
Abstract
Brugada syndrome (BrS) is characterized by ST-segment elevation in the right precordial leads and is associated with increased risk of sudden cardiac death. We have recently reported families with BrS and SCN5A mutations where some affected members do not carry the familial mutation. We evaluated the involvement of additional genetic determinants for BrS in an affected family. We identified three distinct gene variants within a family presenting BrS (5 individuals), cardiac conduction defects (CCD, 3 individuals) and shortened QT interval (4 individuals). The first mutation is nonsense, p.Q1695*, lying within the SCN5A gene, which encodes for NaV1.5, the α–subunit of the cardiac Na+ channel. The second mutation is missense, p.N300D, and alters the CACNA1C gene, which encodes the α–subunit CaV1.2 of the L-type cardiac Ca2+ channel. The SCN5A mutation strictly segregates with CCD. Four out of the 5 BrS patients carry the CACNA1C variant, and three of them present shortened QT interval. One of the BrS patients carries none of these mutations but a rare variant located in the ABCC9 gene as well as his asymptomatic mother. Patch-clamp studies identified a loss-of-function of the mutated CaV1.2 channel. Western-blot experiments showed a global expression defect while increased mobility of CaV1.2 channels on cell surface was revealed by FRAP experiments. Finally, computer simulations of the two mutations recapitulated patient phenotypes. We report a rare CACNA1C mutation as causing BrS and/or shortened QT interval in a family also carrying a SCN5A stop mutation, but which does not segregate with BrS. This study underlies the complexity of BrS inheritance and its pre-symptomatic genetic screening interpretation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sudden cardiac death (SCD) is a leading cause of mortality, with an incidence close to 1 per 1,000 inhabitants each year in Western societies. In most cases, SCD results from ventricular fibrillation, occurring as the initial manifestation of a previously ignored cardiovascular disease. However, in 5–10 % of cases, SCD occurred in the absence of ischemic or identifiable structural heart disease [9]. Among these cases, an 18 % prevalence of a family history of premature sudden death and unexplained accidents has been estimated, raising the possibility of underlying heritable cardiac disease [2]. Among these disorders, Brugada Syndrome (BrS) is characterized by a specific electrocardiographic (ECG) pattern of “coved-type” ST-segment elevation in the right precordial leads (above 2 mm) and associated with high risk of SCD before the age of 50 [7, 25]. BrS, the prevalence of which is estimated from 1 to 5 in 10,000 in Western countries to 12 in 10,000 in Southeast Asia [11], was originally described as a monogenic disease with autosomal dominant transmission.
Most mutations identified to date reside in SCN5A, the gene encoding NaV1.5, the α-subunit of the major cardiac sodium channel. These mutations are usually private and induce a loss of function of NaV1.5. However, SCN5A mutations are detected in only 20–25 % of BrS patients [11] and are characterized by a low penetrance (~20 %) when found in large affected families [27]. SCN5A mutations have also been associated with other cardiac electrical anomalies, as long-QT syndrome type 3, conduction disease and sinus node dysfunction [4, 30, 34]. The same SCN5A mutation can lead to different cardiac arrhythmia phenotypes in the same family or even in a single patient, suggesting that genetic background and environment could play an important role in the occurrence of BrS [26, 32]. Furthermore, we have recently showed that in a subset of BrS families apparently segregating a SCN5A gene defect with proven pathogenicity, some BrS patients do not carry the SCN5A mutation, suggesting that this gene defect is not directly causal to the BrS occurrence [27].
Loss-of-function mutations in CACNA1C and CACNB2 genes encoding CaV1.2 and CaVβ2, the α and regulatory subunits of the L-type calcium channel, respectively, have been associated with a combination of BrS phenotype and shortened QT interval [1]. Other genes have been linked to BrS but their prevalence remains low [5].
Here we report a family with BrS, in which one stop-mutation has been found in SCN5A but does not segregate with BrS. Further genetic investigations revealed a loss-of-function mutation in CACNA1C, carried by four family members presenting BrS and shortened QT interval duration. However, we found one member affected by BrS who does not carry any of the mutations but in which we detected a variant of ABCC9, recently described as BrS susceptibility gene [16]. Our results, in line with recent reports on other familial cases, indicate that BrS is a rather complex disease, with multifactorial inheritance.
Methods
Patient phenotyping
This study conforms to the Declaration of Helsinki. It was conducted in accordance with the French guidelines for genetic research and with the approval of the local medical ethics committees. Informed written consent was obtained from all patients. Baseline 12-lead ECGs were recorded at a paper speed of 25 mm/s. According to the current guidelines, BrS-ECG was defined as an ST-segment elevation with type-1 morphology >2 mm in, at least, 1 lead among the right precordial leads V1, V2, positioned in the 2nd, 3rd or 4th intercostal space, occurring either spontaneously or after provocative drug test with intravenous administration of Class I antiarrhythmic drugs [25]. Ajmaline (1 mg/kg body weight) was used for drug testing. An electrophysiological study (EPS) was carried out in selected cases. Echocardiography, chest roentgenogram and exercise testing excluded any underlying structural heart disease. Laboratory tests excluded acute ischemia and metabolic or electrolyte disturbances. ECG parameters were assessed: heart rate (HR), PQ interval in lead II, QRS duration, maximal ST elevation in right precordial leads and QTc duration in V4 (Bazett formula), blinded to the genetic status.
Genetic analysis
Genomic DNA was extracted from peripheral blood leukocytes using standard protocols. All SCN5A and CACNA1C coding regions and exon boundaries were sequenced for individual III-2 (Fig. 2a). Coding and splice-sites regions of SCN5A, KCNH2, GPD1L, CACNA1C, CACNB2b, SCN1B, KCNE3, SCN3B, MOG1, KCNE5, KCND3 and ABCC9 were sequenced for individuals II-2 and II-3 (Fig. 2a). DNA variants were considered as disease-causing rather than polymorphisms if: (1) absent in ≥190 Caucasian control alleles and in the 1,000 Genomes exome database (http://www.1000genomes.org) (2) below 1 % in the Exome Variant Server (EVS) of the NHLBI Exome Sequencing Project, as EVS includes patients with cardiac diseases [http://evs.gs.washington.edu/EVS/ (02-2013 accessed)] and (3) present in highly conserved regions. Gene mutations were annotated based on the following cDNA reference sequences from Ensembl: SCN5A, ENST00000413689; CACNA1C, ENST00000399655.
cDNA constructs
The four pcDNA3 plasmids containing the cDNA of human CACNA1C with exon 8, CACNA1C with exon 8A fused with the yellow fluorescent protein (clone (EYFP)Nα1C,77), CACNB2b, or CACNA2D1, were gifts from Drs Nikolai Soldatov (NIH, Baltimore, MD, USA) and Charles Antzelevitch (Masonic Medical Research Laboratory, Utica, NY, USA). For CACNA1C mutation, site-directed mutagenesis was performed with QuickChange II XL site-directed (Stratagene, CA, USA) on pcDNA3-CACNA1C and pcDNA3-CACNA1C clone [(EYFP)Nα1C,77] according to the manufacturer’s instructions. The constructs were re-sequenced to ensure that there was no other mutation.
Transfections
The African green monkey kidney fibroblast-like cell line (COS-7) was obtained from the American Type Culture Collection and cultured as previously described [3]. For electrophysiology and biochemistry experiments, the cells were co-transfected using FuGene6 (Roche Diagnostics, USA) with a 1:1:1 molar ratio of WT or mutant human CACNA1C, WT CACNB2b and WT CACNA2D1 plasmids (1 µg of each subunit plasmid). In addition, 0.4 µg of plasmid encoding CD8 antigen was added to all transfections as a reporter gene. For confocal microscopy, the cells were co-transfected with the EYFP-tagged WT or mutant human CACNA1C cDNA-containing plasmids. Cells were used 72–96 h after transfection. The ratio cDNAs/FuGene6 was 3.4 µg cDNA/7.5 µL FuGene6. Previous studies have shown that neither the fused yellow fluorescent protein [(EYFP)Nα1C,77] nor the exon 8A influenced CaV1.2 channel expression [18].
Electrophysiology
Whole-cell currents were recorded using the patch-clamp technique (24 °C). The cells were continuously superfused with Tyrode solution containing (in mmol/L): NaCl 145, KCl 4, MgCl2 1, CaCl2 1, HEPES 5, glucose 5, pH adjusted to 7.4 with NaOH. Wax-coated pipettes (tip resistance: 1.8 to 3 MΩ) were filled with intracellular medium containing (in mmol/L): CsCl 130, HEPES 10, EGTA 10, CaCl2 2, MgCl2 1, MgATP 4, TrisGTP 0.3, pH adjusted to 7.2 with CsOH. Transfected cells were detected using Dynabeads® CD8 (Invitogen, Life Technologies). During data recording, the studied cell was locally superfused with extracellular medium containing (in mmol/L): NaCl 135, TEACl 20, HEPES 10, MgCl2 1, CaCl2 2, mannitol 20, pH adjusted to 7.3 with NaOH. All products were purchased from Sigma. Stimulation, data recording through an A/D converter (Tecmar TM100 Labmaster, Scientific Solutions; 5 kHz filtering), and analysis were performed with Acquis1 software (Bio-Logic). All current measurements were normalized using the cell capacitance. Series resistances were compensated to obtain minimal contribution of capacitive transients using a VE-2 amplifier (Alembic Instrument, Qc, Canada).
Western blots
COS-7 cells of two 6-cm Petri dishes where lysed in 500 µL of lysis buffer (150 mM NaCl, 20 mM Tris pH 7.5, 1 mM EDTA, 1 % Triton X-100 supplemented with complete protease inhibitor mixture—Roche Diagnostics). The cellular lysate was centrifuged 10 min at 1,000 g. Protein concentration was systematically determined by performing a Bradford assay (Biorad). Fifteen µg of protein were denatured for 10 min at 50 °C in Laemli sample buffer, separated by SDS-PAGE on 8 %-gels and transferred to nitrocellulose membranes (Amersham). 5 % fat-free milk was used to block the nitrocellulose membrane. The blots were probed with anti-CaV1.2 (1:1,000) and anti-GAPDH (1:1,000) and secondary HRP-coupled antibody (mouse: 1:10,000, rabbit: 1:5,000). The blots were then revealed using two different enhanced chemi-luminescence kits: super signal west femto maximum sensitivity substrate (Pierce) for the CaV1.2 revelation, and ECL plus western blotting detection system (Amersham) for the others proteins revelation and analyzed with camera ImageQuant RT ECL (GE Healthcare). The ImageQuantL software (GE Healthcare) was used for image quantification. Signals were normalized with respect to GAPDH staining and then normalized the control condition.
Cell membrane biotinylation
Cells were treated for 1 h at 4 °C with 1 mL biotin per 6-cm dish (EZ link Sulfo-NHS-SS-Biotin; Pierce), washed once with cold PBS 1× containing 100 mM glycine, then twice with cold PBS 1× and lysed with 500 µL/dish of lysis buffer. One hundred microliters of streptavidin-agarose beads (Pierce) were added to 800 µg of cell lysate and incubated overnight on a wheel at 4 °C. The beads were washed three times with lysis buffer and the proteins were cleaved and denatured for 30 min at 37 °C in Laemli sample buffer containing DTT. Eluted proteins were analyzed by Western blot. Signals were normalized with respect to transferrin receptor staining and then normalized the control condition.
Antibodies
Anti-CaV1.2 Ab was obtained from Millipore (rabbit, AB5156); anti-GAPDH Ab (mouse, SC-32233), anti-rabbit horseradish peroxidase Ab (SC-2054), and anti-mouse horseradish peroxidase Ab (SC-2055) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA); anti-transferrin receptor Ab was obtained from Invitrogen (mouse, 13-6890).
FRAP analysis
Fluorescence recovery after photobleaching (FRAP) experiments were performed on 2–3 day-old EYFP-Cav1.2 WT- or N300D-transfected COS-7 cells using a confocal microscope Nikon A1R-Si (Nikon, France) equipped with a ×60, N.A. 1.4 oil-immersion lens. Environment was controlled in an incubation chamber (Life Imaging Service, Switzerland) at 37 °C and 5 % of CO2. Cells were mounted in a 4-well Lab-Tek chamber in Opti-MEM (Invitrogen). One rectangular 1-µm2 ROI along the cell surface was photo-bleached by scanning with the 488 nm line of the argon laser and recorded at a bandwidth of 500–550 nm. For the pre-bleach phases (30 s), one image was recorded every 5 s, with the laser set to 5 %. The bleach was performed for 1 s with the laser set to 100 %. For the post-bleach phases, 1 image every 5 s for 1 min and then 1 image per 10 s for 1 min were recorded with the laser set to 5 %. The analysis of fluorescence was performed using NIS element 3.2 and MetaMorph software’s. Average fluorescence in ROIs was measured, background subtracted and corrected for overall photobleaching in each time frame. The average fluorescence within the ROI was double-normalized so that the pre-bleach intensity was set to one and the first frame after photobleaching to zero and plotted as a function of time. The fluorescence recovery was fitted by a double-exponential function.
Statistical analysis
Data are presented as mean ± SEM. The statistical analyses were achieved using SigmaPlot (Systat Software Inc.). Unpaired, two-tailed Student t test was used for group comparison when normal distribution was positively tested using Shapiro–Wilk test. Alternatively, Mann–Whitney rank sum test was used. Wilcoxon signed rank test was used for WB paired measurements. P < 0.05 was considered significant.
Computer simulations
For the right-ventricle model, pseudo-ECG was calculated as in Gima and Rudy [13], by simulating the heterogeneous transmural wedge (right ventricular outflow tract, 1 Hz, 100th beat shown). Mutations Q1695* and N300D were represented by 50 and 44 % reductions in conductance of fast Na+ current and L-type Ca2+ current, respectively. For the left-ventricle model, the single-cell model of human left ventricular myocytes was used [17]. The model was modified from the CellMl.org model (Jul 20, 2012) to minimize the stimulation artefact (stimulation amplitude: −50 µA/µF; stimulation duration: 0.5 ms; GNa 100 mS/µF). Equations corresponding to the Na+ and the Ca2+ currents were modified to reproduce the experimental data. As for the right-ventricle model, mutations Q1695* and N300D were represented by 50 and 44 % reductions in conductance of fast Na+ current and L-type Ca2+ current, respectively.
Results
Family recruitment
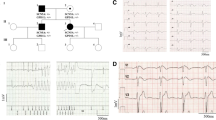
A 22-year-old male (III-2), who experienced aborted SCD from ventricular fibrillation, presented with a prolonged PR interval (206 ms) and a mildly prolonged QRS interval (108 ms) on the ECG at rest (Fig. 1 and Supplementary Figure 1; Table 1). The ECG showed only a minor ST-segment elevation in the right precordial leads. QTc duration was slightly shortened and T-wave showed high amplitude. Ajmaline challenge induced first severe prolongations of PR (240 ms) and QRS durations (Fig. 1, ajmaline 5 min; Table 1). Eleven minutes after ajmaline injection, there was an improvement of the conduction and a type-1 Brugada ECG appeared in the upper precordial leads with a 3-mm ST-segment elevation (Fig. 1, ajmaline 11 min, Supplementary Figure 1; Table 1). No morphological abnormalities were found during the echocardiography and electrophysiological studies gave normal results. Because of the SCD event, an ICD was implanted.

ECG of the 22-year-old proband (proband III-2), showing ECG at rest, at 5 and 11 min after ajmaline injection start (1 mg/kg body weight at a rate of 0.5 mg/kg per min). Ajmaline test induced major conduction disturbance. The J point elevation and the coved type ST-segment elevation observed in high precordial lead (V1H, V2H, V3H recorded at the third ICS) remained significant after the test and after correction of conduction disturbance
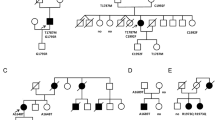
Familial investigations led to the identification of four other family members (II-2, II-3, III-1 and III-3, Fig. 2a) showing an ECG pattern during ajmaline challenge indicating BrS (Fig. 2b, Supplementary Figure 2 to 5; Table 1). The aspect of Patient II-2 ECG during ajmaline challenge appeared to be different from that of his BrS relatives, with a 6-mm ST-segment elevation. For patient III-1, the challenge induced also severe prolongations of QRS duration (200 ms, Supplementary Figure 6) and HV interval: from 119 to 154 ms at 9 min of ajmaline. During the ajmaline test, patient III-4 presented with a saddle-back pattern and a minor ST-segment elevation at the ECG which did not reach BrS diagnostic criteria (ST-segment elevation ≤2 mm, Fig. 2b) [25]. The patients II-1, II-3, III-3 and III-4 presented with a shortened QTc duration at the ECG (≤350 ms) associated with tall T-wave except for patient II-3 (Fig. 2b). Left anterior hemi-block and incomplete right-bundle block were also observed for the patients II-4 and III-1, respectively (Fig. 2b). Ventricular tachycardia was observed in II-4 during ajmaline challenge with no ST-elevation (Supplementary Figure 7).

a Pedigree, phenotype and genotype of the family. Males and females are indicated by squares and circles, respectively. Phenotypes presented by the family members are depicted by a blue top left square for a QTc duration ≤360 ms and/or tall T-wave, yellow top right square for cardiac conduction defects, red bottom left square for a Brugada syndrome. Question mark: ECG with a saddle-back pattern. Arrow proband. Diagonal line indicates the deceased individuals. ICD implantable cardioverter defibrillator. b Representative surface ECG of individuals II-1, II-2, II-3, II-4, III-1, III-3 and III-4 before (top) and during (bottom) ajmaline test
Molecular genetics analysis
BrS-ECG pattern carried by the male proband (III-2) led us to sequence coding regions of the SCN5A gene in first intention. One substitution was detected at the genomic position (hg19, GRCh37) chr3:38592780 (c.5083 C > T; Fig. 3a), which introduces a premature stop codon in the extracellular loop connecting segment 5 and 6 of Nav1.5 domain IV (p.Q1695*; Fig. 3b). Segregation analysis revealed that the SCN5A p.Q1695* mutation, also carried by the proband’s brother (III-1), is inherited from the mother (II-4). The mutation could neither be found in ethnicity-matched controls (190 alleles were tested) nor in the Exome Variant Server (EVS: evs.gs.washington.edu/EVS; ~6,500 individuals). The two other siblings, despite presenting with shortened QT interval and/or BrS, do not carry the familial SCN5A mutation (Fig. 2a). In consequence, because the association of BrS with shortened QTc interval has been causally related to mutations in the calcium channel CaV1.2 complex [1], we also sequenced the proband’s CACNA1C and CACNB2b genes, encoding the α subunit CaV1.2 and the β2 accessory subunit of the L-type Ca2+ channel, respectively. A missense variant was found in CACNA1C, at the genomic position chr12:2595410 (c.898 A>G; Fig. 3c). The corresponding substitution targets a highly conserved asparagine (Fig. 3d) to an aspartic acid in the extracellular loop connecting segment five and six of CaV1.2 domain I (p.N300D; Fig. 3e). This missense variant, which was not found among 576 ethnicity-matched control alleles and had an allele frequency of 0.03 % (4/12,974 alleles) in EVS, was detected in five other family members (II-1, II-3, III-1, III-3 and III-4; Fig. 2a), all presenting with shortened QTc interval and/or BrS. Neither SCN5A nor CACNA1C variants were found in patient II-2. Since ABCC9 gene, encoding SUR2A, the regulatory sulfonylurea receptor of the cardiac ATP-sensitive K+ channel, has been most recently identified as a BrS susceptibility gene [16], we sequenced this gene. In this patient, we found a rare SNP (rs149325742, MAF = 0.0077 %) corresponding to the substitution of A>T at the position chr12:22070007, leading to an amino acid changing an isoleucine to an asparagine in position 146 (p.I146N). This substitution is predicted to be damaging and possibly-damaging for the biological function of the protein by the bioinformatics tools SIFT [22] and Polyphen2 [28], respectively. This variant was found in his unaffected mother (I-2) but not in his two affected brothers (II-1 and II-3).

Genetic analysis (electropherogram) revealed a heterozygous c.5083 C>T SCN5A missense mutation (a) and a heterozygous c.898 A>G CACNA1C missense mutation (c). b and e Location of the resulting change on the predicted channel structures. d Evolutionary conservation of the N300 amino-acid in mammalian CaV1.2
Functional analysis
To determine a possible pathogenic role of the N300D variant in CaV1.2, in vitro analyses were performed in COS-7 cells, by co-transfection of vectors expressing WT or N300D CaV1.2, CaVβ2b and CaVα2δ1.
First, calcium currents (ICa,L) were recorded. Amplitude of the current density was drastically reduced (~88 %) in the presence of N300D versus WT CaV1.2 (Fig. 4a). Since ICa,L was hardly measurable with N300D CaV1.2, Ba2+ was substituted to Ca2+ as permeant ion to further investigate CaV1.2-mediated currents. Comparison of measurable N300D versus WT currents in these new conditions shows that, despite the current reduction, the voltage sensitivity of N300D CaV1.2 remained unchanged as depicted in the inset (Fig. 4b).

a Mean peak Ca2+ current density recorded in COS-7 cells transfected with WT or N300D CACNA1C, CACNB2b, CACNA2D1 and CD8 cDNAs, at 0 mV (holding potential: −80 mV; 200-ms pulse; 0.2 Hz). ***P < 0.001. Inset stimulation protocol and representative Ca2+ current recordings. b Peak Ba2+ current density versus voltage for WT (filled circles) and N300D (open circles) CaV1.2 channels. Left inset normalized I/V curves. Right inset stimulation protocol: holding potential at −80 mV; 200-ms pulses from −60 to +80 mV; 0.2 Hz, and representative whole-cell Ba2+ currents. Red Ba2+ currents at 0 mV
To understand the mechanisms underlying the strong reduction in ICa,L, we analyzed the global expression of CaV1.2 and its expression at the cell surface by Western blot and surface biotinylation assays. We found that the global expression of N300D CaV1.2 was reduced by more than 50 % when compared with the WT CaV1.2 (Fig. 5a), its expression at the cell surface being reduced by 30 % (Fig. 5b).

Biochemical assays were performed on COS-7 cells transfected with WT or N300D CACNA1C, CACNB2b and CACNA2D1 cDNAs. Western blots of whole-cell lysates (a) or of the biotinylated fraction (c), and expression of CaV1.2 normalized with GAPDH signal and relative to WT (b, 6 independent experiments; Wilcoxon signed rank test before normalization to WT condition, *P < 0.05), or normalized with transferrin receptor signal and relative to WT (d, 3 independent experiments)
This expression decrease lead us to investigate the causes of these defects at the membrane by using FRAP assays on EYFP tagged WT/N300D CaV1.2 channels. This technique allows evaluating the channel mobility. We photo-bleached the tagged proteins and measured the rate of fluorescence recovery, which we used as an index of the net movement of the unbleached CaV1.2 EYFP proteins in the plasma membrane of COS-7 cells expressing CaV1.2 (Fig. 6). Quantitative FRAP analysis at 126 s post-bleach revealed a decreased immobile fraction for the N300D CaV1.2 compared to the WT (29 ± 4 and 45 ± 3 %, n = 15 − 9, for WT and N300D, respectively). The fluorescence recovery included a fast and a slow phase. The relative contribution of the fast and slow phases was unchanged in N300D CaV1.2-expressing cells (fast phase: 0.15 ± 0.03 and 0.19 ± 0.05 for WT and N300D, respectively; slow phase 0.79 ± 0.05 and 0.71 ± 0.06 for WT and N300D, respectively). The fast time constants were also similar (12.3 ± 3.3 and 9.9 ± 2.3 s, for WT and N300D, respectively). On the contrary, the slow time constant was reduced in cells expressing N300D CaV1.2 (103.0 ± 15.4 and 200.0 ± 25.9 s for N300D and WT, respectively) indicating a lower stability of the mutated channel at the cell surface.

a Representative images of COS-7 cells expressing EYFP-CaV1.2 WT acquired 33 s before (a, Prebleach), 0 s (b, Bleach), and 126 s (c, Recovery) after photobleach. Red rectangle photo-bleached region. b Mean normalized fluorescence recovery after photobleach for WT (filled circles) and N300D (open circles) EYFP-tagged CaV1.2. c Mean immobile fraction at 126 s, d rapid- and slow-phase contributions to the fluorescence recovery, and e respective time constants. Three independent transfections; t test versus WT, *P < 0.05, **P < 0.01, ns non-significant
To evaluate to which extent the impaired L-type Ca2+ current can modify the action potential, we implemented in silico models [13, 17] with CaV1.2 N300D and NaV1.5 Q1695* currents. As illustrated in Fig. 7a, an accentuation of the epicardial right-ventricle action potential notch was detected when the L-type Ca2+ current was decreased. In addition, we observed that the same L-type Ca2+ current decrease induced a reduction of the left ventricle AP duration, a proposed mechanism responsible for the shortening of the QT interval (Fig. 7b). When the Na+ current was decreased by Q1695* mutation in the right-ventricle AP model, we observed a wide QRS in pseudo-ECG (Fig. 7a). Finally, when we implemented the Na+ current change in addition to the Ca2+ current change, we partly normalized the AP duration in both models.

a Pseudo-ECG (top) and transmural right-ventricle action potentials (bottom thin endocardium, dashed mid-myocardium, thick epicardium) from WT (black), Q1695* (blue), N300D (red), and N300D + Q1695* (green). Q1695* had wide pseudo-QRS duration. N300D had ST elevation and inverted T-wave and shorter QT interval compared with other cases. The double mutant had ST elevation and inverted T-wave, but normalized QT interval. b The effects of the two mutations introduced in a single-cell left ventricle AP computer model [17]. WT (black), N300D (red), Q1695* (blue), N300D + Q1695* (green) and superimposed (bottom) APs. Stimulation: 1 Hz
Discussion
In this study, we have identified two genetic mutations within a family with BrS, shortened QT interval and CCD. It is likely that the familial CACNA1C loss-of-function mutation contributes to BrS phenotype and shorter QTc interval while the SCN5A mutation is associated with severe CCD.
Here, a default of co-segregation is observed between the SCN5A mutation and the BrS phenotype. Indeed, out of five family members affected by the BrS, only two carry the mutation. Furthermore the SCN5A mutation was found in a member without BrS. However, all 3 SCN5A mutation carriers present CCD, with PR and QRS intervals above normal values comprised between 120 and 200 ms, and between 60 and 120 ms, respectively. In addition, transmural conduction defect is observed when the Na+ current was decreased in the right-ventricle AP model, correlating with the severe CCD phenotype of all the SCN5A mutation carriers.
Previous nonsense mutations have been reported in the same region of NaV1.5 (p.S1710 + 75* and p.S1812*), leading to isolated CCD [30, 31]. We hypothesis that the p.Q1695* mutation leads to total loss of function without dominant-negative effect. To support this idea, the reported stop mutation p.S1812*, localized in the same region of NaV1.5, enhances a total loss of function due to a defect of protein expression [14]. Furthermore, BrS-related (p.D1816VfsX7) or atrial-fibrillation related (R1860Gfs*12) C-terminus truncated mutants did not displayed any dominant negative activity [12, 21]. To date, C-terminus truncated Nav1.5 mutants with dominant-negative effect have not been described, yet and the known dominant-negative mutations are substitutions [10, 15, 24].
The N300D CACNA1C variant is carried by six out seven patients presenting BrS and/or a shortened QT interval. A QTc interval value below 360 ms is considered as shortened, and when below 330 ms, short QT syndrome is diagnosed [25]. Heterologous expression of N300D CaV1.2 leads to a major loss of function consistent with BrS phenotype associated with a shorter than normal QT interval as demonstrated in previous studies [1, 8]. This supports the hypothesis that CaV1.2 p.N300D variant plays a main role in BrS and/or shorter QT outcome, here. The lower CaV1.2global expression and its larger mobility at the cell surface suggest acceleration of CaV1.2 turn-over by destabilization which may lead to a quicker degradation of the protein. However, further experiments are needed to unveil the molecular mechanisms responsible for these effects.
In BrS, one of the mechanisms proposed is an imbalance of inward (I Na, I Ca,L) and outward (I to, I Kr, I Ks) currents that may cause apparition of a characteristically pronounced notch on the epicardial right-ventricle AP [5]. This provides a possibility of phase-2 re-entry and to ventricular arrhythmias. Computer simulations results are in agreement with BrS phenotype and shortened QT interval originating from CACNA1C. Dominant negative CaV2.1 constructs have been described [20, 23] and a dominant negative CaV1.2 splice variant has also been observed in rat [33]. Therefore, a possible dominant-negative effect of CaV1.2 p.N300D variant on WT CaV1.2 inducing a larger than 44 % current decrease in heterozygous conditions cannot be excluded. However, the simulation results obtained with a 44 % reduction only are already in favor of a causative CACNA1C mutation.
Finally, implementation of the Na+ current change in addition to the Ca2+ current change, mimicking the presence of both mutations, partly normalized the AP duration in both models, in agreement with the two double-carriers’ phenotype in this family, presenting moderate or no QT shortening.
In summary, we identified a pathogenic variant in the CACNA1C gene, as cause of BrS and/or shortened QT interval, in a family carrying a stop mutation in the SCN5A gene which does not segregate with the phenotype. We observed that the CACNA1C mutation has been identified in 4 patients out of 6,489 (MAF: 0.03 %) from the NHLBI GO Exome Sequencing Project aimed “to discover novel genes and mechanisms contributing to heart, lung and blood disorders”. Hence, we can speculate that the CACNA1C mutation has a significant prevalence in this cardiopathy-dedicated database as other proved BrS mutations found in this database such as GPD1L A280 V [29]. This is in line with a potential underestimation of BrS prevalence as discussed in Letsas et al. [19] study. Alternatively, we can also speculate that low frequency variants play a role in the pathophysiological mechanism of BrS.
Despite clear electrophysiological, molecular evidences and subsequent in silico modelling results, regarding the effects of Cav1.2 p.N300D channel impairment, two carriers did not present clear or any BrS trait (III-4 and II-1). This mutation shows an incomplete penetrance in this BrS family as previously reported in other BrS families [1, 8, 27]. This observation is in line with the hypothesis we previously formulated, speculating on a significant impact of the genetic background in the pathophysiology of BrS [27]. This hypothesis is stressed by the individual II-2 presenting a typical BrS-ECG pattern. He harbours a rare variant in ABCC9, described as a potential BrS susceptibility gene [16], however his mother who carries the ABCC9 variant, does not present a BrS-ECG pattern even after drug challenge. Variants in other genes or in non-coding region could explain this phenocopy and, more generally, the variability of expressivity found in this family. Indeed, through a case–control genome-wide association study led on BrS, we have recently reported common variants with large cumulative effects [6]. The genotyping of the six risk alleles was performed but only one risk allele was found in individual II-2.
Our study also points to the complex interpretation of BrS genetic testing and more specifically for the SCN5A gene. Indeed, based on the SCN5A molecular diagnosis usually proposed to the first degree relatives, patient II-3 or his siblings would not have been diagnosed for BrS.
As in this family, we do need to revisit the genetic model and the molecular diagnostics of BrS patients/families and search for new variants in other candidate genes to improve (1) the genotype/phenotype relationship of this syndrome (2) the pre-symptomatic detection within relatives and (3) the risk stratification.
References
Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP Jr, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haïssaguerre M, Schimpf R, Borggrefe M, Wolpert C (2007) Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115:442–449. doi:10.1161/CIRCULATIONAHA.106.668392
Behr ER, Casey A, Sheppard M, Wright M, Bowker TJ, Davies MJ, McKenna WJ, Wood DA (2007) Sudden arrhythmic death syndrome: a national survey of sudden unexplained cardiac death. Heart 93:601–605. doi:10.1136/hrt.2006.099598
Bellocq C, Wilders R, Schott JJ, Louérat-Oriou B, Boisseau P, Le Marec H, Escande D, Baró I (2004) A common antitussive drug, clobutinol, precipitates the long QT syndrome 2. Mol Pharmacol 66:1093–1102. doi:10.1124/mol.104.001065
Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, Rhodes TH, George AL Jr (2003) Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest 112:1019–1028. doi:10.1172/JCI18062
Berne P, Brugada J (2012) Brugada syndrome 2012. Circ J 76:1563–1571. doi:10.1253/circj.CJ-12-0717
Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, Verkerk AO, Schwartz PJ, Crotti L, Dagradi F, Guicheney P, Fressart V, Leenhardt A, Antzelevitch C, Bartkowiak S, Borggrefe M, Schimpf R, Schulze-Bahr E, Zumhagen S, Behr ER, Bastiaenen R, Tfelt-Hansen J, Olesen MS, Kääb S, Beckmann BM, Weeke P, Watanabe H, Endo N, Minamino T, Horie M, Ohno S, Hasegawa K, Makita N, Nogami A, Shimizu W, Aiba T, Froguel P, Balkau B, Lantieri O, Torchio M, Wiese C, Weber D, Wolswinkel R, Coronel R, Boukens BJ, Bézieau S, Charpentier E, Chatel S, Despres A, Gros F, Kyndt F, Lecointe S, Lindenbaum P, Portero V, Violleau J, Gessler M, Tan HL, Roden DM, Christoffels VM, Le Marec H, Wilde AA, Probst V, Schott JJ, Dina C, Redon R (2013) Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet 45:1044–1049. doi:10.1038/ng.2712
Brugada P, Brugada J (1992) Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol 20:1391–1396. doi:10.1016/0735-1097(92)90253-J
Burashnikov E, Pfeiffer R, Barajas-Martinez H, Delpón E, Hu D, Desai M, Borggrefe M, Häissaguerre M, Kanter R, Pollevick GD, Guerchicoff A, Laiño R, Marieb M, Nademanee K, Nam GB, Robles R, Schimpf R, Stapleton DD, Viskin S, Winters S, Wolpert C, Zimmern S, Veltmann C, Antzelevitch C (2010) Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 7:1872–1882. doi:10.1016/j.hrthm.2010.08.026
Chugh SS, Kelly KL, Titus JL (2000) Sudden cardiac death with apparently normal heart. Circulation 102:649–654. doi:10.1161/01.CIR.102.6.649
Clatot J, Ziyadeh-Isleem A, Maugenre S, Denjoy I, Liu H, Dilanian G, Hatem SN, Deschênes I, Coulombe A, Guicheney P, Neyroud N (2012) Dominant-negative effect of SCN5A N-terminal mutations through the interaction of Nav1.5 alpha-subunits. Cardiovasc Res 96:53–63. doi:10.1093/cvr/cvs211
Dolmatova E, Mahida S, Ellinor PT, Lubitz SA (2013) Genetic etiology and evaluation of sudden cardiac death. Curr Cardiol Rep 15:389. doi:10.1007/s11886-013-0389-8
Dolz-Gaitón P, Núñez M, Núñez L, Barana A, Amorós I, Matamoros M, Pérez-Hernández M, González de la Fuente M, Alvarez-López M, Macías-Ruiz R, Tercedor-Sánchez L, Jiménez-Jáimez J, Delpón E, Caballero R, Tamargo J (2013) Functional characterization of a novel frameshift mutation in the C-terminus of the Nav1.5 channel underlying a Brugada syndrome with variable expression in a Spanish family. PLoS One 8:e81493. doi:10.1371/journal.pone.0081493
Gima K, Rudy Y (2002) Ionic current basis of electrocardiographic waveforms: a model study. Circ Res 90:889–896. doi:10.1161/01.RES.0000016960.61087.86
Herfst LJ, Potet F, Bezzina CR, Groenewegen WA, Le Marec H, Hoorntje TM, Demolombe S, Baró I, Escande D, Jongsma HJ, Wilde AA, Rook MB (2003) Na+ channel mutation leading to loss of function and non-progressive cardiac conduction defects. J Mol Cell Cardiol 35:549–557. doi:10.1016/S0022-2828(03)00078-6
Hoshi M, Du XX, Shinlapawittayatorn K, Liu H, Chai S, Wan X, Ficker E, Deschênes I (2014) Brugada syndrome disease phenotype explained in apparently benign sodium channel mutations. Circ Cardiovasc Genet 7:123–131. doi:10.1161/CIRCGENETICS.113.000292
Hu D, Barajas-Martínez H, Terzic A, Park S, Pfeiffer R, Burashnikov E, Wu Y, Borggrefe M, Veltmann C, Schimpf R, Cai JJ, Nam GB, Deshmukh P, Scheinman M, Preminger M, Steinberg J, López-Izquierdo A, Ponce-Balbuena D, Wolpert C, Haïssaguerre M, Sánchez-Chapula JA, Antzelevitch C (2014) ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int J Cardiol 171:431–442. doi:10.1016/j.ijcard.2013.12.084
Iyer V, Mazhari R, Winslow RL (2004) A computational model of the human left-ventricular epicardial myocyte. Biophys J 87:1507–1525. doi:10.1529/biophysj.104.043299
Kobrinsky E, Tiwari S, Maltsev VA, Harry JB, Lakatta E, Abernethy DR, Soldatov NM (2005) Differential role of the alpha1C subunit tails in regulation of the CaV1.2 channel by membrane potential, beta subunits, and Ca2+ ions. J Biol Chem 280:12474–12485. doi:10.1074/jbc.M412140200
Letsas KP, Gavrielatos G, Efremidis M, Kounas SP, Filippatos GS, Sideris A, Kardaras F (2007) Prevalence of Brugada sign in a Greek tertiary hospital population. Europace 9:1077–1080. doi:10.1093/europace/eum221
Mezghrani A, Monteil A, Watschinger K, Sinnegger-Brauns MJ, Barrère C, Bourinet E, Nargeot J, Striessnig J, Lory P (2008) A destructive interaction mechanism accounts for dominant-negative effects of misfolded mutants of voltage-gated calcium channels. J Neurosci 28:4501–4511. doi:10.1523/JNEUROSCI.2844-07.2008
Neyroud N, Ziyadeh-Isleem A, Clatot J, Deschenes I, Coulombe A, Guicheney P (2014) Role of the N- and distal C-terminal domains in Nav1.5 alpha-subunit interaction. Cardiovasc Res 103(Suppl 1):S68. doi:10.1093/cvr/cvu091.54
Ng PC, Henikoff S (2001) Predicting deleterious amino acid substitutions. Genome Res 11:863–874. doi:10.1101/gr.176601
Page KM, Heblich F, Margas W, Pratt WS, Nieto-Rostro M, Chaggar K, Sandhu K, Davies A, Dolphin AC (2010) N terminus is key to the dominant negative suppression of Cav2 calcium channels: implications for episodic ataxia type 2. J Biol Chem 285:835–844. doi:10.1074/jbc.M109.065045
Pambrun T, Mercier A, Chatelier A, Patri S, Schott JJ, Le Scouarnec S, Chahine M, Degand B, Bois P (2014) Myotonic dystrophy type 1 mimics and exacerbates Brugada phenotype induced by Nav1.5 sodium channel loss-of-function mutation. Heart Rhythm 11:1393–1400. doi:10.1016/j.hrthm.2014.04.026
Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G, Tracy C (2013) HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 10:1932–1963. doi:10.1016/j.hrthm.2013.05.014
Probst V, Allouis M, Sacher F, Pattier S, Babuty D, Mabo P, Mansourati J, Victor J, Nguyen JM, Schott JJ, Boisseau P, Escande D, Le Marec H (2006) Progressive cardiac conduction defect is the prevailing phenotype in carriers of a Brugada syndrome SCN5A mutation. J Cardiovasc Electrophysiol 17:270–275. doi:10.1111/j.1540-8167.2006.00349.x
Probst V, Wilde AA, Barc J, Sacher F, Babuty D, Mabo P, Mansourati J, Le Scouarnec S, Kyndt F, Le Caignec C, Guicheney P, Gouas L, Albuisson J, Meregalli PG, Le Marec H, Tan HL, Schott JJ (2009) SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ Cardiovasc Genet 2:552–557
Ramensky V, Bork P, Sunyaev S (2002) Human non-synonymous SNPs: server and survey. Nucleic Acids Res 30:3894–3900. doi:10.1093/nar/gkf493
Risgaard B, Jabbari R, Refsgaard L, Holst AG, Haunsø S, Sadjadieh A, Winkel BG, Olesen MS, Tfelt-Hansen J (2013) High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin Genet 84:489–495. doi:10.1111/cge.12126
Schott JJ, Alshinawi C, Kyndt F, Probst V, Hoorntje TM, Hulsbeek M, Wilde AA, Escande D, Mannens MM, Le Marec H (1999) Cardiac conduction defects associate with mutations in SCN5A. Nat Genet 23:20–21. doi:10.1161/CIRCGENETICS.109.853374
Schulze-Bahr E, Eckardt L, Breithardt G, Seidl K, Wichter T, Wolpert C, Borggrefe M, Haverkamp W (2003) Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum Mutat 21:651–652. doi:10.1002/humu.9144
Six I, Hermida JS, Huang H, Gouas L, Fressart V, Benammar N, Hainque B, Denjoy I, Chahine M, Guicheney P (2008) The occurrence of Brugada syndrome and isolated cardiac conductive disease in the same family could be due to a single SCN5A mutation or to the accidental association of both diseases. Europace 10:79–85. doi:10.1093/europace/eum271
Tang ZZ, Liao P, Li G, Jiang FL, Yu D, Hong X, Yong TF, Tan G, Lu S, Wang J, Soong TW (2008) Differential splicing patterns of L-type calcium channel Cav1.2 subunit in hearts of Spontaneously Hypertensive Rats and Wistar Kyoto Rats. Biochim Biophys Acta 1783:118–130. doi:10.1016/j.bbamcr.2007.11.003
Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT (1995) SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80:805–811. doi:10.1016/0092-8674(95)90359-3
Acknowledgments
We are greatly indebted to the patients. We thank Dr Alexandre Duparc (Pôle cardiovasculaire et Métabolique de Cardiologie B, Hôpital de Rangueil, Toulouse, France), Stéphanie Chatel and Christine Fruchet for assistance in the family recruitment; Estelle Baron, Stéphanie Bonnaud, Laëtitia Duboscq-Bidot, Béatrice Leray, and Aurore Girardeau for technical assistance; Jean-Baptiste Chéron for his help in action potential modelling; and Philippe Hulin at Imaging Core Facility (MicroPICell, SFR François Bonamy, University of Nantes) for expert technical assistance with FRAP. We are also grateful to the Genomic Platform of Nantes (Biogenouest Genomics) for technical support, to Raluca Teusan and Pierre Lindenbaum for the bioinformatics support and to Anne Ponchaux, Jerome Buscail, Thierry Marsaud, Prof Stéphane Bézieau (Service de génétique médicale, Nantes) for their assistance in SCN5A sequencing. We are also grateful to the referral centre against inherited cardiac arrhythmias of Nantes and Rennes. This work was supported by the Leducq Foundation [CVD-05; Alliance Against Sudden Cardiac Death]; and by the French Ministry of Health (P.H.R.C.I. DGS2001/0248).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
D. M. Béziau and J. Barc are contributed equally to this work.
J.-J. Schott, V. Probst and I. Baró authors jointly directed this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Béziau, D.M., Barc, J., O’Hara, T. et al. Complex Brugada syndrome inheritance in a family harbouring compound SCN5A and CACNA1C mutations. Basic Res Cardiol 109, 446 (2014). https://doi.org/10.1007/s00395-014-0446-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-014-0446-5