
Abstract
Introduction
Craniopharyngioma constitutes approximately 10% of primary brain tumors in children. It can cause considerable morbidity and mortality due to the local aggressiveness of the tumor itself or its management affecting the hypothalamus-pituitary axis and optic pathway involvement. There is very scarce data available from LMIC which makes the management controversial where multidisciplinary teams are already not available in most of the centers. This is a single-center cross-sectional retrospective review of 20-year record of 49 patients with craniopharyngioma treated between 2001 and 2020 at Aga Khan University Hospital, a tertiary care center in Karachi, Pakistan.
Methods
We have assessed the epidemiological data of children presenting with the diagnosis of craniopharyngioma, treatment modalities used, and neurological, endocrine, and hypothalamic complications in these patients. The assessment involved a retrospective review of medical records and medical follow-up.
Results
Out of a total of 49 patients, 26 (53%) were male, and 23 (46.9%) were female. The mean age was 9.5 years (SD ± 4.5 years). Most common symptoms at initial presentation were headache 41 (83.6%), visual deficit 40 (81.6%), nausea and vomiting 26 (53%), and endocrine abnormalities 16 (32%). Treatment modalities used at our center include gross total resection 11 (22%) and subtotal resection 38 (77%) out of total, while 6 (12.2%) patients received intracystic interferon. Histopathologic findings of the majority of patients (40 (81%)) revealed an adamantinomatous type of tumor. Only 23 (46.9%) children followed in clinic post-op. Median follow-up after craniopharyngioma presentation was 5 years (± 2.1 SD, range: 2–10 years). Pituitary hormone deficiencies (98%) and visual disturbances (75%) were the most common long-term health conditions observed.
Conclusions
Since pituitary hormone deficiencies and visual disturbance were the most common long-term health conditions observed in our study, these patients require a multidisciplinary team follow-up to improve their quality of life.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Craniopharyngiomas are non-malignant tumors that develop from remnants of Rathke’s pouch close to the pituitary gland. While they account for less than 1% of primary CNS tumors, they are the most common non-malignant non-glial tumors in children and represent 5–10% of all intracranial tumors [1]. Children with craniopharyngiomas may experience symptoms such as headaches, vision problems, excessive thirst and urination, weight gain, growth retardation, and premature and/or delayed puberty.
The diagnosis of craniopharyngiomas typically relies on imaging like CT and MRI. These modalities reveal characteristic features of these tumors, including frequent solid cum cystic mass with calcifications, which are instrumental in differentiating craniopharyngiomas from other intracranial neoplasms [2, 3]. Although long-term event-free survival is around 65% in children, there is a risk of significant tumor and therapy-related complications [2,3,4].
The recommended therapy is safe resection while preserving visual, hypothalamic, and pituitary functions. Aggressive surgery, especially in small children, increases the risks of surgically induced deficits mainly hypothalamic, and even after performing gross total resections, there have been reports of recurrence, which heightens the vulnerability of children to surgically induced deficits [5]. Visual impairment, neuropsychological deficits, and endocrine deficits are common complications that significantly affect patients’ quality of life, observed in more than 50% of craniopharyngioma patients. A recent systematic review suggests that GTR is associated with higher rates of post-treatment endocrinopathy compared to STR and STR combined with radiation therapy. For instance, STR reduces the rates of endocrine dysfunction from 55 to 48.9%, diabetes insipidus from 29 to 19.5%, obesity from 6 to 0%, and panhypopituitarism from 22 to 16.1% compared to GTR. Nevertheless, the addition of radiation therapy raises the rates of endocrine dysfunction to 52.4%, obesity to 4%, and panhypopituitarism to 26.4% [6]. With better endocrinological management and new techniques like the use of an operating microscope, operative morbidity and mortality have reduced over time [7]. At our center, a large cohort of children with craniopharyngioma has been followed but their characteristics were not yet evaluated. As survival rates have increased in children with craniopharyngioma, there is growing awareness of the potential for significant complications related to both the tumor and its treatment. To better understand the outcomes of primary surgical treatment in these patients, we conducted a retrospective review of 49 patients who were treated at a single hospital over 20 years. Our evaluation included a comprehensive assessment of presentation, treatment modality used, and tumor control, as well as a thorough analysis of any neurological, endocrine, and hypothalamic complications that may have arisen.
Data collection
Method
Patients were identified by a computer-based search for children and adolescents treated for craniopharyngioma between 2001 and 2020 at Aga Khan University, Karachi, Pakistan. A total of 49 patients under the age of 18 years with a minimum of 1-year follow-up were included in the study. The ethical review committee approved this retrospective record review (ERC no. 2022-7168-20,979). Diagnoses of all craniopharyngiomas were confirmed by histologic assessment of the tumor. Data regarding baseline characteristics including clinical features at presentation, radiological findings (tumor localization and characteristic, hydrocephalus, calcification, and consistency), management in terms of surgery, omaya reservoir (interferon therapy), and radiation was collected. Endocrine and vision status before and after surgery and outcome were collected from the medical and electronic record. Long-term health conditions including periods of follow-up and disease/treatment-related morbidities (visual problems, hypothalamic disorders, and endocrinopathies) were also recorded. The determination of the resection extent was guided by the neurosurgeon’s evaluation and corroborated by postoperative imaging, when accessible. The term gross total resection (GTR) refers to the complete excision of the tumor as per the documentation of the operating neurosurgeon (microscopic examination), a fact that is further corroborated by subsequent neuroimaging. Conversely, subtotal resection (STR) indicates an incomplete tumor removal, a status that is declared by the treating neurosurgeon and subsequently confirmed via neuroimaging. Body mass index was subdivided into four categories (low, normal, overweight, and obese). BMI more than the 95th age- and sex-adjusted centile was defined as obesity. A body height of less than the − 2SDrd age-adjusted centile was defined as short stature. The visual problem at the time of presentation was assessed by different categories, normal, mild to moderate visual field defects, unilateral and bilateral blindness, and hypothalamic disorder was assessed by reviewing records regarding a complaint of mood disorder, sleep disturbance, increased appetite, and temperature instability. Pituitary function was assessed by checking pre- and post-op pituitary hormonal profile.
Statistical analysis
Data was analyzed by using SPSS (Statistical Package for Social Science for Windows version 20.0). Tables and charts were created on SPSS version 20 and Microsoft Excel version 2010. Descriptive statistics were reported as mean/median standard deviation/interquartile range for quantitative variables depending on data distribution and frequency (percentage) for qualitative variables.
Results
Demographic variables
A total of 49 records were reviewed of patients presenting with craniopharyngioma between 2001 and 2020. The mean age of patients at the time of the initial diagnosis was 9.5 years (SD ± 4.5 years, range 3–18 years); 8 (16%) were < 5 years of age, 19 (38.8%) were between 5 and 10 years, and 22 (44.9%) were > 10 years of age (Table 1). Twenty-six patients were male and 23 were female. In 9 (18%) craniopharyngioma children, the height at the time of diagnosis was < − 2SDS. The mean duration of symptoms was 11.4 months (1–48 months). The most common symptoms at initial presentation were headache 41 (83.6%), visual deficit 40 (81.6%), nausea and vomiting 26 (53%), and endocrine abnormalities 16 (32%) (Table 1).
Characteristics of tumor in imaging
MRI investigations were conducted for 44 patients (89%). Approximately 69% (34 cases) had lesions located in the suprasellar region. 14.2% (7 cases) showed involvement of both the suprasellar and sellar regions, and 1 case had an extension into the parasellar region. Only one patient had a lesion confined to the sellar region, another was involved in both the sellar and infrasellar regions, and one patient was involved in the sellar region with an extension into the parasellar region. Around 4% (2 cases) had lesions that encompassed all areas, including the sellar, suprasellar, and infrasellar regions. Additionally, 3 cases (6%) presented with extensive tumors, one extending into the cerebral region. One case exhibited tumor extension to the nasopharynx, sphenoidal sinuses, and nasal cavity, while the third case showed tumor extension to the interpeduncular cistern and the foramen of Monro. Among these, 34 (69%) presented with a tumor exhibiting a combination of solid and cystic consistency. A subset of 14 patients (28%) demonstrated predominantly cystic characteristics, and a solitary case (2%) exhibited a purely solid tumor consistency. Most patients harbored tumors that measured between 4.1 and 6 cm, accounting for 22 cases (44%). This was followed by 20 patients (40%) whose tumor sizes ranged from 2.1 to 4 cm, 4 patients (8%) with tumor sizes greater than 6 cm, and 3 patients (6.1%) presenting with tumors less than 2 cm in size. Calcifications were visible within the tumors of 32 patients (65%), and 24 cases (48%) were accompanied by hydrocephalus, as evident from imaging studies. Hypothalamic involvement was discernible on initial imaging for a significant proportion of patients, encompassing 42 cases (85%) (refer to Table 2 for further details). In 32 (65%) patients, tumor calcification was seen and 24 (48%) had hydrocephalus on imaging and almost all of these had 42 (85%) hypothalamic involvement on initial imaging (Table 2).
Surgical technique and outcome
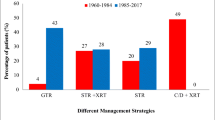
The mean duration between initial imaging and the time of surgery was 3.2 months (0–24 months). In 13 (26%), initial surgery was performed at other institutes. The initial treatment was STR (subtotal resection) in 38 (77%) patients. Out of which, 11 (28%) received radiotherapy and 27 (72%) did not receive radiotherapy. Gross total resection (GTR) was done in 11 (22%) patients. The surgical approach was craniotomy in 39 (80%) patients and transsphenoidal route in 9 (18.3%) patients; in one patient, biopsy was done, and a VP shunt was placed. The mean follow-up time of all patients after primary surgery was 5 years (± 2.1SD, range: 2–10 years). Residual disease in the post-op scan was observed in 38 (77%) patients, and about 21 (42%) had repeated surgeries within few months of initial surgery while 26 (53%) had multiple surgeries. Recurrence after complete resection and tumor progression of residual tumor was observed in 3 (27%) of 11 patients who underwent GTR. Patients with subtotal had tumor progression; 17 (44%) out of 38 children underwent STR.
Histopathologic findings of the majority 40 (81%) revealed adamantinomatous type; 3 results were inconclusive. In 5 patients, histopathology was not sent and only one patient was found to have papillary type in histopathology. Omaya reservoir was placed in 11 (22%) patients; 6 (12.2%) patients received interferon therapy. VP shunt was placed in 20 (40%) patients. Table 2 summarizes this information.
Endocrine complications
Preoperative endocrine hormone evaluation was done in 27 (55%) patients, 10 (20%) were hypothyroid, 5 (10%) had adrenal insufficiency, and 4 (8%) had DI. Four (8%) pubertal children had biochemically proven hypogonadism, 4 (8%) were growth hormone deficient, and a total of 6 (12%) had panhypopituitarism pre-op. After primary surgery, 37 (75%) of 49 children required hormonal replacement therapy; record of 3 patients was missing. Hypothyroidism is the most common endocrine abnormality found in our patient 37 (75%) followed by DI 34 (69%) and hypercortisolism 33 (79%) was identified in the immediate postoperative period; 37 (75%) patients were panhypopituitarism after 1 year of surgery and required long-term hormonal replacement. Summary is in Table 3.
Neurological and visual complications
One child presented with cranial nerve palsy, 10 (20%) had seizures and 6 (12%) presented with drowsiness. Forty-one (83.6%) developed postoperative deterioration of visual and neurological complications including total blindness (n = 7), unilateral loss of vision (n = 15), strabismus or mild visual field defect (n = 19), and hemiparesis (n = 5) (Table 3).
Hypothalamic symptoms
Nine (18.3%) patients presented with obesity while 3 had body weight > 95% centile and all three had hypothalamic involvement on their initial MRI scans. They had an increased propensity for higher BMI postoperatively as well. Post-surgery, many children developed polyphagia and about 14 (28%) and 4 (8.1%) were overweight and developed hypothalamic obesity. About half of the patients 25 (51%) were lost to follow-up so it is difficult to estimate the true number. Preoperatively, 7 (14%) had hypothalamic symptoms like increased appetite in 3 (6%) patients and altered sleep in 1 patient, and 3 (6%) patients had more than one hypothalamic symptom including mood changes. Postoperatively, 18 (36%) developed hypothalamic symptoms at 1-year follow-up (Table 3). We could not find a significant relationship between gross total resection and the manifestation of hypothalamic symptoms, as indicated by a p-value of 0.06. This lack of significance may be attributed to the presence of hypothalamic involvement of the tumor at the time of diagnosis.
Survival and lost to follow-up
About 19 (38%) patients followed in clinic with mean follow-up duration of 5 years (± 2.1SD, range: 2–10 years). However, approximately 10% (5 individuals) sadly passed away, and it is important to note that none of these deaths occurred during the surgery itself or in the immediate perioperative period. We cannot predict the true survival rate because 25 patients were lost to follow-up (Table 3).
Discussion
This study offers an extensive overview of the epidemiological characteristics, clinical presentation, and treatment outcomes of children with craniopharyngioma at a tertiary care hospital in Pakistan over the last 20 years. Prior studies have concentrated on the clinicopathological aspects and surgical approach and outcomes of patients of all ages who had craniopharyngioma [8, 9]. In contrast to prior publications, which have reported cumulative data for both adults and children, our study focuses specifically on the demographic and tumor characteristics, surgical outcomes, and complications in pediatric patients. By analyzing a cohort of 49 pediatric patients, our study provides a distinct and targeted perspective of this population in Pakistan, offering valuable insight into this understudied group [10].
The age of the patient is a crucial demographic feature in craniopharyngioma due to its bimodal age distribution. Our cohort showed 38.8% of the patients were aged between 5 and 10 years, and 44.9% were over the age of 10. These results are in line with previous studies that have reported that children comprise approximately half of all craniopharyngioma cases, with the highest incidence observed between the ages of 5 and 14 years [11].
The duration of symptoms in children varies widely, and it can be challenging to estimate precisely in our setting because children present late and parents are usually unaware of the morbidities they have developed with the progression of their tumors. A study published in the Journal of Pediatric Endocrinology and Metabolism found that children with craniopharyngioma had a longer duration of symptoms; mean was 14.4 months [12]. It should be emphasized that these are only mean values and individual cases may differ significantly. Moreover, some children may not display any symptoms until the tumor has progressed to a more advanced stage. The length of time that symptoms have been present is a crucial factor in the diagnosis of craniopharyngioma, and it is frequently correlated with the tumor’s size and location [12].
We confirm that similar to other research studies [13, 14], headache was the most prevalent symptom, and younger age was more commonly associated with neurological symptoms, while older age as an adolescent was linked to growth failure. Our study found that 83% of patients presented with headaches, which is consistent with previous studies that have shown that headache is the most common presenting complaint in patients with craniopharyngioma owing to the tumor location and mass effects.
The initial BMI of patients with craniopharyngioma is an important aspect to consider due to the tumor’s impact on the hypothalamus. Studies also have indicated that over 50% of children with craniopharyngioma may develop morbid obesity after undergoing radical surgery, which may lead to further weight gain or obesity [15]. Our study found that 9 patients (8.1%) were overweight, and 3 patients (6.1%) were obese at presentation. After surgery and 1-year follow-up, some patients experienced polyphagia, and approximately 4 patients (8.1%) developed morbid obesity, 14 patients (28%) being overweight. Unfortunately, half of the patients were lost to follow-up, so we anticipate that there may be more cases of obesity among them.
Other studies have found higher rates of visual dysfunction in children with craniopharyngioma at presentation (55–75%), emphasizing the importance of assessing visual function whenever a serious intracranial process is suspected [14]. However, the rate of similar complications was greater in our setting, with 83.6% having neurological and eye symptoms in long-term follow-up postoperatively, and this is comparable to other studies also [4, 14, 16, 17].
Craniopharyngioma can have significant effects on the endocrine system due to its location in the pituitary gland and hypothalamus. Also the extent of surgery strongly influences the degree of hypothalamic–pituitary dysfunction [18]. Our study demonstrates the high prevalence of endocrine complications associated with this type of tumor. A preoperative endocrine hormone evaluation was conducted on 27 patients, representing approximately 55% of the total. However, it is important to note that there were cases where data on preoperative evaluations were missing. This was primarily due to the lack of established practices among treating neurosurgeons regarding referrals for preoperative hormonal assessments to pediatric endocrinologists. It is worth mentioning that significant improvements have been made in recent years, and currently, all patients are evaluated before undergoing surgery.
It is important to note that the evaluation revealed significant endocrine abnormalities in a substantial proportion of patients. These findings are consistent with previous studies that have shown a high prevalence of endocrine abnormalities in patients with craniopharyngioma before surgery [19]. After primary surgery, 75% of children required hormonal replacement therapy, which also highlights the significant impact of surgery on the endocrine system. Similar findings were confirmed by other studies [20]. Among all, hypothyroidism was the most common endocrine abnormality found in our patient population. Which is also consistent with the previous studies [21]. However, some studies found growth hormone and hypogonadism as the most common endocrine abnormalities [22].
One year after surgery, 37 (75%) patients were found to have panhypopituitarism and required long-term hormonal replacement therapy. This finding is consistent with previous research that has shown that craniopharyngioma is associated with a high risk of irreversible panhypopituitarism following surgery [4, 23]. Our study emphasizes the substantial impact of craniopharyngioma on the endocrine system, as evidenced by the high incidence of preoperative endocrine abnormalities and the necessity for long-term hormonal replacement therapy after surgery. These findings highlight the importance of continuous follow-up care for patients with craniopharyngioma and provide valuable insights for clinicians to improve patient care and outcomes.
Although surgical management is the main modality of management in craniopharyngioma, there is an ongoing debate regarding the most effective treatment approach for craniopharyngioma in children, specifically whether primary radical surgery or conservative surgery followed by radiation therapy offers the best chance of cure with the lowest risk of long-term complications [4]. STR is associated with a higher risk of recurrence and may require additional treatment modalities such as radiotherapy. On the other hand, some studies confirm that radical surgery results in a high probability of tumor control. However, such surgery often leads to significant damage to visual, hypothalamic, endocrine, and neurocognitive functions [4]. The findings of our study indicate that the majority of patients underwent STR 38 (77%), while GTR was performed in 11 (22%) of cases of them. When complete resection of a tumor poses a significant risk of complications due to its location and size, STR may be preferred. However, Diana et al. found that there was no significant difference in overall survival between patients who underwent GTR and STR followed by postoperative radiotherapy, highlighting the importance of individualized treatment plans for patients with craniopharyngioma [24]. Moreover, in treating craniopharyngioma, the choice of surgical approach is crucial for achieving the best outcome. In our cohort, the most common surgical approach used was craniotomy in 80%, while the transsphenoidal technique was used in 18.3% of patients. Craniotomy is a more invasive approach associated with a higher risk of complications, while the transsphenoidal approach is minimally invasive and can reduce the risk of complications. Previous studies also confirm that the choice of surgical approach depends on the tumor’s size and location and the surgeon’s expertise. However, the transsphenoidal approach is found to be safer than the craniotomy in literature [25]. It later became the standard of choice surgical technique for children presented with craniopharyngioma in our institute.
Our study depicted that 77% of patients had residual disease after surgery, and 42% required additional surgeries within a few months of their initial operation. Additionally, we observed a higher recurrence rate with 27% of patients after GTR experiencing recurrence after complete resection. These results emphasize more about the importance of long-term monitoring and surveillance after the surgical procedure. Furthermore, although the average duration of follow-up after the initial surgery was 5 years, a concerning 51% (25 patients) did not attend follow-up appointments at the clinic. Unfortunately, the return for observation and surveillance of craniopharyngioma patients and radiotherapy after STR is limited due to many factors such as inaccessibility of healthcare facilities at their respective areas, lack of awareness, inefficient health surveillance systems, and limited tertiary care facilities with multidisciplinary care. These challenges hinder regular follow-up and care. In addition to these factors, there are several other challenges like lack of insurance coverage, which makes this treatment option inaccessible for many needed highly specialized services. Moreover, philosophical or religious beliefs are often driven by a lack of awareness about the condition and its treatment options. These socioeconomic and geographic factors contribute to disparities in treatment access and long-term morbidity outcomes among craniopharyngioma patients in low/middle-income countries. Addressing these issues requires improving healthcare access, increasing awareness, reducing costs, strengthening surveillance systems, and enhancing healthcare professional training.
Conclusion
In summary, the results of the study on demographic characteristics of craniopharyngioma patients are consistent with existing literature on the presentation and epidemiology of this rare brain tumor. The findings highlight the importance of understanding the demographic characteristics of individuals with craniopharyngioma to develop multidisciplinary teams, site-specific tumor boards, and appropriate treatment plans and improve patient outcomes.
Limitations and future perspective
We acknowledge that our data collection is not sufficient to fully assess the long-term outcomes of children with craniopharyngioma. During the review, we identified several gaps, such as inadequate documentation and records, especially on hypothalamic symptoms, inadequate preop endocrine evaluation, and disorganized care in the early years of the study. Additionally, many patients were lost to follow-up after surgery. We have since made significant improvements, including the evaluation of patients by a neurooncologist and endocrinologist before surgery, the implementation of multidisciplinary team meetings to develop effective management plans, and increased compliance with postoperative follow-up appointments with pediatric endocrinologists. However, we believe that further prospective studies are necessary to investigate other aspects of craniopharyngioma like molecular studies, and outcomes, particularly concerning hypothalamic symptoms, quality of life, cognitive function, behavioral and occupational problems, school performance, and long-term survival rates.
Abbreviations
- STR:
-
Subtotal resection
- GTR:
-
Gross total resection
- LMIC:
-
Low- or middle-income country
- BMI:
-
Body mass index
References
Hoffman HJ (1994) Surgical management of craniopharyngioma. Pediatr Neurosurg 21(Suppl. 1):44–49
Muller HL (2008) Childhood craniopharyngioma. Recent advances in diagnosis, treatment, and follow-up. Hormone Res 69(4):193–202
Müller HL (2010) Childhood craniopharyngioma—current concepts in diagnosis, therapy, and follow-up. Nat Rev Endocrinol 6(11):609–618
Poretti A, Grotzer MA, Ribi K, Schönle E, Boltshauser E (2004) Outcome of craniopharyngioma in children: long-term complications and quality of life. Dev Med Child Neurol 46(4):220–229
Fischer EG, Welch K, Shillito J, Winston KR, Tarbell NJ (1990) Craniopharyngiomas in children: long-term effects of conservative surgical procedures combined with radiation therapy. J Neurosurg 73(4):534–540
Grewal MR, Spielman DB, Safi C, Overdevest JB, Otten M, Bruce J et al (2020) Gross total versus subtotal surgical resection in the management of craniopharyngiomas. Allergy Rhinol 11:2152656720964158
Hoffman HJ, Hendrick EB, Humphreys RP, Buncic JR, Armstrong DL, Jenkin RD (1977) Management of craniopharyngioma in children. J Neurosurg 47(2):218–227
Haroon S, Afzal A, Zia S, Ali SJ, Zia F, Shamail F et al (2021) Clinicopathological features of craniopharyngioma: a 15-year study from a tertiary care center in Pakistan. Cureus 13(3):e14153. https://doi.org/10.7759/cureus.14153
Khalid MU, Shah MM, Bajwa MH, Mirza FA, Laghari AA (2022) Craniopharyngioma: a lower-middle-income-country epidemiology perspective. J Pak Med Assoc 72(Suppl 4)(11):S61–S67
Mazher S, Ashraf J (2015) Craniopharyngioma total or near total surgical resection: an outcome-based learning experience. Pak J Med Dent 4(01):10
Drapeau A, Walz PC, Eide JG, Rugino AJ, Shaikhouni A, Mohyeldin A et al (2019) Pediatric craniopharyngioma. Child’s Nervous. System 35:2133–2145
Lithgow K, Hamblin R, Pohl U, Karavitaki N (2022) Craniopharyngiomas. Endotext. http://www.endotext.org/
Hoffmann A, Boekhoff S, Gebhardt U, Sterkenburg AS, Daubenbüchel AM, Eveslage M et al (2015) History before diagnosis in childhood craniopharyngioma: associations with initial presentation and long-term prognosis. Eur J Endocrinol 173(6):853–862
Hoffman HJ, De Silva M, Humphreys RP, Drake JM, Smith ML, Blaser SI (1992) Aggressive surgical management of craniopharyngiomas in children. J Neurosurg 76(1):47–52
Müller H, Gebhardt U, Maroske J, Hanisch E (2011) Long-term follow-up of morbidly obese patients with childhood craniopharyngioma after laparoscopic adjustable gastric banding (LAGB). Klin Padiatr 223(06):372–373
Yaşargil MG, Curcic M, Kis M, Siegenthaler G, Teddy PJ, Roth P (1990) Total removal of craniopharyngiomas: approaches and long-term results in 144 patients. J Neurosurg 73(1):3–11
Abrams LS, Repka MX (1997) Visual outcome of craniopharyngioma in children. J Pediatr Ophthalmol Strabismus 34(4):223–228
Mortini P, Losa M, Pozzobon G, Barzaghi R, Riva M, Acerno S et al (2011) Neurosurgical treatment of craniopharyngioma in adults and children: early and long-term results in a large case series. J Neurosurg 114(5):1350–1359
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35(6):1547
DeVile CJ, Grant DB, Hayward RD, Stanhope R (1996) Growth and endocrine sequelae of craniopharyngioma. Arch Dis Child 75(2):108–114
Daneman D, Hoffman HJ, Ehrlich RM (1994) The endocrine outcome after surgical removal of craniopharyngiomas. Pediatr Neurosurg 21(1):24–27
Gonc EN, Yordam N, Ozon A, Alikasifoglu A, Kandemir N (2004) Endocrinological outcome of different treatment options in children with craniopharyngioma: a retrospective analysis of 66 cases. Pediatr Neurosurg 40(3):112–119
Karavitaki N, Brufani C, Warner J, Adams C, Richards P, Ansorge O et al (2005) Craniopharyngiomas in children and adults: systematic analysis of 121 cases with long-term follow-up. Clin Endocrinol 62(4):397–409
Stripp DC, Maity A, Janss AJ, Belasco JB, Tochner ZA, Goldwein JW et al (2004) Surgery with or without radiation therapy in the management of craniopharyngiomas in children and young adults. Int J Radiat Oncol Biol Phys 58(3):714–20
Webb KL, Pruter WW, Hinkle ML, Walsh MT (2023) Comparing surgical approaches for craniopharyngioma resection among adults and children: a meta-analysis and systematic review. World Neurosurg 175:e876–e896
Acknowledgements
No individuals other than the listed co-authors contributed to this publication.
Author information
Authors and Affiliations
Contributions
Dr Naureen Mushtaq and Dr Fozia Memon wrote the main manuscript text. Dr Khadija Nuzhat Humayun conceptualized the idea and did proofreading. Dr Kiran Hilal added the radiology section and reviewed the article. Dr Altaf Laghari and Dr Muzna Arif reviewed the article and did the final correction. All authors read and approved the final manuscript with corrections.
Corresponding author
Ethics declarations
Ethics approval
The retrospective data collection of this study has been approved by the institutional ethics committee (2022-7168-20979).
Consent for publication
All authors gave their consent for the publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Memon, F., Humayun, K.N., Riaz, Q. et al. Pediatric craniopharyngioma: a 20-year study on epidemiological features, clinical presentation, and survival outcomes in a tertiary care center from LMIC. Childs Nerv Syst 40, 427–434 (2024). https://doi.org/10.1007/s00381-023-06177-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-023-06177-8