
Abstract
Purpose
Several underlying conditions of moyamoya syndrome (MMS) are well established, but so far, D-2-hydroxyglutaric aciduria (D-2-HGA) has not been mentioned. We are the first to describe a case of a patient suffering from D-2-HGA developing MMS.
Methods
The co-occurrence of D-2-HGA and MMS in a patient is reported. Furthermore, we describe the neurosurgical revascularization procedure performed and report on the follow-up.
Results
A 7-year-old girl suffering from D-2-HGA developed two transient ischemic attacks (TIAs). Using MRI/MRA and invasive angiography MMS was diagnosed. We performed an encephalo-duro-arterio-myo-synangiosis (EDAMS) as an indirect revascularization procedure first on the right and 2 months later on the left hemisphere. We have followed her up until the age of 10. Since the second surgery, she has not suffered further TIAs and is in a better general medical condition.
Conclusion
Even though children with D-2-HGA often suffer epileptic attacks, every new (transient) neurological deficit should be followed up by an MRI/MRA so as not to oversee a possible underlying MMS. After diagnosis, EDAMS in combination with acetylsalicylic acid (ASA) is recommended to prevent further ischemic events.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Moyamoya disease (MMD) was first described in 1957 and named by Suzuki et. al in 1961 [1]. While moyamoya angiopathy is most common in Japan (incidence rate 0.54 per 100,000) the occurrence in Western countries is 10 times less frequent [2]. Autopsy studies revealed a thickened intima in affected arteries leading to stenosis of their lumen [3]. The Japanese term moyamoya is translated as “puff of smoke,” which refers to the angiographic appearance of collaterals sprouting to compensate for the reduced blood supply to the brain tissue [4]. These characteristic vessels can appear idiopathically (this is called MMD), but they have also been found in connection with several underlying extrinsic and/or intrinsic conditions (the disease is then called moyamoya syndrome (MMS)) [5,6,7]. In the literature, the rare D-2-hydroxyglutaric aciduria (D-2-HGA, incidence rate < 1:1,000,000 [8]) has not yet been described in the context of MMS.
Case
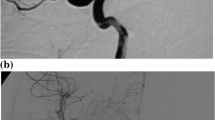
Our patient is a 7-year-old girl with a weight of 22.8 kg. Prenatal sonography showed enlargement of the bowel, the left kidney, and the ventricles. While in postnatal sonography, the ventricles were not enlarged anymore and she developed microcephaly within 3 months. At five months, she presented persisting vomiting due to a gastric emptying disorder. A laparotomy showed pylorus hypertrophy and an annular pancreas. This constellation led to further testing. Urine tests showed elevated levels of 2-hydroxyglutaric acid and a skin biopsy verified D-2-HGA type II with p.Arg140Gln mutation in the IDH2 gene. The parents did not show the mutation. She suffers from global retardation and primary generalized tonic–clonic seizures and visits a school for mentally disabled children. At the age of 7 1/2 years, she developed two episodes of sudden onset hemiparesis affecting the arms. The strokes ceased spontaneously after a few minutes—first a left-sided hemiparesis and later on a (weaker) right-sided hemiparesis. A seizure was excluded by electroencephalography (EEG). An MRI/MRA scan in an external hospital showed bilateral stenosis of the distal internal carotid arteries with moyamoya suspect vessels. In addition, a digital subtraction angiography was performed which confirmed the suspected diagnosis (Fig. 1).

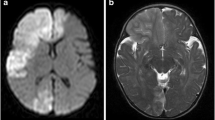
Preoperative MR-imaging of our patient A demonstrates periventricular lesions of the white matter as a common finding in D-2-HGA. The lesions are hyperintense in T2-weighted sequences (A1) and hypointense in T1-series without contrast enhancement (A2). B shows the non-invasive angiography (TOF-MRA) of the left (B1) and of the right (B2) hemisphere. Note the moyamoya typical changes in the marked areas
As a result of the clinical presentation with transient ischemic attacks (TIAs) and the radiographic diagnosis, we decided to perform bilateral revascularization surgery—first on the right hemisphere, since the left hemiparesis had been more pronounced. Encephalo-duro-arterio-myo-synangiosis (EDAMS) as an indirect revascularization procedure has been performed. The surgery was uneventful. Seventy-five milligrams of acetylsalicylic acid (ASA) a day was prescribed after surgery. Two weeks after surgery, the girl developed again a transient right-sided hemiparesis lasting 3 min, accompanied by a reduction of vigilance and followed by headache and vomiting. The neurological symptoms subsided spontaneously. The patient was admitted to an external hospital close to home. Cranial MRI showed no new infarction or bleeding, and the EEG excluded a seizure. Using Doppler sonography and MRA stenosis of the recently transposed right superficial temporal artery was excluded so that a third TIA was diagnosed. Two months after the first surgery, we performed the left-sided EDAMS. The second surgery was uneventful as well.
At the last follow-up, at the age of 10 years, the girl showed no new neurological symptoms and a better general medical condition compared to her preoperative state. A follow-up cerebral MRI with non-invasive angiography showed no signs of new infarction. The mother did not observe further TIAs.
Discussion
2-HGA is a very rare medical condition with elevated levels of D-2-hydroxyglutarate acid in the urine, plasma, and cerebrospinal fluid. This causes neurometabolic disorders which can be classified by their stereo-isometric geometry into three types that share similarities but refer to different diseases due to different genetic mutations [9,10,11,12]. Each of the following entities is caused by mutations of genes coding mitochondrial proteins [9]: the more common L-2-HGA [13, 14], the rare D-2-HGA [10, 15, 16], and the combined D, L-2-HGA [17]. D-2-HGA can be further divided into two subtypes due to different mutations: homozygous mutations of D-2-hydroxyglutarate dehydrogenase (type I) [16] or heterozygous mutations of isocitrate dehydrogenase 2 (type II) [8]. While type II, which affected our patient, is commonly more severe and has a neonatal onset, clinical manifestation varies tremendously and includes developmental delay, hypotension, and epilepsy [9, 18].
Our patient with D-2-HGA type II developed at the age of 7 recurrent TIAs with hemiparesis on both sides as clinical symptoms of MMS.
One autopsy case of a child suffering D-2-HGA described multiple vascular anomalies in several organs [19]. In the central nervous system, there were multiple bilateral aneurysms of the middle cerebral arteries, as well as occasional thickening of the walls of some arterioles in the white matter of the brain. Arteries of the lung and kidneys showed a loss of smooth muscle cells and fibrosis in the intima and media. Even though the exact histology of vascular lesions in D-2-HGA patients and MMD patients differ, high levels of D-2-hydroxyglutaric acid seem to affect vessels pathologically. This might be a pathophysiological link between D-2-HGA and MMS. It is likely that the change in hemodynamics caused by MMS affects the brains of D-2-HGA patients even more as a result of the above-mentioned possibly pre-existing macro- and microvascular pathologies. Clinically, this could result in more and earlier ischemic events or hemorrhages in D-2-HGA patients.
There is no curative therapeutical approach to MMS [20]. Usually, it is reasonable to treat the underlying disease, but in the special case of D-2-HGA, this is not possible [9]. Secondary prophylactic measures include ASA and neurosurgery [21]. Even though data show continuous uncertainty over the benefits and risks of antiplatelet therapy, the expert consensus of the European Stroke Organization suggests the use of ASA and assesses it as safe during surgery [2]. Surgical techniques can be categorized into direct and indirect procedures [23]. Looking at the Japanese guidelines on the management of MMD in adults, indirect vascularization alone is considered inferior to direct vascularization or combined procedures [20, 22]. Up to now, in children, there is no superior method to improve clinical outcomes [20, 23]. In line with the Japanese guidelines on the management of MMD, we chose a surgical approach, because of the recurring bilateral TIAs with high-grade hemiparesis and a high risk of recurrence.
While evaluating the appropriate surgical method (direct vs. indirect) in 2-HGA patients, possible pre-existing macrovascular abnormalities (e.g., aneurysms) should be considered and detected radiologically. We performed an EDAMS as a multi-layered indirect revascularization procedure, in which part of the temporalis muscle and branches of the superficial temporal artery are directly laid down on the brain surface and dural/cortical vessels. We chose the EDAMS procedure, because we are very familiar with the surgery and have had very favorable clinical outcomes in our patients. Finally, we would like to emphasize that this indirect procedure is based on several kinds of tissues (muscles and vessels) that start collateralization.
After surgery, the TIAs ceased, and an improvement in activity was observed, which is known to be associated with moyamoya surgery in children [24].
Recommendations and conclusion
D-2-HGA type II is a neurometabolic disorder with a broad spectrum of symptoms. It is often impossible to distinguish whether a new neurological event (e.g., epilepsy) is due to the underlying condition or is a symptom of a second neurological disease, like MMS. We suggest that the basic diagnostic procedures for patients suffering from D-2-HGA with new neurological symptoms should include MRI scans with non-invasive angiographic techniques. Diagnosing MMS after the first symptomatic event and thus initiating secondary prophylactic measures are crucial to prevent further brain damage. Performing bilateral EDAMS surgery in two separate procedures and initiating ASA treatment were safe for our patient and prevented a recurrence of TIAs. Moreover, an improvement in activities was observed by her pediatric caregivers.
Data availability
The data are not publicly available due to information that could compromise the privacy of the patient. Data that support the findings of this study are available on request from the corresponding author.
References
Suzuki J, Takaku A (1969) Cerebrovascular “moyamoya” disease. Disease showing abnormal net-like vessels in base of brain Arch Neurol 20(3):288–299. https://doi.org/10.1001/archneur.1969.00480090076012
Bersan A, Khan N, Fuentes B, Acerbi F, CanaveroI T-L, Vajcoczy P, Zedde ML, Hussain S, Lémere S, Kraemer M, Herve D (2023) European Stroke Organisation (ESO) guidelines on moyamoya angiopathy. Eur Stroke J 8(1):55–84. https://doi.org/10.1177/23969873221144089
Takekawa Y, Umezawa T, Ueno Y, Sawada T, Kobayashi M (2004) Pathological and immunohistochemical findings of an autopsy case of adult moyamoya disease. Neuropathology 24(3):236–242. https://doi.org/10.1111/j.1440-1789.2004.00550.x
Suzuki J, Kodama N (1983) Moyamoya disease–a review. Stroke 14(1):104–109. https://doi.org/10.1161/01.str.14.1.104
Göksel BK, Ozdogu H, Yildirim T, Oğuzkurt L, Asma S (2010) Beta-thalassemia intermedia associated with moyamoya syndrome. J Clin Neurosci 17(7):919–920. https://doi.org/10.1016/j.jocn.2009.10.023
Phi JH, Wang KC, Lee JY, Kim SK (2015) Moyamoya syndrome: a window of moyamoya disease. J Korean Neurosurg Soc 57(6):408–414. https://doi.org/10.3340/jkns.2015.57.6.408
Scott RM, Smith ER (2009) Moyamoya disease and moyamoya syndrome. N Engl J Med 360(12):1226–1237. https://doi.org/10.1056/NEJMra0804622
Salomons GS, Struys EA (2012) D-2-Hydroxy-glutarazidurie. In: Orphanet. Available via https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=DE&Expert=79315. Accessed 12 Feb 2024
Kranendijk M, Struys EA, Salomons GS, Van der Knaap MS, Jakobs C (2012) Progress in understanding 2-hydroxyglutaric acidurias. J Inherit Metab Dis 35(4):571–587. https://doi.org/10.1007/s10545-012-9462-5
Kranendijk M, Struys EA, van Schaftingen E, Gibson KM, Kanhai WA, van der Knaap MS, Amiel J, Buist NR, Das AM, de Klerk JB, Feigenbaum AS, Grange DK, Hofstede FC, Holme E, Kirk EP, Korman SH, Morava E, Morris A, Smeitink J, Sukhai RN, Vallance H, Jakobs C, Salomons GS (2010) IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science 330(6002):336. https://doi.org/10.1126/science.1192632
Kranendijk M, Struys EA, Gibson KM, Wickenhagen WV, Abdenur JE, Buechner J, Christensen E, de Kremer RD, Errami A, Gissen P, Gradowska W, Hobson E, Islam L, Korman SH, Kurczynski T, Maranda B, Meli C, Rizzo C, Sansaricq C, Trefz FK, Webster R, Jakobs C, Salomons GS (2010) Evidence for genetic heterogeneity in D-2-hydroxyglutaric aciduria. Hum Mutat 31(3):279–283. https://doi.org/10.1002/humu.21186
Struys EA, Gibson KM, Jakobs C (2007) Novel insights into L-2-hydroxyglutaric aciduria: mass isotopomer studies reveal 2-oxoglutaric acid as the metabolic precursor of L-2-hydroxyglutaric acid. J Inherit Metab Dis 30(5):690–693. https://doi.org/10.1007/s10545-007-0697-5
Duran M, Kamerling JP, Bakker HD, van Gennip AH, Wadman SK (1980) L-2-Hydroxyglutaric aciduria: an inborn error of metabolism? J Inherit Metab Dis 3(4):109–112. https://doi.org/10.1007/BF02312543
Topçu M, Jobard F, Halliez S, Coskun T, Yalçinkayal C, Gerceker FO, Wanders RJ, Prud’homme JF, Lathrop M, Ozguc M, Fischer J (2004) L-2-Hydroxyglutaric aciduria: identification of a mutant gene C14orf160, localized on chromosome 14q22.1. Hum Mol Genet 13(22):2803–11. https://doi.org/10.1093/hmg/ddh300
Chalmers RA, Lawson AM, Watts RW, Tavill AS, Kamerling JP, Hey E, Ogilvie D (1980) D-2-hydroxyglutaric aciduria: case report and biochemical studies. J Inherit Metab Dis 3(1):11–15. https://doi.org/10.1007/BF02312516
Struys EA, Korman SH, Salomons GS, Darmin PS, Achouri Y, van Schaftingen E, Verhoeven NM, Jakobs C (2005) Mutations in phenotypically mild D-2-hydroxyglutaric aciduria. Ann Neurol 58(4):626–630. https://doi.org/10.1002/ana.20559
Muntau AC, Röschinger W, Merkenschlager A, van der Knaap MS, Jakobs C, Duran M, Hoffmann GF, Roscher AA (2000) Combined D-2- and L-2-hydroxyglutaric aciduria with neonatal onset encephalopathy: a third biochemical variant of 2-hydroxyglutaric aciduria? Neuropediatrics 31(3):137–140. https://doi.org/10.1055/s-2000-7497
Baker NS, Sarnat HB, Jack RM, Patterson K, Shaw DW, Herndon SP (1997) D-2-Hydroxyglutaric aciduria: hypotonia, cortical blindness, seizures, cardiomyopathy, and cylindrical spirals in skeletal muscle. J Child Neurol 12(1):31–36. https://doi.org/10.1177/088307389701200105
Eeg-Olofsson O, Wei W, Olsson Y, Jagell S, Hagenfeldt L (2000) D-2-Hydroxyglutaric aciduria with cerebral, vascular, and muscular abnormalities in a 14-year-old boy. J Child Neurol 15(7):488–492. https://doi.org/10.1177/088307380001500714
Fujimura M, Tominaga T, Kuroda S, Takahashi JC, Endo H, Ogasawara K, Miyamoto S (2022) 2021 Japanese Guidelines for the Management of Moyamoya Disease: guidelines from the Research Committee on Moyamoya Disease and Japan Stroke Society. Neurol Med Chir (Tokyo) 62(4):165–170. https://doi.org/10.2176/jns-nmc.2021-0382
Yamada S, Oki K, Itoh Y, Kuroda S, Houkin K, Tominaga T, Miyamoto S, Hashimoto N, Suzuki N (2016) Effects of surgery and antiplatelet therapy in ten-year follow-up from the registry study of Research Committee on Moyamoya Disease in Japan. J Stroke Cerebrovasc Dis 25(2):340–349. https://doi.org/10.1016/j.jstrokecerebrovasdis.2015.10.003
Jeon JP, Kim JE, Cho WS, Bang JS, Son YJ, Oh CW (2018) Meta-analysis of the surgical outcomes of symptomatic moyamoya disease in adults. J Neurosurg 128(3):793799. https://doi.org/10.3171/2016.11.JNS161688
Matsushima T, Inoue T, Suzuki SO, Fujii K, Fukui M, Hasuo K (1992) Surgical treatment of moyamoya disease in pediatric patients–comparison between the results of indirect and direct revascularization procedures. Neurosurgery 31(3):401–405. https://doi.org/10.1227/00006123-199209000-00003
Choi JU, Kim DS, Kim EY, Lee KC (1997) Natural history of moyamoya disease: comparison of activity of daily living in surgery and non surgery groups. Clin Neurol Neurosurg Suppl 2:S11–S18. https://doi.org/10.1016/s0303-8467(97)00033-4
Author information
Authors and Affiliations
Contributions
All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kühnl, T., Januschek, E. & Offenbach, S.K. Moyamoya syndrome in a patient with D-2-hydroxyglutaric aciduria type II: a rare association. Childs Nerv Syst 40, 2241–2244 (2024). https://doi.org/10.1007/s00381-024-06340-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-024-06340-9