayuga PC, Zhao X, Yano J, Lio WM, Trinidad P, Honjo T, Cercek B, Shah PK, Chyu KY (2014) CD8(+)CD25(+) T cells reduce atherosclerosis in apoE(−/−) mice. Biochem Biophys Res Commun 443:864–870. doi:10.1016/j.bbrc.2013.12.057
Zhou X, Stemme S, Hansson GK (1996) Evidence for a local immune response in atherosclerosis. CD4+ T cells infiltrate lesions of apolipoprotein-E-deficient mice. Am J Pathol 149:359–366
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (SFB688 TPA22).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Cochain, C., Zernecke, A. Protective and pathogenic roles of CD8+ T cells in atherosclerosis. Basic Res Cardiol 111, 71 (2016). https://doi.org/10.1007/s00395-016-0589-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-016-0589-7