
Abstract
Pulmonary hypertension (PH) is a common phenomenon that may occur as a consequence of various diseases (e.g., heart failure, chronic lung diseases, and pulmonary embolism), as a distinct disease of the small pulmonary arterioles, or a combination of both. Independently from the origin, PH has important impact on patient´s symptoms and life expectancy. The establishment of an exact diagnosis and classification, as well as the understanding of the hemodynamic interrelations, provides the basis for often challenging treatment decisions. Recently, the 5th World Symposium on PH took place in Nice, France, where important standards and definitions were specified. Furthermore, the results of recent phase III trials have led to the approval of new targeted therapies. The most relevant developments including the rating of novel treatment options are summarized in this article.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite significant improvements in the diagnosis and treatment of pulmonary hypertension (PH), this disease remains to be associated with a profound reduction of quality of life and survival. The timely establishment of the diagnosis and the precise subclassification of patients according to the clinical classification of PH (Nice 2013) are of great importance, particularly because targeted therapy of pulmonary arterial hypertension (PAH) differs from the treatment approaches in other forms of PH [1]. Due to the complexity of the pathophysiological interrelations and because individual treatment decisions are often challenging, patients with PH should be treated in specialized centers [1].
During the last decades, novel developments and improvements in this field were constantly revisited at World Symposia, which have been held every 5 years. The respective recommendations provided the basis for national and international guidelines. The standards and definitions of PH were last revisited in 2008 in Dana Point. Recently, the “5th World Symposium on Pulmonary Hypertension” (WSPH) was held in Nice, France, where important modifications were established. The respective results were now published in a supplement of the Journal of the American College of Cardiology (JACC). Furthermore, the results of recent phase III trials have led to the approval of new targeted therapies. This article summarizes the most important aspects and provides a detailed overview of the definitions, classification, terminology, and treatment of PH.
Classification and pathobiology
The current Nice classification of PH remains to distinguish five subgroups of the disease [2]. These are pulmonary arterial hypertension (PAH; group 1), PH due to left heart disease (group 2), PH due to lung diseases and/or hypoxia (group 3), chronic thromboembolic PH (CTEPH, group 4) and PH with unclear multifactorial mechanisms (group 5). This assorting is similar to the previous Dana Point classification [3]. However, novel insights have led to modifications of this classification within the main groups (Table 1, main modifications highlighted in red). For instance, recently identified gene defects (Smad9, CAV1, and KCNK3) have been added to heritable PAH, and group 2 was extended by congenital or acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathies. With regard to the pathobiology of PAH, the disease is increasingly recognized as a proliferative and inflammatory disorder [4, 5], which has important impact on the identification of novel targets and the development of new treatment strategies. In addition, the significance of pathogenic changes within the venous part of the pulmonary circuit is increasingly recognized [4].
Change of phenotype and improved prognosis
Current registry data have provided important information about the epidemiology and phenotype of patients with PAH. Interestingly, profound changes of the PAH phenotype have been observed during the past decades. These include substantial changes in age, gender, comorbidities, and survival [6]. Whereas the mean age in patients with idiopathic PAH (IPAH; then termed primary PH) in the initial NIH registry (data acquisition 1981–1982) was 36 ± 15 years [7, 8], PAH is nowadays frequently diagnosed in elderly patients, so that the mean age at diagnosis in current registries is between 50 ± 14 and 65 ± 15 years [9, 10]. A possible explanation for this development may be the increased awareness for PAH in the modern management era, since effective therapies are now available. PAH may indeed be detected more frequently in elderly patients, as the population of most western countries is aging. However, potential misclassifications between PAH and non-PAH PH may also be considered, particularly in patients with heart failure with preserved ejection fraction (HFpEF), which may occur as a result of uncertainties in the current definitions and difficulties in the measurement of the pulmonary arterial wedge pressure (PAWP) [11, 12], that are especially relevant in elderly patients.
A further profound change is the improved survival of patients with PAH. Whereas the median survival time after establishment of the diagnosis in the early registries (NIH) was limited to only 2.8 years [7, 8], current registry data show a 3-year survival of up to 83 % [10]. These data implicate that improved care of PAH patients including the treatment with targeted PAH drugs has substantially improved quality of life and survival. Of note, elderly patients (>65 years) diagnosed with PAH display a worse prognosis as compared to younger patients, despite less pronounced impairment of pulmonary hemodynamics [10].
Definitions and standardization of diagnostic procedures
In light of the complexity of the hemodynamic interrelations between left heart, pulmonary circulation, and right heart, the establishment and confirmation of the diagnosis “pulmonary hypertension” and the proper classification of the disease (groups 1–5) are of major importance, particularly with regard to treatment decisions. A diagnostic algorithm aiming to ensure a proper work-up is shown in Fig. 1 [13]. The PH World Symposium in Nice also aimed to overcome a lack of standardization of diagnostic procedures, particularly right heart catheterization (RHC). This is particularly related to the measurement of the PAWP, which is frequently used to make the distinction between pre- and postcapillary PH and thus has direct therapeutic implications. The Nice recommendations [13] suggest to measure PAWP, which is influenced by respiratory swings, at the end of normal expiration, because this method showed best agreement with the direct measurement of left ventricular end-diastolic pressure (LVEDP) [11], although this remains a matter of debate [12]. The Nice recommendation is in accordance with existing german recommendations for RHC in PH [14]. Furthermore, due to current scientific data [15], there was agreement to set the “zero point” for all invasive hemodynamic measurements at the mid-thoracic level [13].

Diagnostic algorithm for the detection and classification of pulmonary hypertension (PH) according to the 5th World Symposium on PH 2013 in Nice (modified from [13]). BGA blood gas analysis, CHD congenital heart disease, CLD chronic lung disease, CPET cardiopulmonary exercise testing, CTD connective tissue disease, CTEPH chronic thromboembolic pulmonary hypertension, DLCO diffusion capacity for carbon monoxide, HPAH heritable PAH, HR-CT high-resolution computed tomography, IPAH idiopathic PAH, LHD left heart disease, PFT pulmonary function testing, PoPH portopulmonary hypertension, PVOD pulmonary veno-occlusive disease, V/Q scan ventilation/perfusion scintigraphy
Treatment of PAH (group 1): novel compounds
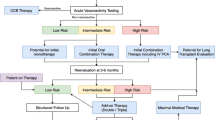
A number of compounds belonging to three distinct drug classes have been approved for the treatment of PAH. These include endothelin receptor antagonists (ERAs) such as bosentan and ambrisentan, phosphodiesterase type 5 (PDE5) inhibitors such as sildenafil and tadalafil, and prostacyclin analogues such as epoprostenol, iloprost, and treprostinil. In accordance with clinical trial results and the recommendations of the World Symposium in Dana Point, the treatment algorithm of the current European guidelines (ESC/ERS 2009) recommends the use of these compounds with various levels of evidence, based on the clinical severity of the disease (WHO functional class) [1, 16]. However, despite significant improvements the treatment options in PAH remain limited at present. Although the establishment of the current treatments has led to a substantial improvement of quality of life and survival, there is no cure for the disease, and mortality remains unacceptably high (5–10 % per year) [8–10]. Hence, it is essential to develop additional treatment options which further improve outcome. Recently, several new drugs have been developed, and their efficacy and safety has been investigated in randomized, controlled phase III trials. The updated treatment algorithm (World Symposium Nice 2013) now includes two novel compounds (macitentan and riociguat), which were recently approved by the EMA and the FDA for the treatment of PAH (Fig. 2) [17].

Updated treatment algorithm for PAH according to the 5th World Symposium on PH 2013 in Nice (modified from [17]). BSA balloon atrial septostomy, CCB calcium channel blocker, ERA endothelin receptor antagonist, PDE-5i phosphodiesterase type 5 inhibitor, sGCS soluble guanylate cyclase stimulator, WHO-FC World Health Organization functional class
The novel ERA macitentan was particularly developed to improve tissue penetration, reach a higher affinity for endothelin receptors, and to limit the potential for drug interactions [18–20]. The efficacy and safety of macitentan was now investigated in 742 patients with symptomatic PAH in the multicenter, double-blind, placebo-controlled SERAPHIN study (Study with Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome). In this trial, patients received two distinct dosages of macitentan (3 and 10 mg once daily) or placebo (randomized 1:1:1). This event-driven study, which was the first PAH study to use a combined morbidity/mortality endpoint, demonstrated a significant risk reduction of the composite primary study endpoint (death, atrial septostomy, lung transplantation, initiation of intravenous/subcutaneous prostanoid therapy or worsening of PAH) by macitentan by 30 % in the 3 mg arm (p = 0.008) and by 45 % in the 10 mg arm (p < 0.0001) [21]. The treatment duration until the predefined number of events was reached was up to three and a half years (mean time 85.3 weeks with placebo, 99.5 weeks in the macitentan 3 mg and 103.9 weeks in the macitentan 10 mg arms). The impact of macitentan on the primary endpoint was independent from pre-existing treatments with other targeted PAH therapies such as PDE5 inhibitors or prostanoids. Thus, it was shown for the first time that a targeted PAH drug improved a combined morbidity/mortality endpoint in an event-driven RCT, both in treatment-naïve and pretreated patients. Furthermore, there were no signs for liver toxicity of macitentan, as was previously reported for other ERAs [21]. Observed side effects included headache, nasopharyngitis, and anemia.
Riociguat is a stimulator of soluble guanylate cyclase (sGC), which induces the liberation of cyclic guanosine monophosphate (cGMP) and thereby promotes vasorelaxation. Riociguat stimulates the enzyme directly in an NO-independent manner and furthermore increases the sensitivity of sGC toward endogenous NO [22]. It thus represents the first member of a new class of drugs, which—similar to PDE5 inhibitors—affects the NO pathway. The concomitant use of sGC stimulators and PDE5 inhibitors is therefore contraindicated. The results of an open-label phase II study implicated that riociguat improves exercise tolerance and pulmonary hemodynamics in patients with PAH and CTEPH [23].
The efficacy and safety of riociguat was now investigated in two multicenter, randomized, double-blind, placebo-controlled phase III studies in patients with PAH (PATENT trial) and CTEPH (CHEST trial) [24, 25]. The PATENT 1 trial (Pulmonary Arterial Hypertension sGC-Stimulator Trial) recruited 443 patients, of which 317 were randomized to receive riociguat (254 individual titration up to 2.5 mg tid, 63 explorative dosing up to 1.5 mg tid). When compared to placebo, riociguat led to a significant improvement of the primary study endpoint (6-min walk distance) by 36 m (95 % CI 20–52 m, p < 0.001) at 12 weeks [24]. A prespecified subgroup analysis revealed that riociguat improved the primary endpoint both in treatment-naïve patients and in patients with pre-existing targeted PAH therapy with either ERAs or prostanoids (improvement of 6MWD by 38 vs. 36 m). In patients who received riociguat, pulmonary vascular resistance (PVR) was reduced by 223 dyn × s × cm−5 as compared to 9 dyn × s × cm−5 in the placebo group (p < 0.0001). Likewise, riociguat led to significant improvements of the mean pulmonary artery pressure (PAP) (p = 0.0002), cardiac output (p < 0.0001), NTproBNP serum levels (p < 0.001), WHO functional class (p = 0.003), and time to clinical worsening (p = 0.005). A current interim analysis of the open-label PATENT-2 extension study revealed that the improvement of the 6MWD by riociguat persists for at least 1 year and may even be further improved, if the treatment is constantly continued [24]. Significant adverse events included the occurence of syncope (4 vs. 1 %), and hemoptyses.
Based on the recent evidence from the above RCTs and approval by the regulatory agencies, macitentan and riociguat were included in the updated treatment algorithm (Fig. 2) [17]. A further compound, the prostanoid receptor agonist selexipag [26], also improved clinical outcome in an event-driven RCT and may await approval for PAH [27]. In contrast, the tyrosine kinase inhibitor imatinib [28], which has also proven effective for the treatment of PAH in the IMPRES trial [29], will not be approved for this indication due to safety concerns.
The updated PAH treatment algorithm (Nice) suggests that initial combination therapy may be considered in patients presenting in WHO-FC II or III (Fig. 2). This concept is supported by the recent AMBITION (first-line combination therapy with AMBbrIsentan and Tadalafil in patients with pulmonary arterial hypertensION) trial, which demonstrated that upfront combination therapy with the PDE5i tadalafil and the ERA ambrisentan was superior to either compound alone in preventing morbidity and mortality events [30, 31]. In this randomized, double-blind, multicenter study, 500 patients with PAH were randomized (2:1:1) to receive first-line treatment with ambrisentan and tadalafil (n = 253), or monotherapy with ambrisentan (n = 126) or tadalafil (n = 121). Upfront combination reduced the primary endpoint of clinical failure (defined as time from randomization to the first occurrence of death, hospitalization for worsening PAH, disease progression or unsatisfactory long-term clinical response) by 50 % compared to the pooled ambrisentan and tadalafil monotherapy arm (HR = 0.502; 95 % CI 0.348–0.724; p = 0.0002), and this reduction of the primary endpoint was also statistically significant versus the individual ambrisentan and tadalafil monotherapy groups (p < 0.01) [30, 31]. This is in line with the observations that novel therapies (macitentan, selexipag) were also able to prevent future morbidity/mortality events when given in addition to other PAH drugs [21, 27], and that riociguat has also proven effective in pretreated patients [24]. Together, these results challenge the current concept of target-oriented, sequential combination therapy and favor early combination treatment in the majority of patients with PAH. This is also supported by recent data from a pilot study demonstrating that upfront triple combination therapy (epoprostenol, bosentan, sildenafil) in patients with very severe PAH (WHO-FC III/IV; PVR >1.500 dyn × s × cm−5) was associated with substantial improvement of hemodynamics and excellent survival [32].
In addition to targeted PAH therapies, supportive care is an integral component of PAH therapy [1, 17]. The current ESC/ERS guidelines for the diagnosis and treatment of PH recommend the use of therapeutic anticoagulation in patients with idiopathic PAH (IPAH) [1]. However, scientific data on this topic remain sparse, and it is unclear, whether this historical recommendation holds true in the modern era of PAH therapy. A recent analysis of the COMPERA registry investigated the impact of anticoagulation on survival in 1.283 consecutively included patients with newly diagnosed PAH. Anticoagulation was given in 66 % of 800 patients with IPAH and in 43 % of the 483 patients with other forms of PAH [33]. The outcome analysis revealed that the 3-year survival rate of anticoagulated IPAH patients was significantly better than in patients without anticoagulation (p = 0.006), although the group of patients on anticoagulation had more severe disease at entry into the registry. In contrast, anticoagulation was not associated with a survival benefit in other forms of PAH.
Recent studies have demonstrated that iron deficiency (ID) is frequent and may be important in patients with PAH, as it correlates with disease severity and impacts on survival in various forms of PAH [34, 35]. ID in PAH may be caused by impaired iron absorption from the gut, which is due to increased levels of the main regulator of iron homeostasis, hepcidin [34]. In a pilot study, intravenous iron supplementation with ferric carboxymaltose was shown to markedly improve exercise capacity (increase of the 6MWD by 36 m) and quality of life [36]. While these results have to be viewed as preliminary, the importance of ID in PAH and the potential long-term benefit of iron treatment have to be investigated in randomized controlled trials which are currently under way.
Pulmonary hypertension owing to left heart disease (PH-LHD): group 2
Pulmonary hypertension is a frequent phenomenon in patients with left heart disease. This was established for both systolic [heart failure with reduced ejection fraction (HFrEF)] and diastolic [heart failure with preserved ejection fraction (HFpEF)] heart failure, as well as valvular heart disease. Numerous trials have consistently shown that the extent of PH and right ventricular dysfunction have important impact on survival in patients with left heart failure (both HFpEF and HFrEF) [37–41], and also determine the risk of patients with left-sided valve disease [42, 43]. Furthermore, the CHAMPION trial demonstrated that continuous monitoring of PAP and its consideration as a treatment target in heart failure was able to significantly lower the rate of heart failure-associated hospitalizations [44].
Among the patients diagnosed with PH, pulmonary hypertension owing to left heart disease (Nice classification group 2) by far represents the most common type of PH. According to the hemodynamic definition, patients have a mean PAP ≥25 mmHg and an increased PAWP of >15 mmHg [1, 13]. However, it is important to note that the PAWP may be “artificially” normalized in patients with left heart failure, if patients are pretreated with diuretics. Based on the transpulmonary pressure gradient (TPG = PAPmean−PAWP), the current PH guidelines distinguish between passive (TPG <12 mmHg) and reactive or “out of proportion” postcapillary PH (TPG ≥12 mmHg) [1]. However, current studies indicate that the PVR and PA compliance, but not the TPG are prognostically relevant in HFrEF [41], and that the TPG is highly dependent on volume load, left-sided filling pressure, and stroke volume, whereas the diastolic pressure difference (DPD = PAPdiast−PAWP) is much less influenced by these variables and furthermore appears to be a prognostic indicator in patients with postcapillary PH (Table 2a) [45, 46].
Due to the high prevalence and the prognostic impact of PH owing to left heart failure, it may be important to consider therapeutic consequences such as the use of PAH-approved drugs. To this end, based on the current evidence the Cologne Consensus Conference recommended, that targeted treatment of PH owing to left heart disease should only be considered, if appropriate diagnostic work-up reveals that a precapillary component is foregrounded to the disease, and other potential causes of PH have been excluded [47]. In agreement with this recommendation, the 5th PH World Symposium in Nice decided to abandon the terms “active“ or “out of proportion“ postcapillary PH, and—based on the DPD—define isolated postcapillary PH (IpcPH) and combined pre- and postcapillary PH (CpcPH). This new classification (Table 2b) has now replaced the previous terminology [48]. When PAH drugs are considered, treatment decisions should exclusively be made in expert centers with expertise in both heart failure and PH, and whenever possible, patients should be included into clinical trials. ERA and epoprostenol have not proven effective in heart failure [48], although it should be noted that these drugs have never been studied for PH owing to left heart failure. Small studies indicated that patients with HFpEF or HFrEF with significant PH may benefit from treatment with PDE-5 inhibitors [49, 50]. However, the RELAX trial recently demonstrated that patients with HFpEF but without pronounced PH do not benefit from additional treatment with the PDE-5 inhibitor sildenafil [51]. Likewise, the LEPHT study has shown that the sGC stimulator riociguat was not able to significantly lower PAP as compared to placebo in patients with HFrEF, although riociguat led to a substantial increase of cardiac index and a reduction of PVR [52]. Based on the current evidence, the use of targeted PAH therapies is not recommended in patients with heart failure and PH. Further studies are required to investigate the safety and the potential therapeutic efficacy of single compounds for the treatment of PH in hemodynamically well-characterized patients with PH owing to HFrEF or HFpEF.
Pulmonary hypertension due to chronic lung diseases (PH-CLD): group 3
Chronic lung diseases such as chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF) and combined pulmonary fibrosis and emphysema (CPFE) are frequently associated with PH, which has important impact on clinical symptoms and survival. While moderate increases of PAP and PVR do not represent indications for targeted PAH therapies, it may be difficult to interpret and classify severe PH in patients with co-existing lung diseases, and resulting treatment decisions may be challenging. Based on the NETT registry (National Emphysema Treatment Trial), the Cologne Consensus Conference in 2010 defined criteria, that suggest severe pulmonary vascular disease in group 3 PH and may thus justify a treatment attempt with targeted PAH drugs (Table 3a) [53]. These considerations were confirmed in a recent study investigating the role of PH for ventilatory and cardiocirculatory exercise profiles in COPD. This study demonstrated that patients with COPD and without or with mild PH are mainly characterized by ventilatory limitation, whereas patients with COPD and severe PH are mainly limited by an exhausted circulatory reserve (Table 3b) [54]. This important finding may have impact on the classification of PH due to pulmonary disease and potential therapeutic consequences and was thus considered in the current Nice recommendations for PH associated with chronic lung disease [55]. The suggested terminology, which is based on the mean PAP and cardiac index (CI), is summarized in Table 4a. With regard to treatment decisions, both the degree of PH and the severity of the underlying lung disease must be considered (Table 4b). Particularly in patients with severe PH, a definitive discrimination might be impossible and the PH classification may remain unclear. Based on various criteria, it must be decided whether PAH (group 1) with concomitant lung disease or PH due to lung disease (group 3) is present (Table 4b; Fig. 1). Such patients should be referred to an expert center with expertise in both PAH and lung disease. Since sufficient evidence from clinical trials is lacking in this group of patients but prognosis is poor, individualized treatment decisions and patient care are required, and patients should be included in ongoing or future clinical studies [55].
Chronic thromboembolic pulmonary hypertension (CTEPH): group 4
An important cause of PH, that is frequently overlooked or diagnosed with significant delay, is chronic or recurrent pulmonary embolism (PE). Ventilation/perfusion scintigraphy of the lung remains the gold standard for the detection of CTEPH in patients with PH (Fig. 1) [56, 57]. In contrast to PAH, the primary treatment option in patients with CTEPH, who harbor a poor prognosis if left untreated, is the surgical removal of the thromboembolic lesions by pulmonary endarterectomy (PEA). In properly selected patients, this procedure is able to substantially improve or even normalize pulmonary hemodynamics [56–58]. According to the current recommendations of the World Symposium in Nice, the diagnostic work-up and assessment of operability must be accomplished by a distinguished CTEPH team which includes an experienced PEA surgeon [57]. Registry data have shown that PVR is nearly normalized in the majority of operated patients, which is due to both reduction of PAP and a substantial improvement of cardiac output. In many cases, this is associated with normalization of right ventricular function and hence restoration of normal life expectancy. This complex intervention, which is mostly performed in deep hypothermia cardiac arrest, thus represents a potentially curative approach. Perioperative mortality is highly dependent on disease severity as reflected by the preoperative PVR and on the experience of the PEA surgeon as well as the perioperative management. According to recent data from case series and an international CTEPH registry, in-hospital mortality is between 2.2 and 4.7 % [57, 58].
However, if the thromboembolic lesions are located in the distal parts of the pulmonary vascular bed, they may not be accessible for surgical interventions, and patients may be judged as technically inoperable. Therefore, only approximately 2/3 of patients with CTEPH are candidates for successful surgery [57]. Furthermore, PH may persist or recur even after successful PEA. In these cases, targeted medical therapy of PH may be considered. However, there has been no approved medical treatment for this condition thus far. Preliminary studies suggested that the sGC stimulator riociguat—in addition to PAH—may also be effective in CTEPH [23]. The efficacy and safety of riociguat in patients with CTEPH, who were not candidates for PEA (technical inoperability) or had persistent or recurrent PH after PEA, was now investigated in a double-blind, randomized, controlled phase 3 trial (CHEST; chronic thromboembolic pulmonary hypertension soluble guanylate cyclase-stimulator trial) [25]. A total of 261 patients were randomized, of which 173 received riociguat. The majority of patients were in WHO-FC III at baseline. At 16 weeks, the 6MWD (primary endpoint) improved by 39 m in patients treated with riociguat versus −6 m in the placebo group, resulting in a net increase of 45 m (p < 0.001). In patients on riociguat, PVR decreased by 262 dyn × s × cm−5, as compared to 23 dyn × s × cm−5 in the placebo group (p < 0.001). Likewise, riociguat led to significant improvements of the mean PAP, the mean systemic blood pressure, cardiac output, Borg dyspnea index, quality of life, and WHO-FC. Preliminary data from the CHEST-2 open-label extension study implicate that the improvement of the 6MWD by riociguat persists for at least 1 year if patients remain on treatment. Further compounds such as the ERAs ambrisentan (AMBER-1) and macitentan (MERIT-1) are currently investigated in clinical trials. Despite the positive trial results and the approval of riociguat for inoperable CTEPH, medical treatment of CTEPH must not be viewed as an alternative for surgery. PEA remains the primary treatment option, and the assessment of operability is a central task of the multidisciplinary CTEPH team in each patient [57]. In addition to PEA and medical treatment, pulmonary balloon angioplasty (PBA) may develop as an emerging treatment option in patients with inoperable CTEPH, as it was shown to markedly improve pulmonary hemodynamics and long-term prognosis in such patients [59]. However, this method, which is associated with significant risks such as pulmonary hemorrhage, is not established yet and awaits further evaluation.
Pulmonary hypertension with unclear multifactorial mechanisms: group 5
Group 5 of the Nice classification summarizes other forms of PH, which are characterized by unclear multifactorial mechanisms [2]. These include hematologic, systemic, or metabolic disorders, and other entities that may be associated with PH (Table 1). In the updated clinical classification, chronic hemolytic anemia as occurring in sickle cell disease, thalassemia, spherocytosis, and stomatocytosis, was moved from group 1 to group 5, because the cause of PH is often unclear, and recent studies have shown that precapillary PH associated with chronic hemolytic anemia appears significantly different from other forms of PAH with regard to pathological findings, hemodynamic characteristics and response to PAH therapies [2]. While targeted therapies are approved for PAH (group 1), only small studies are available in various subpopulations of group 5, which precludes a proper analysis of their efficacy and safety. Therefore, treatment decisions in patients with PH categorized into group 5 have to be made on an individual basis.
References
Galiè N, Hoeper MM, Humbert M et al (2009) Guidelines for the diagnosis and treatment of pulmonary hypertension: the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung transplantation (ISHLT). Eur Heart J 30:2493–2537
Simonneau G, Gatzoulis MA, Adatia I et al (2013) Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 62(Suppl D):D34–D41
Simonneau G, Robbins IM, Beghetti M et al (2009) Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 54(Suppl S):S43–S54
Tuder RM, Archer SL, Dorfmüller P et al (2013) Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 62(Suppl D):D4–D12
Soubrier F, Chung WK, Machado R et al (2013) Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol 63(Suppl D):D13–D21
McGoon M, Benza RL, Escribano-Subias P et al (2013) Pulmonary arterial hypertension: epidemiology and registries. J Am Coll Cardiol 62(Suppl D):D51–D59
Rich S, Dantzker DR, Ayres SM et al (1987) Primary pulmonary hypertension: a national prospective study. Ann Intern Med 107:216–223
D’Alonzo GE, Barst RJ, Ayres SM et al (1991) Survival in patients with primary pulmonary hypertension. Ann Intern Med 115:343–349
Humbert M, Sitbon O, Chaouat A et al (2010) Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 122:156–163
Hoeper MM, Huscher D, Ghofrani HA et al (2013) Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: results from the COMPERA registry. Int J Cardiol 168:871–880
Ryan JJ, Rich JD, Thiruvoipati T et al (2012) Current practice for determining pulmonary capillary wedge pressure predisposes to serious errors in the classification of patients with pulmonary hypertension. Am Heart J 163:589–594
LeVarge BL, Pomerantsev E, Channick RN (2014) Reliance on end-expiratory wedge pressure leads to misclassification of pulmonary hypertension. Eur Respir J 43:425 (Epub ahead of print)
Hoeper M, Bogaard HJ, Condliffe R et al (2013) Definition and diagnosis of pulmonary hypertension. J Am Coll Cardiol 62(Suppl D):D42–D50
Rosenkranz S, Behr J, Ewert R, Ghofrani HA et al (2011) Rechtherzkatheter-Untersuchung bei pulmonaler Hypertonie. Dtsch Med Wochenschr 136:2601–2620
Kovacs G, Avian A, Olschewski A, Olschewski H (2013) Zero reference level for right heart catheterization. Eur Respir J 42:1586–1594
Barst RJ, Gibbs SR, Ghofrani HA et al (2009) Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol 54(Suppl S):S78–S84
Galiè N, Corris PA, Frost A et al (2013) Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol 62(Suppl D):D60–D72
Bolli MH, Boss C, Binkert C et al (2012) The discovery of N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-propyl-sulfamide (Macitentan), an orally active, potent dual endothelin receptor antagonist. J Med Chem 55:7849–7861
Gatfield J, Mueller Grandjean C, Sasse T, Clozel M, Nayler O (2012) Slow receptor dissociation kinetics differentiate macitentan from other endothelin receptor antagonists in pulmonary arterial smooth muscle cells. PLoS One 7:e47662
Iglarz M, Binkert C, Morrison K et al (2008) Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther 327:736–745
Pulido T, Adzerikho I, Channick RN, For the SERAPHIN Investigators et al (2013) Macitentan and morbidity and mortality in pulmonary arterial hypertension. New Engl J Med 369:809–818
Evgenov OV, Pacher P, Schmidt PM et al (2006) NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov 5:755–768
Ghofrani HA, Hoeper M, Halank M et al (2010) Riociguat for chronic thromboembolic pulmonary hypertension and pulmonary arterial hypertension. A phase II study. Eur Respir J 36:792–799
Ghofrani HA, Galiè N, Grimminger F, For the PATENT Investigators et al (2013) Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 369:330–340
Ghofrani HA, D`Armini AM, Grimminger F, For the CHEST Investigators et al (2013) Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 369:319–329
Simonneau G, Lang I, Torbicki A, Hoeper MM, Delcrox M, Karlocai K, Galiè N (2012) Selexipag, an oral, selective IP receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J 40:874–880
Actelion press release: selexipag meets primary endpoint in pivotal phase III GRIPHON outcome study in patients with pulmonary arterial hypertension (2014) (Published June 16)
Freyhaus T, Dumitrescu D, Berghausen E, Vantler M, Caglayan E, Rosenkranz S (2012) Imatinib mesylate for pulmonary arterial hypertension. Exp Opin Investig Drugs 21:119–134
Hoeper MM, Barst RJ, Bourge RC et al (2013) Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 127:1128–1138
GlaxoSmithKline/Gilead press release: first-line combination of ambrisentan and tadalafil reduces risk of clinical failure compared to monotherapy in pulmonary arterial hypertension outcomes study (2014) (Published September 8)
Galiè N, for the AMBITION Investigators (2014) The AMBITION study: design and results. Presented at ERS International Congress 2014 (abstract #2916)
Sitbon O, Jais X, Savale L et al (2014) Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study. Eur Respir J 43:1691–1697
Olsson KM, Delcroix M, Ghofrani HA et al (2014) Anticoagulation and survival in pulmonary arterial hypertension: results from the COMPERA registry. Circulation 129:57–65
Rhodes CJ, Howard LS, Busbridge M et al (2011) Iron deficiency and raised hepcidin in idiopathic pulmonary arterial hypertension: clinical prevalence, outcomes and mechanistic insights. J Am Coll Cardiol 58:300–309
Ruiter G, Lanser IJ, de Man FS et al (2014) Iron deficiency in systemic sclerosis patients with and without pulmonary hypertension. Rheumatology 53:285–292
Viethen T, Gerhardt F, Dumitrescu D, Knoop-Busch S, ten Freyhaus H, Rudolph TK, Baldus S, Rosenkranz S (2014) Ferric carboxymaltose improves exercise capacity and quality of life in patients with pulmonary arterial hypertension and iron deficiency: a pilot study. Int J Cardiol. doi:10.1016/j.ijcard.2014.04.233 (Epub ahead of print)
Bursi F, McNallan SM, Redfield MM et al (2012) Pulmonary pressures and death in heart failure. J Am Coll Cardiol 59:222–231
Lam CS, Roger VL, Rodeheffer RJ et al (2009) Pulmonary hypertension in heart failure with preserved ejection fraction: a community-based study. J Am Coll Cardiol 53:1119–1126
Ghio S, Gavazzi A, Campana C et al (2001) Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J Am Coll Cardiol 37:183–188
Melenovsky V, Hwang SJ, Lin G, Redfield MM, Borlaug BA (2014) Right heart dysfunction in heart failure with preserved ejection fraction. Eur Heart J [Epub ahead of print]
Miller WL, Grill DE, Borlaug BA (2013) Clinical features, hemodynamics, and outcomes of pulmonary hypertension due to chronic heart failure with reduced ejection fraction. JACC Heart Fail 1:290–299
Seiffert M, Sinning JM, Meyer A et al (2014) Development of a risk score for outcome after transcatheter aortic valve implantation. Clin Res Cardiol 103:631–640
Zuern CS, Eick C, Rizas K et al (2012) Prognostic value of mild-to-moderate pulmonary hypertension in patients with severe aortic valve stenosis undergoing aortic valve replacement. Clin Res Cardiol 101:81–88
Abraham WT, Adamson PB, Bourge RC et al (2011) Wireless pulmonary artery haemodynamic monitoring in chronic heart failure: a randomised controlled trial. Lancet 377:658–666
Naejie R, Vachiéry JL, Vanderpool R (2013) The transpulmonary pressure gradient for the diagnoisis of pulmonary vascular disease. Eur Respir J 41:217–223
Gerges C, Gerges M, Lang MB et al (2013) Diastolic pulmonary vascular pressure gradient. A predictor of prognosis in out-of-proportion pulmonary hypertension. Chest 143:758–766
Rosenkranz S, Bondermann D, Buerke M et al (2011) Pulmonary hypertension due to left heart disease: updated recommendations of the Cologne Consensus Conference 2011. Int J Cardiol 154(Suppl):S34–S44
Vachiery JL, Adir Y, Barbera JA et al (2013) Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol 62(Suppl D):D100–D108
Guazzi M, Vicenzi M, Arena R, Guazzi MD (2011) Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study. Circulation 124:164–174
Dumitrescu D, Seck C, Möhle L, Erdmann E, Rosenkranz S (2012) Therapeutic potential of sildenafil in patients with heart failure and reactive pulmonary hypertension—results of compassionate care treatment. Int J Cardiol 154:205–206
Redfield MM, Chen HH, Borlaug BA et al (2013) Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 309:1268–1277
Bonderman D, Ghio S, Felix SB, For the LEPHT Investigators et al (2013) Riociguat for patients with pulmonary hypertension caused by systolic left ventricular dysfunction: a phase IIb double-blind, randomized, placebo-controlled, dose-ranging hemodynamic study. Circulation 128:502–511
Hoeper MM, Andreas S, Bastian A et al (2011) Pulmonary hypertension due to chronic lung disease. Updated recommendations of the cologne consensus conference 2011. Int J Cardiol 154(Suppl):S45–S53
Boerrigter BG, Bogaard HJ, Trip P et al (2012) Ventilatory and cardiocirculatory exercise profiles in COPD: the role of pulmonary hypertension. Chest 142:1166–1174
Seeger W, Adir Y, Barbera JA et al (2013) Pulmonary hypertension in chronic lung diseases. J Am Coll Cardiol 62(Suppl D):D109–D116
Mayer E, Jenkins D, Lindner J et al (2011) Surgical management and outcome of patients with chronic thromboembolic pulmonary hypertension: results from an international prospective registry. J Thorac Cardiovasc Surg 141:702–710
Kim NH, Delcroix M, Jenkins DP et al (2013) Chronic thromboembolic pulmonary hypertension. J Am Coll Cardiol 62(Suppl D):D92–D99
Madani MM, Auger WR, Pretorius V et al (2012) Pulmonary endarterectomy: recent changes in a single institution´s experience of more than 2,700 patients. Ann Thorac Surg 94:97–103
Sugimura K, Fukumoto Y, Satoh K et al (2012) Percutaneous transluminal pulmonary angioplasty markedly improves pulmonary hemodynamics and long-term prognosis in patients with chronic thromboembolic pulmonary hypertension. Circ J 76:485–488
Acknowledgments
The technical assistance by Linda Esin and Alice Dittmann is greatly acknowledged.
Conflict of interest
The author has received remunerations for lectures and/or consultancy for Actavis, Actelion, Bayer, Gilead, GSK, Lilly, Novartis, Pfizer, and United Therapeutics. In addition, he has received research grants from Actelion, AOP, Bayer, Novartis, Pfizer, and United Therapeutics.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rosenkranz, S. Pulmonary hypertension 2015: current definitions, terminology, and novel treatment options. Clin Res Cardiol 104, 197–207 (2015). https://doi.org/10.1007/s00392-014-0765-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00392-014-0765-4