
Abstract
Purpose
Infant gut microbiota which plays an important role in long-term health is mainly shaped by early life nutrition. However, the effect of nutrients on infants gut microbiota is less researched. Here, we present a study aiming to investigate in vitro a modified formula that is supplemented with milk fat globule membrane (MFGM) that were missing in common formulas when compared with human milk and to assess the impact of feeding scheme on microbiota and metabolism.
Methods
A total of 44 infants including 16 from breast milk feeding, 13 from common formula feeding and 15 from modified formula feeding were analyzed, and A cross-sectional sampling of fecal and urine was done at 1 month-of-age. Stool microbiota composition was characterized using high-throughput DNA sequencing, and urinary metabolome was profiled by nuclear magnetic resonance (NMR). In vitro growth experiment of Bifidobacterium with key components from MFGM was performed and analyzed by both DNA and RNA.
Results
Stool samples from the infants who were breastfed had a higher relative abundance of Bifidobacterium and a lower relative abundance of Escherichia than the formula-fed infants. The stool microbiome shifts were associated with urine metabolites changes. Three substances including lactadherin, sialic acid and phospholipid, key components of MFGM were significantly positively correlated to Bifidobacterium of stool samples from infants, and stimulated the growth rate of Bifidobacterium significantly by provided energy in vitro growth experiment with RNA analysis.
Conclusions
These findings suggest that the key components from MFGM could improve infants’ health by modulating the gut microbiome, and possibly supporting the growth of Bifidobacterium.
Registration
Clinicaltrials.gov NCT02658500 (registered on January 20, 2016).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gut microbiome’s major role in human health is being increasingly understood and emphasized in recent years. Various gut microbes are essential for the normal function of host immune system [1]. In adults, many disease-oriented studies illustrated associations between nutrition and gut microbiota or metabolites of the gut microbiota [2,3,4]. In contrast, more studies are needed to understand the structuring forces of the infant gut microbiome [5, 6].
Infants acquire gut microbiome at birth, and studies of infant meconium suggest that bacteria are present in the foetal gut prior to birth [7]. The infant’s gastrointestinal microbiome gradually matures over time after birth. This period is also recognized as a key window of immune system development, during which immune tolerance is built for common microbes. It has been reported that a dysbiosis in the gut microbiome may underlie the development of type I diabetes and allergies before 4 years old [8]. Understanding the structuring factors and the possible modulation of the gut microbiome development in infants would thus be vitally important for improving infant health and preventing certain diseases, even into adulthood.
Patterns of infant feeding would impact the development of gut microbiome [9,10,11]. Human milk contains essential nutrients including protein, fat, carbohydrates and vitamins primarily for the infants, and some of them would be utilized by microbes. Milk fat globule membrane (MFGM) is a unique three-layered biological membrane surrounding the milk fat globule. There are approx 76% protein, 7% phospholipids and 1.9% sialic acid in MFGM with bioactivity of anti-adhesion of bacteria and viruses, which are beneficial for the infant gut microbiome [12, 13]. In contrast, common formulas may lack those key components with essential functions in modulating gut microbiome, and could be accounted for the observed differences between breast-fed and formula-fed infant’s gut microbiome such as Bifidobacterium [14,15,16]. More importantly, the health consequences associated with feeding pattern and gut microbiome differences call for a mechanistic investigation and potential interventions in infants, as it has been demonstrated that formula-fed infants have a higher tendency in developing asthma, type I diabetes compared to those breastfed [17, 18]. However, the effect of MFGM on Bifidobacterium which is beneficial for infant health such as antiallergy and antidiabetic activity has not been proven [19, 20].
Here, we studied a modified formula that is supplemented with specific proteins and fat that were from MFGM but missing in common formulas when compared to human milk. We hypothesize that MFGM components could promote gut Bifidobacterium growth in infant. The effect of modified formula on stool microbiome was analyzed. The influence on infant metabolism was investigated by relation analysis between stool microbiome and urine metabolites. The mechanism was researched by in vitro growth experiment (Supplemental Fig. 1).
Methods
Study design
We have administrated three different feeding strategies to infants from birth to 1 month of age, including breastfeeding (BF), common formula feeding (FF1) and modified formula feeding (FF2), the average nutrient compositions of which are listed in Table 1. MFGM, rich in lipids, is applied in the modified formula, leading to higher content of phospolipid and sfingolipid compared to breast milk. Formula-fed infants were randomized to FF1 or FF2. Formula taken records and the empty formula box were collected to ensure the fidelity of the intervention.
Infant study population
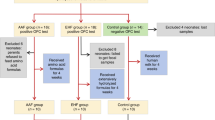
The Ethics Committee of Beijing Ditan Hospital affiliated with Capital Medical University approved all aspects of the study (#2015-027-01), and informed consent was obtained from all of the subjects. The procedures were in accordance with the ethical standards. Study subjects were recruited from Tongzhou Maternal and Child Health Hospital (Tongzhou, Beijing, China) and Maternal and Child Health Hospital of Liuyang (Liuyang, Hunan, China).
The primary outcome measure was the relative abundance of gut Bifidobacterium in stool samples between human milk-fed infant and formula0fed infant at 1 month. To demonstrate a clinically significant difference in the relative abundance of Bifidobacterium by 76% between two groups (i.e. from 54 to 13%) with 90% power and an alpha value of 0.05, we estimated a sample size of 13 infants in each group for a total of 39 infants [14].
This report, which is a cross-sectional analysis of a single time point, was comprised of 44 participants (16 from BF, 13 from FF1 and 15 from FF2 group) whose data passed all quality control criteria (See details below). All analyses involving stool microbiome and urine metabolites data were restricted to 44 individuals (Supplemental Fig. 1 and Supplemental Table 1).
Criteria
Newborns were eligible to be included if they were: healthy term newborns (gestational age ranges from 37 to 41 weeks) with a birth weight range from 2500 to 4000 g appropriate for gestational age and Apgar scores larger than 7. Infants were excluded from the entire study if any the following exclusion criteria were met at any time of the study: diagnosis of a significant chronic medical condition including: HIV infection, cancer, bone marrow or organ transplantation, blood product administration within the last 3 months, bleeding disorder, known congenital malformation or genetic disorder; if the parent or legal guardian was unable to read and/or comprehend Chinese; if the family moved outside of the city they lived during the study period; if infant take antibiotics during the research period. Feeding practices were assessed by careful chart review and classified as exclusive formula or breast-fed within the first month of life.
Sampling and growth data collecting
Parents were trained for fecal and urine collection when they agreed to participate in the research. At the age of 1 month (± 2 days), fecal samples were collected by parents at home from diapers after defaecation, immediately put into the stool collection tubes with stool DNA stabilizer (Stratec, Germany) and stored at − 20 °C until they were transported (in dry ice, within 24 h) to hospital. Urine was collected by parents at home with urine collection bag which was sticked on the uriding area and transferred into sterile tubes and stored at − 20 °C until they were transported to the hospital (in dry ice, within 24 h). Then the samples were stored at − 80 °C until analyses. Growth data including height, weight, and disease frequency were also collected at the same time.
High-throughput sequencing of the 16S rRNA gene of the stool microbiome
Genomic DNA was extracted from 100 mg of homogenized fecal samples using the QIAamp Fast DNA Stool Mini Kit (Qiagen, GmbH, Hilden, Germany) following the manufacturer’s protocol. PCR amplification of the V3-V4 region of 16S rRNA genes (primers 341F: CCTACGGGNGGCWGCAG and 805R: GACTACHVGGGTATCTAATCC) was performed with 10 ng DNA as a template, using 15 μL of Phusion High-Fidelity PCR Master Mix (New England Biolabs), 0.2 μM of forward and reverse primers in a 30 μL total reaction volume. The PCR program included 3 min at 95 °C, 25 cycles of 30 s at 95 °C and 30 s at 55 °C, and then 30 s at 72 °C. Sequencing libraries were generated using NEB Next Ultra DNA Library Prep Kit for Illumina (NEB, USA) following the manufacturer’s recommendations. Amplicons were sequenced using 2 × 250 bp paired-end protocol by Illumina HiSeq 2500 according to the manufacturer’s instructions.
Infant urine NMR metabolites
Samples were centrifuged at 15,870 rcf for 2 min at 4 °C, the 540 μL aqueous layer was transferred to a clean 2 mL centrifuge tube. 60 μL 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (DSS) standard solution (Anachro, Canada) was added. Samples were mixed well before transfer to 5 mm NMR tube (Norwell, USA). Spectra were collected using a nuclear magnetic resonance (NMR) spectroscopy (Bruker AV III 600 MHz). The first increment of a 2D-1H, 1H-NOESY pulse sequence was utilized for the acquisition of 1H NMR data and for suppressing the solvent signal. Experiments used a 100 ms mixing time along with a 990 ms pre-saturation (~ 80 Hz gamma B1). Spectra were collected at 25 °C, with a total of 128 scans over a period of 15 min. The collected Free Induction Decay (FID) signal was automatically zero filled and Fourier transform in Processing module in Chenomx NMR Suite 8.1 (Chenomx Inc., Edmonton, Canada). The data was then carefully phased and baseline corrected in Chenomx Processor. All the spectra were referenced to the internal standard, DSS and analyzed against Chenomx Compound Library. All metabolites’ concentration was normalized by weight across all parallel samples before used in the later on multivariable analysis.
A total of 77 metabolites concentrations and derived measures were obtained from the NMR spectroscopy platform. These metabolites cover several types of metabolite classes, including vitamin/cofactors, organic acids, amino acids, amino acid derivatives, ketones, nucleic acid components, ammoniums compounds, amines, sugars, alcohols, and amides.
In vitro growth experiment
Bacteria strain B. longum subsp. infantis DSMZ20090, provided by China General Microbiological Culture Collection Center (CGMCC) (http://www.cgmcc.net/) was used for the in vitro growth experiment. B. infantis were first cultured in TPY (Trypticase Soy-Yeast) liquid broth under anaerobic conditions with Anaerocult A system (Merck, Darmstadt, Germany) for 48 h, and then diluted to 105 cells/mL in each media. Key MFGM components (lactadherin, L; sialic acid, S; phospholipid, P) were provided by Arla Foods (Denmark), and added to TPY medium alone (L, S, P), or combining two out of three (L + S, L + P, S + P), or all three components (L + S + P), with concentration identical to that of BF provided in Table 1. TPY medium without additives was used as a control. All groups were grown in triplicate under anaerobic conditions in a modular atmosphere-controlled system (Davidson and Hardy, Belfast, Ireland) at 37 °C for 18 h to reach mid-exponential phase, and the optical density at 600 nm (OD600) was determined, using a Shimadzu UV-1280 UV-399 VIS Spectrophotometer (Shimadzu Corp., Kyoto, Japan) directly after inoculation as well as after 18 h; bacterial growth was measured as changes in OD600. After growth, tubes were centrifuged at 10,000×g for 5 min, supernatant was removed, and the pellet was re-suspended in 1 mL of Trizol, and the RNA was extracted and purified using RNeasy Mini Kit (Qiagen, Hilden, Germany, 74104) following the instructions. RNA libraries were prepared using a TruSeq Stranded Total Library Preparation Kit (Illumina, USA) following the instructions. Samples were sequenced at the Institute of Microbiology, Chinese Academy of Sciences, using 150-bp pair-end reads chemistry on a HiSeq 2500 instrument.
Bioinformatic analysis
USEARCH [21] and VSEARCH [22] were used to quality filter, cluster, and removing chimera of demultiplexed 16S rRNA raw sequencing data of stool sample. The clustered sequences at 97% similarity level were utilized to construct operational taxonomy unit (OTU) tables and representative sequences were classified into the respective taxonomical level from phylum to species based on the RDP 16S rRNA gene database [23].
All statistical analyses were performed in the statistical computing language R [24]. For 16S rRNA data, total sum normalization (TSS) was firstly used to normalize count data by dividing feature read counts by the total number of reads in each sample. Rarefaction analysis, principal coordinates analysis (PCoA) using Bray–Curtis distance of stool microbiome data were performed using Phyloseq [25] with vegan (https://cran.r-project.org/web/packages/vegan/index.html) and ade4 [26] packages. Boxplots with significance level were plotted using “ggpubr” package version 0.1.6 (https://cran.r-project.org/web/packages/ggpubr/index.html). Correlation of microbes and metabolites were performed using Spearman correlation in the R package “neuropsychology” package 0.5.0 [27], 16S rRNA abundance data were TSS and square root transformed. Urinary metabolites data were log2-transformed, and the p value was corrected by false discovery rate (FDR) (< 0.01). RNA-Seq raw reads were quality control using FastQC [28], then trimmed into clean reads using Trim-Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Next, clean reads were aligned to the reference genome (NCBI Accession ID: GCF_000020425.1) with HISAT2 [29], then.sam files were converted to.bam files using SamTools [30], and an expression table was generated using featureCounts of Subread [31]. Differential expression analysis was performed using DESeq2 [32]. KEGG pathway annotation was performed using clusterProfiler [33] and DOSE [34] packages in R.
Results
Feeding strategies lead to different microbiome in infants
We first analyzed the alpha-diversity of each group, both Simpson index and Shannon evenness were higher in the BF group than FF1 and FF2 (Supplemental Fig. 2), while Chao 1 index was higher in FF1 group than BF and FF2. Further beta-diversity analysis revealed that stool microbiota of BF group was also distinct from that of FF1 and FF2 (Supplemental Fig. 3). Overall, 40.28% of the total microbiota variations could be explained by the feeding scheme.
We further examined the urine metabolites, anthropometric measures as well as clinical parameters among the three groups. In urine metabolites, 10 out of 77 tested metabolites showed significant differences among the three groups (FDR < 10%) (Fig. 1). Hippurate was significantly lower in FF2 group than that in FF1 group but was not different significantly from that in BF group. Creatine was significantly lower in BF group than that in both FF2 group and FF1 group.

Differential urinary metabolites of three feeding schemes. The analysis was performed using Kruskal–Wallis test with posthoc analysis by Wilcoxon-test (0.05 level). Black dot show the result of each sample. BF breastfeeding, FF1 common formula feeding, FF2 modified formula feeding
Among the infants’ anthropometric measures as well as clinical parameters of infant, FF2 group and FF1 group were significantly lower in height and weight compared to BF both at birth and 1 month, while the growth of height and weight and other anthropometric measures, clinical parameters at 1 month were not significantly different from BF (Supplemental Table 2).
Bifidobacterium as key modulator of stool microbiome
We then have performed further compositional analysis of the stool microbiota from different groups, especially at the genus level. Among all major genera (average relative abundance > 1%), Bifidobacterium was found to be the main source of differences in infants fed different strategies. The average relative abundance of Bifidobacterium for three groups reaches 12% in our study, and 29.89%, 2.66% and 2.27% in BF, FF1 and FF2 groups, respectively (Supplemental Fig. 4). Negatively correlated with Bifidobacterium, Veillonella was among the major genera that are different among the three groups, accounting for 0.26%, 6.80% and 5.06% in BF, FF1 and FF2 groups, respectively; and Escherichia/Shigella accounting for 0.03%, 8.48% and 8.03% in BF, FF1 and FF2 groups, respectively.
We further performed an association analysis between the different nutrients measured within BF, FF1 and FF2 formula and the relative abundances of the major genera, especially Bifidobacterium (Fig. 2). Three nutrient components, lactadherin, sialic acid and phospholipid showed significant positive correlations to the relative abundance of Bifidobacterium and consequently negative correlation to that of Veillonella and Escherichia/Shigella (Figs. 2 and 3); they are the major active components within MFGM, a complex structure surrounding milk fat globules secreted in human and other mammals’ milk. Other components including mucin, lactoferrin, IgG and sphingomyelins demonstrated a negative correlation with Veillonella and Escherichia/Shigella as well, yet they do not correlate with Bifidobacterium. Mucin and lactoferrin are negatively correlated with Bacteroides as well as Clostridium, and despite they are usually key members of adult gut microbiota, in infants their abundances are relatively low (9% and 16% on average, respectively, Supplemental Fig. 4).

Association analysis and heatmap visualization of nutrients measured within formula and the relative abundances of the gut microbial genera. The red and blue color demonstrated a significant positive and negative association between the nutrients and the microbiome, respectively

Regression of three nutrient components, lactadherin, sialic acid and phospholipid with the relative abundances of Bifidobacterium and Escherichia/Shigella. Black dot show the result of each sample. X axis stands for the content of nutrient component, Y axis stands for the relative abundance of microbial genera
Lastly, we investigated the potential sources of differential metabolites within the infants’ urine by performing an association analysis of stool microbiota members to that of metabolites (Supplemental Table 3). In total, 40 correlations, including 24 positive correlations and 16 negative correlations were found with a FDR of 10%. Of all metabolites with significant differences among the three groups, creatine and benzoate showed a positive correlation with Veillonella. None of any metabolites were found to significantly correlate with both Bifidobacterium and Escherichia/Shigella. Benzoate was positively correlated with Veillonella, as well as other short-chain fatty acids (e.g., propionate).
MFGM components stimulates Bifidobacterium growth in vitro
Since the concentrations of lactadherin, sialic acid and phospholipid are correlated within human milk as well as in modified formula, it is yet difficult to elucidate the effects of the three components simply from infants’ stool microbiota data. Moreover, the effect of three components in modified formula on infant stool microbiota is not yet clear. To investigate whether the Bifidobacterium enrichment effect comes from single components or rather a combination and the effect of three components in FF2 formula, we have used one Bifidobacterium longum subsp. infantis strain isolated from infant fecal samples, and tested its growth on simple TPY (Trypticase Soy-Yeast extract) liquid medium versus addition of one, two or all three components, of which the concentrations reflect those in FF2 formula, each with three technical replicates. After 18 h of anaerobic growth under the same temperature (37 °C), bacterial growth was measured with OD600 absorption (with blank medium as background). Interestingly, the only medium with all three added showed significantly higher bacterial cell density compared to the control medium while adding one or two components did not lead to a significant increase in cell density (Fig. 4).

OD600 nm absorption of bacterial growth under different combinations of nutrient components. Control stand for control TPY medium. Black dots show three replicates of each group. MFGM components (lactadherin, sialic acid, phospholipid). The analysis was performed using t test (0.05 level), which compared each group with control separately
Finally, to investigate the potential mechanism underlying the Bifidobacterium enrichment under MFGM components, we isolated total RNA from strains grown in a simple medium of triplicate as well as medium with all three components added, and carried out transcriptome analysis with RNA-Seq on Illumina HiSeq. Data were analyzed with Hisat2 using B. infantis reference genome sequences, and differential genes were defined as adjusted p < 0.05 in paired t tests. This analysis revealed 74 genes that were differentially expressed between the two culture conditions, and top enriched genes with all three components included 2-hydroxyacid dehydrogenase (gene2390), 4-hydroxy-tetrahydrodipicolinate synthase (gene2170), and ABC transporter ATP-binding protein (gene2468) (Fig. 5). A gene enrichment analysis revealed that glyoxylate and dicarboxylate metabolism pathway (p = 0.0001) is among the top enriched pathways in Bifidobacterium with MFGM components (Supplemental Fig. 5).

Heatmap visualization of FPKM (Fragments Per Kilobase per Million mapped reads) abundances of differentially expressed genes in Bifidobacterium grown in medium with or without all three components (lactadherin-L, sialic acid-S, phospholipid-S) added. “1” and “2” stand for technical replicates of RNA sequencing of the two groups
Discussion
Diet is known to be a potent and persistent modifier of the microbiota in both adults and children [35, 36] and breastfeeding is one of the main factors shaping infants’ gut microbiota [37]. Previous studies showed that breastfeeding versus non-breastfeeding significantly influenced the gut microbiome [38, 39]. Digestion and metabolism of nutrients are likely influenced by the intestinal microbiome [37], and there is a well-established connection between breast feeding and lower risk for childhood and adult-onset obesity possibly mediated in part by the microbiome in early life [40]. Compared with formula-fed infants, breastfed infants showed an impact on several gut microbes, such as Escherichia coli, Veillonella dispar and Ruminococcus gnavus as well as Klebsiella, known to be pro-inflammatory or opportunistic pathogens, and Bifidobacterium longum, who plays an important role in maintaining a healthy gut environment especially in infants, as reported by previous studies [11, 35]. In most of the cases, no significant alpha-diversity was found between breast- and formula-fed infants [10]. Some cases found that the alpha-diversity of gut mirobiota of breast-fed infant was higher than that of formula-fend infants [10]. This might be due to the components of infant formula.
In this study, we studied a modified formula that is partially replenished with specific protein and fat components that were missing in common formulas when compared with human milk. Three nutrient components added in the modified formula were positively related to Bifidobacterium and really enriched Bifidobacterium in vitro experiment.
Adding three nutrient components, lactadherin, sialic acid and phospholipid shifts stool microbiome of three feeding strategies
Assessment of association between major genera of stool microbiome and nutrition components revealed that those three nutrient components, lactadherin, sialic acid and phospholipid were significantly and positively correlated with Bifidobacterium and negatively correlated with Veillonella and Escherichia/Shigella. MFGM components were reported to resist several pathogens in vitro, such as Campylobacter jejuni, Salmonella enteriditis, and Listeria monocytogenes [41], and also protect against pathogens in vivo, such as L. monocytogenes. Addition of MFGM in formula also showed many beneficial effects, such as leading to a microbiome more similar to that of mother’s milk-fed rat, protecting against C. difficile toxins [13]. MFGM also modulates microbiota in mouse studies, but no effect on Bifidobacterium is mentioned [13].
In this study, it was shown that Bifidobacterium was stimulated in vitro by the combination of lactadherin, sialic acid and phospholipid. Thus, the combination of lactadherin, sialic acid and phospholipid is required for the enrichment of Bifidobacterium. Three components may also enrich Bifidobacterium of infant. In-depth RNA-Seq sequencing showed that 2-hydroxyacid dehydrogenase, 4-hydroxy-tetrahydrodipicolinate synthase, ABC transporter ATP-binding protein genes were differentially expressed in the existence of these three components. These differentially expressed genes were significantly enriched in glyoxylate and dicarboxylate metabolism pathway. This pathway belongs to energy and carbohydrate metabolism, both of which are critical for the tricarboxylic acid cycle (TCA cycle), and therefore, it could be a potential mechanism for stimulating the growth of Bifidobacterium [42]. It might be those components that provide energy specifically towards Bifidobacterium. However, the abundance of Bifidobacterium in FF2 group was not higher than that of FF1 group. A possible reason could be interactions between MFGM and other components of FF2.
Change of microbiota correlated with metabolites and health
Urine contains metabolic breakdown products from various sources, such as foods, drinks, and microbial by-products [43]. Metabolites, which were affected by gut microbiota, are directly associated with health conditions and disease phenotypes [44].
Hippurate content in three group increased as BF < FF2 < FF1. The difference was significant between BF and FF1, FF2 and FF1 but not between FF2 and BF (Fig. 1). Hippurate is synthetized by the liver and kidney with glycine and benzoic acid. Benzoic acid is a metabolite produced by intestinal bacteria from aromatic compounds of diet [45]. Therefore, hippurate, together with Trimethylamine N-oxide, acetate and formate, were generally accepted as metabolites sourced from the gut microbiome [46]. It could be deduced that FF2 alters the microbiome regarding hippureate in a way that it becomes more similar to that of breastfed infants.
Creatine content in three groups increased as BF < FF2 < FF1. The difference was not significant between FF2 and FF1 but significant between FF2 and BF as well as FF1 and BF. Creatine is an amino acid derivative participated in cellular energy production, which is known as an anti-inflammation agent [47] and can improve maintenance of ATP content and reduce oxidative stress [48]. Creatine was positively associated with Veillonella (the correlation coefficient was 0.48) in this study. Veillonella, which was 0.26%, 6.80% and 5.06% in BF, FF1, FF2, respectively, participated in the modulation of linked pathways including short-chain fatty acids production [49]. It is resident of oral cavity, genito-urinary, respiratory, and intestinal tracts of humans, and can cause severe human infections such as endocarditis, osteomyelitis, and prosthetic joint infection [50]. Lower content of creatine and Veillonella demonstrate better health status in BF and FF2 group.
Taken together, this study suggests that the key components from MFGM could improve infants’ health by modulating the gut microbiome, and possibly supporting the growth of Bifidobacterium. These results offer a first step in changing formula for the good of children. Our follow-on studies will expand the number of recruits from more areas, potentially follow the infants for a longer time, and employ metagenomic sequencing to further profile the metabolic functions.
Data availability
The datasets generated and/or analyzed during the current study are not publicly available due to ethical and privacy considerations but are available from the corresponding author on reasonable request.
Code availability
Not applicable.
References
Human Microbiome Project Consortium T (2012) Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. https://doi.org/10.1038/nature11234
Qin JJ, Li YR, Cai ZM et al (2012) A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490:55–60. https://doi.org/10.1038/nature11450
Schulz MD, Atay Ç, Heringer J et al (2014) High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 514:508–512. https://doi.org/10.1038/nature13398
Ridaura VK, Faith JJ, Rey FE et al (2013) Gut microbiota from twins discordant for obesity modulate metabolism in XZmice. Science 341:1241214. https://doi.org/10.1126/science.1241214
Zimmermann P, Curtis N (2020) Breast milk microbiota: a review of the factors that influence composition. J Infect 81:17–47. https://doi.org/10.1016/j.jinf.2020.01.023
Sindi AS, Geddes DT, Wlodek ME et al (2021) Can we modulate the breastfed infant gut microbiota through maternal diet? FEMS Microbiol Rev. https://doi.org/10.1093/femsre/fuab011
Walker RW, Clemente JC, Peter I, Loos RJF (2017) The prenatal gut microbiome: are we colonized with bacteria in utero ? Pediatr Obes 12:3–17. https://doi.org/10.1111/ijpo.12217
Kostic ADD, Gevers D, Siljander H et al (2015) The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 17:260–273. https://doi.org/10.1016/j.chom.2015.01.001
Le Huërou-Luron I, Blat S, Boudry G (2010) Breast- v. formula-feeding: impacts on the digestive tract and immediate and long-term health effects. Nutr Res Rev 23:23–36. https://doi.org/10.1017/S0954422410000065
Guaraldi F, Salvatori G (2012) Effect of breast and formula feeding on gut microbiota shaping in newborns. Front Cell Infect Microbiol 2:1–4. https://doi.org/10.3389/fcimb.2012.00094
Bezirtzoglou E, Tsiotsias A, Welling GW (2011) Microbiota profile in feces of breast- and formula-fed newborns by using fluorescence in situ hybridization (FISH). Anaerobe 17:478–482. https://doi.org/10.1016/j.anaerobe.2011.03.009
Zhao LL, Du M, Gao J et al (2019) Label-free quantitative proteomic analysis of milk fat globule membrane proteins of yak and cow and identification of proteins associated with glucose and lipid metabolism. Food Chem 275:59–68. https://doi.org/10.1016/j.foodchem.2018.09.04413
Bhinder G, Allaire JM, Garcia C et al (2017) Milk fat globule membrane supplementation in formula modulates the neonatal gut microbiome and normalizes intestinal development. Sci Rep 7:45274. https://doi.org/10.1038/srep45274
Aimee MBD, Alaric WDS, Phillip IT, Barbara BW, Gautam D (2018) Infant diet and maternal gestational weight gain predict early metabolic maturation of gut microbiomes. Nat med 24:1822–1829. https://doi.org/10.1038/s41591-018-0216-2
Martin CR, Ling PR, Blackburn GL (2016) Review of infant feeding: key features of breast milk and infant formula. Nutrients 8:279. https://doi.org/10.3390/nu8050279
Brink LR, Katelin M, Piccolo BD et al (2019) Neonatal diet impacts bioregional microbiota composition in piglets fed human breast milk or infant formula. J Nutr 149:2236–2246. https://doi.org/10.1093/jn/nxz170
Johnson CC, Ownby DR (2017) The infant gut bacterial microbiota and risk of pediatric asthma and allergic diseases. Transl Res 179:60–70. https://doi.org/10.1016/j.trsl.2016.06.010
Stewart CJ, Ajami NJ, O’Brien JL et al (2018) Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 7728:583–588. https://doi.org/10.1038/s41586-018-0617-x
Cukrowska B, Biera JB, Zakrzewska M et al (2020) The Relationship between the infant gut microbiota and allergy. The role of Bifidobacterium breve and prebiotic oligosaccharides in the activation of anti-allergic mechanisms in early life. Nutrients 12:946. https://doi.org/10.3390/nu12040946
Li T, Yang JJ, Zhang HX et al (2020) Bifidobacterium from breastfed infant faeces prevent high-fat-diet-induced glucose tolerance impairment, mediated by the modulation of glucose intake and the incretin hormone secretion axis. J Sci Food Agric 100:3308–3318. https://doi.org/10.1002/jsfa.10360
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. https://doi.org/10.1093/bioinformatics/btq461
Rognes T, Flouri T, Nichols B, Quince C, Mahé F (2016) VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2409v1. https://doi.org/10.7717/peerj.2584
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. https://doi.org/10.1128/AEM.00062-0
R Core Team (2015) R: a language and environment for statistical computing. R Foundation for Statistical Computing. Available from http://www.r-project.org/
McMurdie PJ, Holmes S (2013) phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217. https://doi.org/10.1371/journal.pone.0061217
Dray S, Dufour A-BB (2007) The ade4 package: implementing the duality diagram for ecologists. J Stat Softw 22:1–20. https://doi.org/10.18637/jss.v022.i04
Makowski D (2016) Package “neuropsychology”: an R toolbox for psychologists, neuropsychologists and neuroscientists. Available from: https://github.com/neuropsychology/neuropsychology.R Accessed 30 June 2019
Patel RK, Jain M (2012) NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 7:e30619. https://doi.org/10.1371/journal.pone.0030619
Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL (2016) Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11:1650–1667. https://doi.org/10.1038/nprot.2016.095
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Liao Y, Smyth GK, Shi W (2013) The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41:e108–e108. https://doi.org/10.1093/nar/gkt214
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:1–34. https://doi.org/10.1186/s13059-014-0550-8
Yu G, Wang LG, Han Y, He QY (2012) ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16:284–287. https://doi.org/10.1089/omi.2011.0118
Yu G, Wang LG, Yan GR, He QY (2015) DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 31:608–609. https://doi.org/10.1093/bioinformatics/btu684
Cani PD, Hul MV (2020) Mediterranean diet, gut microbiota and health: when age and calories do not add up. Gut 69:1167–1168. https://doi.org/10.1136/gutjnl-2020-320781
Forbes JD, Azad MB, Lorena V et al (2018) Association of exposure to formula in the hospital and subsequent infant feeding practices with gut microbiota and risk of overweight in the first year of life. JAMA Pediatr 172:e181161. https://doi.org/10.1001/jamapediatrics.2018.1161
Newburg DS, Morelli L (2015) Human milk and infant intestinal mucosal glycans guide succession of the neonatal intestinal microbiota. Pediatr Res 77:115–120. https://doi.org/10.1038/pr.2014.178
Stearns JC, Zulyniak MA, de Souza RJ et al (2017) Ethnic and diet-related differences in the healthy infant microbiome. Genome Med 9:32. https://doi.org/10.1186/s13073-017-0421-5
Li N, Yan F, Wang N et al (2020) Distinct gut microbiota and metabolite profiles induced by different feeding methods in healthy Chinese infants. Front Microbiol 11:714. https://doi.org/10.3389/fmicb.2020.00714
Thompson AL (2012) Developmental origins of obesity: early feeding environments, infant growth, and the intestinal microbiome. Am J Hum Biol 24:350–360. https://doi.org/10.1002/ajhb.22254
Doueellou T, Montel MC, Sergentet DT (2017) Anti-adhesive properties of bovine oligosaccharides and bovine milk fat globule membrane-associated glycoconjugates against bacterial food enteropathogens. J Dairy Sci 100:3348–3359. https://doi.org/10.3168/jds.2016-11611
Underwood MA, German JB, Lebrilla CB, Mills DA (2015) Bifidobacterium longum subspecies infantis: champion colonizer of the infant gut. Pediatr Res 77:229. https://doi.org/10.1038/pr.2014.156
Bouatra S, Aziat F, Mandal R et al (2013) The human urine metabolome. PLoS ONE 8:e73076. https://doi.org/10.1371/journal.pone.0073076
Bowling FG, Thomas M (2014) Analyzing the metabolome. Methods Mol Biol 1168:31–45. https://doi.org/10.1007/978-1-4939-0847-9_3
Gonthier MP, Cheynier V, Donovan JL, Manach C, Morand C, Mila I, Lapierre C, Rémésy C, Scalbert A (2003) Microbial aromatic acid metabolites formed in the gut account for a major fraction of the polyphenols excreted in urine of rats fed red wine polyphenols. J Nutr 133(461–467):42. https://doi.org/10.1046/j.1365-277X.2003.00411.x
O’Sullivan A, He X, McNiven EMS, Hinde K, Haggarty NW, Lönnerdal B, Slupsky CM (2013) Metabolomic phenotyping validates the infant rhesus monkey as a model of human infant metabolism. J Pediatr Gastroenterol Nutr 56:355–363. https://doi.org/10.1097/MPG.0b013e31827e1f07
Riesberg LA, Weed SA, McDonald TL, Eckerson JM, Drescher KM (2016) Beyond muscles: the untapped potential of creatine. Int Immunopharmacol 37:31–42. https://doi.org/10.1016/j.intimp.2015.12.034
Cunha MP, Martin-De-Saavedra MD, Romero A, Javier E, Fabiana KL, Carla IT, Marcelo F, Ana LSR, Manuela GL (2014) Both creatine and its product phosphocreatine reduce oxidative stress and afford neuroprotection in an in vitro Parkinson’s model. ASN Neuro 6:1–6. https://doi.org/10.1177/1759091414554945
Muccioli GG, Naslain D, Bäckhed F, Reigstad CS, Lambert DM, Delzenne NM, Cani PD (2010) The endocannabinoid system links gut microbiota to adipogenesis. Mol Syst Biol 6:392. https://doi.org/10.1038/msb.2010.46
Carlier JP (2015) Veillonella. In: David H (ed) Bergey’s manual of systematics of archaea and bacteria, 2nd edn. John Wiley & Sons, Hoboken, pp 1–11
Acknowledgements
We thank the infants and their parents participating in this study for their trust. We also thank the study team for their involvement in this work, especially nurses and coordinators. We thank Dr Jufang Li and Dr Xueyan Dong for helping collecting samples, and we also thank Dr Jun Wang and Dr Yue Ma for bioinformatic analysis and revision.
Funding
This work was supported by National Natural Science Foundation of China No. 32072191, Beijing Science and Technology Plan No. Z201100008020005, Daxing District Major Scientific and Technological Achievements Transformation Project No. 2020006, National Key Research and Development Program No. 2019YFF0216702, Beijing Science and Technology Plan No. Z201100002620005.
Author information
Authors and Affiliations
Contributions
JZ: investigation, data curation, writing original draft preparation; WY: methodology, investigation, data curation; BL: data curation, writing original draft preparation; YD: methodology, investigation; TJ: methodology, investigation; SC: investigation; JW: investigation; BF: investigation; WQ: investigation; YL: investigation; HZ: investigation; JH: investigation; JH: methodology; LC: conceptualization, methodology, infant formula design, funding acquisition, resources.
Corresponding author
Ethics declarations
Conflict of interest
There was no conflicts of interest/competing interests for this article.
Ethical approval
The Ethics Committee of Beijing Ditan Hospital affiliated to Capital Medical University approved all aspects of the study (#2015-027-01).
Consent to participate
Informed consent was obtained from all of the parents.
Consent for publication
All authors were consent for publicaiton.
Supplementary Information
Below is the link to the electronic supplementary material.
394_2021_2638_MOESM2_ESM.tiff
Supplementary file2 Principle coordinated analysis (unweighted Unifrac distance) of stool microbiome of three feeding schemes including Breast feeding (BF), common formula feeding (FF1) and modified formula feeding (FF2) (TIFF 537 KB)
394_2021_2638_MOESM3_ESM.tif
Supplementary file3 Alpha-diversity analysis of stool microbiome of three feeding schemes. Breast feeding (BF), common formula feeding (FF1) and modified formula feeding (FF2) (TIF 403 KB)
394_2021_2638_MOESM4_ESM.docx
Supplementary file4 Relative abundance of Bifidobacterium, Veillonella and Escherichia/Shigella of three feeding schemes. Breast feeding (BF), common formula feeding (FF1) and modified formula feeding (FF2) (DOCX 1239 KB)
394_2021_2638_MOESM5_ESM.docx
Supplementary file5 Dot plot of gene enrichment analysis of Bifidobacterium cultured with MFGM components in vitro. Supplemental Table 2. Anthropometric measures as well as clinical parameters of 1 month of infant. (DOCX 31 KB)
Rights and permissions
About this article

Cite this article
Zhao, J., Yi, W., Liu, B. et al. MFGM components promote gut Bifidobacterium growth in infant and in vitro. Eur J Nutr 61, 277–288 (2022). https://doi.org/10.1007/s00394-021-02638-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00394-021-02638-5