
Abstract
Atypical teratoid/rhabdoid tumors (ATRTs) are malignant central nervous system tumors that affect early childhood (< 3 years), and mostly located in the infratentorial space. Owing to an infrequent occurrence, their radiological features have not been completely defined. Nevertheless, these are characteristically intra-axial except for few instances in the cerebellopontine angle region. We describe a case of a 10-year-old boy who harbored an extra-axial, dural-based ATRT in the right parietal region. The lesion was totally excised followed by adjuvant chemo-radiotherapy. At 10-month follow-up, he was well with no recurrence. The report intends to highlight an atypical imaging presentation of ATRT in an older child, and adds to the radiological spectrum. This uncommon pathology should be borne in mind, even in a supratentorial dural-based location.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atypical rhabdoid/teratoid tumors (ATRTs), accounting for 1.3% of primary pediatric central nervous system neoplasms, remain one of the most aggressive malignant lesions [1,2,3]. In its typical form, ATRT primarily involves the first 3 years of life (median, 1.3) [3]. However, cases in older children and adults have been increasingly reported [1, 3]. Though in initial reports, the predominant location was the posterior fossa, supratentorial involvement has also been well recognized [1,2,3,4,5]. Irrespective of their localization, ATRTs are predominantly parenchymal lesions. We present a case of an extra-axial, dural-based ATRT in a 10-year-old child and the clinical course along with brief related literature.
Case report
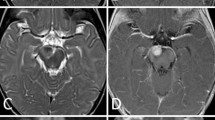
A 10-year-old boy presented with complaint of headache over the right temporo-parietal region of 1-month duration. He also had occasional vomiting episodes. The child had no focal deficits. Systemic examination revealed no organomegaly, bony tenderness, or enlarged lymph nodes. Magnetic resonance imaging (MRI) revealed a 5.6*5.3*4.2 cm extra-axial, dural-based lesion occupying the right parietal region, and causing mass effect (Fig. 1). The tumor was T1 hypo- to hyperintense, T2 hyperintense, and heterogenously contrast-enhancing along with small dural tail. A central T1 hyperintensity with susceptibility on SWI sequence suggested bleed within. On computed tomography (CT), there was no evidence of intratumoral calcification, and calvarium showed no pathological changes.

A–F: Preoperative MRI sections show a right parietal extra-axial, dural-based, T1 hypointense (A), T2 hyperintense (B), FLAIR hyperintense (F) lesion with strong heterogenous enhancement (C–E) and small dural tailing (arrow). G, H: Follow-up contrast MRI shows complete excision of the lesion
Intraoperatively, the overlying bone was healthy. The tumor was multilobulated, soft, suckable, with minimal vascularity but with hemorrhagic necrotic areas. It was stuck to the dura from which it was however easily separable. On deeper surface, it was found to be adherent to a small cortical area. The lesion was totally excised, and postoperative course was uneventful. Based on morphological and immunohistochemical features (loss of nuclear expression for INI-1), a diagnosis of ATRT was rendered (Fig. 2).

A: Neoplastic cells arranged in sheets and loose clusters in a prominent myxoid background (H&E ×200). B: Neoplastic cells arranged around blood vessels (H&E ×200) set against a loose stroma in the background. C: Areas representing solid sheets within a relatively less myxomatous stroma (H&E ×200). D and E: High magnification depicting round to oval cells with variable cytoplasm. No rhabdoid cells were noted. Mitoses are also seen (arrows) (H&E ×400). F: Round to oval cells with vesicular nuclei and conspicuous nucleoli. Mitoses can be also seen (arrow) (H&E ×1000). G: Neoplastic cells negative for GFAP, desmin, SMA, cytokeratin, synaptophysin, S-100 (immunoperoxidase ×400). H: Loss of nuclear expression for INI-1. Endothelial cells act as positive internal control (immunoperoxidase ×400). I: Ki-67 labelling index was high (immunoperoxidase ×200)
The child underwent chemo-radiotherapy (55.5 Gy in 32 fractions with 6 cycles of vincristine, cisplatin, and cyclophosphamide). Follow-up MRI at 10 months showed no recurrence.
Discussion
ATRTs are rare, aggressive embryonal tumors characterized by fast growth potential, a large size at presentation, and, in most cases, an unfavorable outcome [1,2,3,4, 6]. Earlier, many of these cases were misdiagnosed as primitive neuroectodermal tumor (PNET)/medulloblastoma because of potential histological resemblance between the pathologies [2, 7]. Nevertheless, the polyimmunophenotypic nature of the neoplastic cells and aggressive clinical behavior compelled their recognition as a distinct entity, and ATRTs were classified as WHO grade IV embryonal tumors [8]. Mutation or loss of SMARCB1 locus at 22q11.2 identified as the genetic hallmark for this tumor leads to loss of nuclear SMARCB1 protein (INI1) in approximately 95% of cases [9, 10].
Even the imaging of ATRT is heterogenous, and shares many characteristics with PNET causing difficulty in reliably distinguishing the two lesions [1, 2]. Their differentiation however becomes important because the prognosis of ATRT is worse than that of the PNET [2]. ATRTs are usually large, solid, or solid cystic mass with mixed T1/T2 signal intensities, and show heterogenous enhancement due to varied cellularity and presence of features such as hemorrhage, necrosis, and calcification; despite being aggressive tumors with brain invasion and significant mass effect, perilesional edema is usually minimal [1, 2, 6]. These tumors show restricted diffusion, and on spectroscopy demonstrate raised choline and decreased N-acetyl aspartate levels. Occasionally, ATRTs may show leptomeningeal enhancement suggesting disseminated disease [1, 2]. On CT, they are hyperdense, and calcifications can be seen in half of the cases. A tendency to occur in off-midline locations, eccentric cysts, and visible calcifications may favor diagnosis of ATRT over PNET [6].
Typically, the ATRTs are intra-axial, and commonly located in the cerebellar vermis [1, 2]. Rarely, extra-axial tumors can occur in the cerebellopontine angle location [1, 2]. In the supratentorial location, they present as cortical parenchymal mass with predilection for the frontal lobe, and can also arise from midline structures such as the pineal region, third ventricle, and basal ganglia [2,3,4]. A supratentorial occurrence has been noted in up to 41%, and preferentially in children >3 years of age [2, 3]. Though ATRT may involve overlying meninges, predominant extra-axial growth is extremely rare. There has been only a single report of supratentorial extra-axial ATRT in a 22-month-old child in the temporal region [11]. However, this particular case was different. First, the occurrence was in slightly older-aged. Second, the radiology suggested an avidly contrast-enhancing dural-based lesion with a small dural tail mimicking other frequently encountered dural pathologies. In children with such mass, an array of neoplastic differentials exists which include Ewing’s sarcoma/PNET, leukemic deposits, metastatic neuroblastoma, and rarely meningioma and medulloblastoma [12, 13]. Among the differentials, though the tumors with round-cell morphology can be distinguished based on specific molecular abnormalities, meningioma needs special mention. Among its histological variants, chordoid meningioma is the closest mimic as it demonstrates INI-1 loss similar to ATRT; however, these tumors are characterized by cords or trabeculae of small epithelioid, variably vacuolated cells embedded in a mucin-rich matrix [14, 15]. Occasionally, inflammatory pathologies such as Rosai-Dorfman disease can also be dural-based [13].
The histopathology of ATRT is diverse. They typically show small round cells with epithelial and mesenchymal features along with the presence of rhabdoid cells. Notably, rhabdoid cells as a predominant histopathological finding are seen in only a minority, and may be very rare or even completely lacking in some cases, similar to the index case [6, 16]. Immunohistochemically, these tumors are polyimmunophenotypic. Loss of nuclear expression of INI-1 is a highly sensitive marker for the diagnosis of AT/RT [17].
Owing to relative paucity of cases, consensus guidelines for optimal management of ATRT are less established. [4]. Overall, the disease prognosis is poor with mean survival of 8 months [1, 4]. A younger age and leptomeningeal dissemination at presentation mark worse prognosis; in the latest pediatric series, up to 60.5% showed disseminated disease [1, 3]. Although the main treatment modality remains safe surgical resection, a multimodal therapy consisting of aggressive attempt for total resection, chemotherapy, and radiotherapy has recently resulted in better outcomes [3, 4]. The extent of resection has also been associated with improved outcome. However, it is often hampered by tumor occurrence in a very young age and in eloquent locations. In this case, the tumor could be totally resected as it was extra-axial, and was not found to be tightly adhered to the overlying dura with minimal cortical infiltration.
To summarize, the imaging patterns of ATRT are being continually characterized. We describe an unusual form of this tumor that expands its radiological spectrum. We suggest considering ATRT in the differential of dural-based lesions in children in whom the radiology is not typical of other commonly encountered pathologies.
References
Lee IH, Yoo SY, Kim JH, Eo H, Kim OH, Kim IO, Cheon JE, Jung AY, Yoon BJ (2009) Atypical teratoid/rhabdoid tumors of the central nervous system: imaging and clinical findings in 16 children. Clin Radiol 64(3):256–264
Meyers SP, Khademian ZP, Biegel JA, Chuang SH, Korones DN, Zimmerman RA (2006) Primary intracranial atypical teratoid/rhabdoid tumors of infancy and childhood: MRI features and patient outcomes. AJNR Am J Neuroradiol 27(5):962–971
Yamasaki K, Kiyotani C, Terashima K, Watanabe Y, Kanamori M, Koga Y, Hata N, Iwasaki F, Goto H, Koh K, Kurihara J, Tokunaga S, Arakawa Y, Hasegawa D, Kosaka Y, Hara J (2019) Clinical characteristics, treatment, and survival outcome in pediatric patients with atypical teratoid/rhabdoid tumors: a retrospective study by the Japan Children’s Cancer Group. J Neurosurg Pediatr Nov 15:1–10. https://doi.org/10.3171/2019.9.PEDS19367 Epub ahead of print
Chen ML, McComb JG, Krieger MD (2005) Atypical teratoid/rhabdoid tumors of the central nervous system: management and outcomes. Neurosurg Focus 18(6A):E8
Gendle C, Karthigeyan M, Salunke P, Gupta K (2020) Pineal atypical teratoid rhabdoid tumor in a 5-month-old child. Pediatr Neurosurg Dec 10:1–5. https://doi.org/10.1159/000511995 Epub ahead of print
Arslanoglu A, Aygun N, Tekhtani D, Aronson L, Cohen K, Burger PC, Yousem DM (2004) Imaging findings of CNS atypical teratoid/rhabdoid tumors. AJNR Am J Neuroradiol 25(3):476–480
Burger PC, Yu IT, Tihan T, Friedman HS, Strother DR, Kepner JL, Duffner PK, Kun LE, Perlman EJ (1998) Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol 22(9):1083–1092
Ho DM, Hsu CY, Wong TT, Ting LT, Chiang H (2000) Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma. Acta Neuropathol 99(5):482–488
Biegel JA, Rorke LB, Packer RJ, Emanuel BS (1990) Monosomy 22 in rhabdoid or atypical tumors of the brain. J Neurosurg 73(5):710–714
Biegel JA, Tan L, Zhang F, Wainwright L, Russo P, Rorke LB (2002) Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 8(11):3461–3467
Bing F, Nugues F, Grand S, Bessou P, Salon C (2009) Primary intracranial extra-axial and supratentorial atypical rhabdoid tumor. Pediatr Neurol 41(6):453–456
Gupta K, Karthigeyan M, Satapathy A, Salunke P (2018) Synchronous solitary calvarial yolk sac tumor metastasis as the initial presentation of mediastinal germ cell tumor. Childs Nerv Syst 34(2):363–366
Perry A, Dehner LP (2003) Meningeal tumors of childhood and infancy. An update and literature review. Brain Pathol 13(3):386–408
van den Munckhof P, Christiaans I, Kenter SB, Baas F, Hulsebos TJ (2012) Germline SMARCB1 mutation predisposes to multiple meningiomas and schwannomas with preferential location of cranial meningiomas at the falx cerebri. Neurogenetics 13(1):1–7
Couce ME, Aker FV, Scheithauer BW (2000) Chordoid meningioma: a clinicopathologic study of 42 cases. Am J Surg Pathol 24(7):899–905
Haberler C, Laggner U, Slavc I, Czech T, Ambros IM, Ambros PF, Budka H, Hainfellner JA (2006) Immunohistochemical analysis of INI1 protein in malignant pediatric CNS tumors: Lack of INI1 in atypical teratoid/rhabdoid tumors and in a fraction of primitive neuroectodermal tumors without rhabdoid phenotype. Am J Surg Pathol 30(11):1462–1468
Judkins AR, Mauger J, Ht A, Rorke LB, Biegel JA (2004) Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol 28(5):644–650
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Nil
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Karthigeyan, M., Mondal, P., Salunke, P. et al. Extra-axial, dural-based atypical teratoid/rhabdoid tumor. Childs Nerv Syst 38, 655–658 (2022). https://doi.org/10.1007/s00381-021-05196-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-021-05196-7