
Abstract
Background
Primary central nervous system (CNS) neuroblastoma is a rare intracranial tumor affecting children mainly in the first years of life. It is usually a supratentorial tumor with a wide spectrum of clinical presentation, seizures, and focal neurological deficits being the most common presenting signs.
Case description
A 2-year-old child was admitted to our ward after a generalized seizure. Neurological examination was normal. Radiological studies showed a small DWI hyperintense lesion of the right rectus gyrus. Follow-up brain MRI 8 months later showed a huge growth of the tumor (90 × 80 × 65 mm) with polycyclic and apparently defined margins, cystic components, and diffuse contrast enhancement. Complete tumor removal was performed in two planned surgical steps. Histological diagnosis was CNS neuroblastoma. At a follow-up of 8 months, the child is in good clinical and neurological condition and is completing chemotherapy treatment according to the SIOP PNET 4 protocol.
Discussion and conclusion
A thorough review of the literature confirms that primary CNS neuroblastoma has to be considered a distinct entity. The disease related mortality is 12.5%, lower than the one usually reported for other previously described as PNETs tumors. The most relevant factors influencing prognosis are the possibility of obtaining a complete tumor removal and age more than 3 years, which allows to include radiotherapy among treatment options.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the 2016 WHO classification of the CNS tumors, among embryonal tumors primary CNS neuroblastoma is defined as an embryonal tumor characterized by poorly differentiated neuroepithelial cells, groups of neurocytic cells, and variable neuropil-rich stroma [1]. The tumor is relatively rare, and only a limited number of cases have been in detail reported in the literature. Due to its rarity, little is known also about treatment regimens and patients risk stratification; the low age at diagnosis of the majority of the patients represent in this context a limit in the choice of the management protocols and represent a factor which might condition the prognosis.
Background
General epidemiological considerations
From the analysis of the literature primary, CNS neuroblastomas tend to involve preferentially the supratentorial space, with a prevalence of the frontal and the parietal region. The mean age of the reported patients is 5 years confirming that neuroblastoma is substantially a tumor of early infancy. A slight female prevalence has been reported.
Genetic considerations
In the new WHO 2016 classification, CNS-PNET is no longer regarded a single disease but encompassed many distinct molecular entities. A recent study demonstrated that a large number of previously diagnosed CNS-PNETs display molecular profiles indistinguishable from those of various other well-recognized CNS tumors (such as ATRT, ETMR, and H3.3G34-mutated glioma) or other systemic mesenchymal tumors (such as Ewing sarcoma). However, among the remaining putatively “true” CNS-PNETs, the molecular investigation revealed a group of lesion with FOXR2 activation. This group encompassed tumors that would be classified as CNS neuroblastoma or CNS ganglioneuroblastoma in the 2007 WHO classification scheme and have been therefore defined as FOXR2 CNS-neuroblastoma (CNS NB-FOXR2) [2].
Clinical presentation
Primary CNS neuroblastomas may present with a wide spectrum of symptoms due to the variety of possible location of the mass as well as of the size of the lesion.
In line with the preferential supratentorial location, clinical presentation has been reported to vary from focal signs and symptoms due to focal mass effect (focal neurological deficits), to irritative signs (seizures), chronic signs of CNS functions disturbance (neurocognitive impairment), up to signs of increased intracranial pressure due to CSF pathways impairment (headache, vomiting, abnormal head circumference growth) (Table 1) [3,4,5,6,7,8,9,10]. In infants on the other hand, even though the tumor reaches a huge size, no clear sign or symptom might be present, due to compensatory and adaptation mechanisms of the surrounding brain structures.
Metastatic spread of primary CNS neuroblastoma at diagnosis has been reported in only two cases. In one of them, the spreading route was through the CSF pathways and in the other case through the cervical lymph nodes [3].
Diagnosis
The diagnosis of a primary CNS neuroblastoma is most commonly performed on the removed tumor tissue at the time of the histological examination. The basic MR sequences appearance is, indeed, not unique of the disease. In most cases, the tumor appears as heterogeneous due to the common coexistence of solid and cystic components and is apparently separated from the normal CNS tissue. However, pure solid tumors have been described. The solid part is usually isointense in T1 and T2 sequences, slightly hyperintense in DWI and might also not partially enhance after Gadolinium administration. When present that cystic components are usually hyperintense in T2 sequences due to their hyperproteic content. Surrounding edema is inconstant, whereas vascular structures crossing the tumor are not uncommon. Spectroscopy shows an increase in the Choline peak and an inversion of the Cho/NAA rate, indicating an aggressive brain tumor, but again not a unique feature of primary CNS neuroblastomas. Perfusion sequences show elevated values of relative cerebral blood volume (rCBV) with a tendency to a recover of the slope [5].
As for any suspected malignant brain tumor, an enhanced scan of all the neuraxis should be performed and CSF samples should be obtained to search for CSF tumor spreading.
Two are the aspects that need to be respected to define a CNS tumor as a primary neuroblastoma: first of all, the histological findings and second, the exclusion of the presence of a systemic neuroblastoma. There are no tumor blood markers or radiological signs that are pathognomonic and/or might help in the definition of a primary neuroblstoma.
Histology
CNS neuroblastomas are neuro-epithelial/neurocytic tumors with high cellularity and relatively monomorphic cells with a round nucleus that might be irregular or enlarged. Cytoplasm is not abundant and PAS negative. A high number of mitosis is usually present. The cells are Vimentin, GFAP, S-100, and EMA negative while positivity might be found for MAP2, synaptophysin, Olig2, and Neu-N. Homer-Wright rosettes and ganglionic differentiation may occur [10]. The tumor is usually LIN28A negative and expresses INI1.
Management
First line of treatment is represented by surgery which is aimed to a complete tumor resection whenever possible. Surgical treatment for larger sized tumors should take into account a provisional elevated volume of blood loss, which, especially in younger children, should lead the surgeon to a planned staged tumor resection.
Chemotherapy has been the most frequently performed adjuvant treatment. Due to their histological aggressiveness and their “niche” brain location, CNS neuroblastomas are treated according the supratentorial PNET protocols [6]. According to the presence or absence of metastasis at diagnosis, different regimens have been proposed. For non-metastatic tumors, a therapy with a combination of multiple chemotherapeutic agents is considered. The most common ones are vincristine (1.5 mg/m2 (maximum, 2 mg)), lomustine (75 mg/m2,) cisplatin (70 mg/m2), and etoposide. On the other hand, metastatic neuroblastomas are treated with single high-dose chemotherapeutic agents in sequence such as the sequence of cisplatin, methotrexate, etoposide, temozolomide, and steroids.
Radiotherapy has had a limited role as the usual age of primary CNS neuroblastoma patients is less than 3 years old.
It is difficult to compare primary and systemic neuroblastoma from a prognostic point of view as no paper is clearly focused on such a matter. The younger than 3 years age at presentation of most children with primary neuroblastoma exclude in principle the possibility to perform adjunctive radiotherapy, which also is the only reliable combined treatment option after 3 years, leading to a higher rated of possibilities for tumor recurrence and hence for a worse prognosis.
Exemplary case description
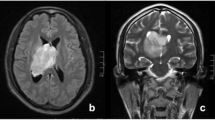
A 2-year-old child was admitted in our ward after a generalized tonic-clonic seizure. On admission, the patient was neurologically intact. A CT scan showed an area of spontaneous hyperdensity on the right rectus gyrus (15 × 10 mm). The brain MRI before and after Gadolinium injection confirmed the alteration seen on the CT scan, only visible on DWI sequences and not enhancing after Gadolinium injection (Fig. 1). Considering the high signal in the DWI sequence, surgery was proposed to the family in order to obtain a histological diagnosis. The family refused surgery. Eight months later, the patient came back to our ward again in good neurological conditions, without any neurological deficit and a negative anamnestic history since the previous admission. A control MR was performed, showing a huge growth of the tumor (90 × 80 × 65 mm) with polycyclic and defined margins, cystic components, and heterogeneous contrast enhancement (Fig. 2). Spectroscopy documented an increase of choline and inversion of the choline/N-acetyl aspartate ratio. Perfusion was clearly increased (2.5:1). This radiological progression is unusual for primary CNS neuroblastomas, none of the previous papers having reported such a fast and clinically asymptomatic tumor growth. A staged tumor removal was planned. The aim of the first surgery was to grossly devascularize the tumor and to remove its front mesial component.

DWI, T2, and enhanced T1 MR scans showing a small DWI hyperintense, slight T1 contrast enhancing oval-shaped lesion of the right frontal gyrus

DWI, T2 inversion recovery, and enhanced T1 MR images after 8 months, showing a huge growth of the tumor. The lesion appears highly vascularized, encasing both anterior cerebral arteries (arrows)
Due to brain parenchyma herniation and the planned second stage of surgery, at the end of this first operation the bone flap was not re-positioned.
The postoperative course was uneventful, the patient remaining in good clinical and neurological conditions. The postoperative MR scan showed a subtotal tumor resection. Few days later, after a good clinical recovery, a second-step surgery was performed completing the tumor removal with back positioning of the bone flap.
The patient had a prompt clinical recovery and did not show any postoperative neurological deficit. The control MRI scan confirmed a complete tumor removal in the absence of complications (Fig. 3). The histological diagnosis was: embryonal neoplasm with neuronal differentiation characteristic of a CNS primary neuroblastoma (WHO 2016 grade IV) [1]. The tumor, which was well demarcated from the surrounding brain parenchyma, showed high cellularity and consisted of proliferation of medium-sized rounded cells which were mostly MAP2+/OLIG+/GFAP− and partially positive for neuronal markers synaptophysin and Neu-N, features that are the most relevant to direct the histological diagnosis to the one of a primary neuroblastoma. Part of the cells cytologically showed neurocytic and ganglionic differentiation. The tumor cells did not display positivity for LIN28A. The expression of INI1 was retained. The tumor showed a high proliferation (MIB1: 20%) and high mitotic index (Fig. 4). The baby was scheduled for chemotherapy. A total body CT scan excluded a systemic involvement. The brain MR before the start of chemotherapy showed the presence of a bilateral subdural hygroma.

DWI, FLAIR, and enhanced T1 MR images after second surgery showing a complete tumor removal

Case sample histological study.
Because of its asymptomatic occurrence, the subdural hygroma was initially treated conservatively. Two months later, the child started to complain headache. A CT scan showed an increase in size of the bilateral subdural hygroma; for this reason, a subduro-peritoneal shunt was implanted (programmable valve set at 10 cm H2O).
Discussion and conclusions
Primary CNS neuroblastomas are a rare condition in the pediatric neurosurgical practice. The histological features and clinical behavior lead to consider them as highly malignant tumors. However, the disease-related mortality reported in the literature is 12.5%, lower than the one usually reported for other embryonal tumors. A dedicated treatment protocol has not been established yet; even if surgery remains the first step of treatment, adjuvant strategies are not specifically tailored to neuroblastomas. A further limiting clue is represented by the fact that radiotherapy is not an option in most cases due to the age lower than 3 years of the majority of the affected children; analyzing data from the literature ( 1), it is possible to see how, in children older than 3 years, in which radiotherapy is a treatment option, the survival rate and the length of the survival is higher. The rarity of the pathology further contributes to render the task of establishing a uniform treatment regimen is not easy though.
Change history
08 March 2019
Unfortunately in the original publication, the affiliation provided for the author G. Tamburrini was incorrect. The correct affiliation for G. Tamburrini should read as follows:
08 March 2019
Unfortunately in the original publication, the affiliation provided for the author G. Tamburrini was incorrect. The correct affiliation for G. Tamburrini should read as follows:
References
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol (Berl) 131:803–820. https://doi.org/10.1007/s00401-016-1545-1
Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D, Sill M, Buchhalter I, Northcott PA, Leis I, Ryzhova M, Koelsche C, Pfaff E, Allen SJ, Balasubramanian G, Worst BC, Pajtler KW, Brabetz S, Johann PD, Sahm F, Reimand J, Mackay A, Carvalho DM, Remke M, Phillips JJ, Perry A, Cowdrey C, Drissi R, Fouladi M, Giangaspero F, Łastowska M, Grajkowska W, Scheurlen W, Pietsch T, Hagel C, Gojo J, Lötsch D, Berger W, Slavc I, Haberler C, Jouvet A, Holm S, Hofer S, Prinz M, Keohane C, Fried I, Mawrin C, Scheie D, Mobley BC, Schniederjan MJ, Santi M, Buccoliero AM, Dahiya S, Kramm CM, von Bueren AO, von Hoff K, Rutkowski S, Herold-Mende C, Frühwald MC, Milde T, Hasselblatt M, Wesseling P, Rößler J, Schüller U, Ebinger M, Schittenhelm J, Frank S, Grobholz R, Vajtai I, Hans V, Schneppenheim R, Zitterbart K, Collins VP, Aronica E, Varlet P, Puget S, Dufour C, Grill J, Figarella-Branger D, Wolter M, Schuhmann MU, Shalaby T, Grotzer M, van Meter T, Monoranu CM, Felsberg J, Reifenberger G, Snuderl M, Forrester LA, Koster J, Versteeg R, Volckmann R, van Sluis P, Wolf S, Mikkelsen T, Gajjar A, Aldape K, Moore AS, Taylor MD, Jones C, Jabado N, Karajannis MA, Eils R, Schlesner M, Lichter P, von Deimling A, Pfister SM, Ellison DW, Korshunov A, Kool M (2016) New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 164:1060–1072. https://doi.org/10.1016/j.cell.2016.01.015
Ahdevaara P, Kalimo H, Törmä T, Haltia M (1977) Differentiating intracerebral neuroblastoma: report of a case and review of the literature. Cancer 40:784–788
Berger MS, Edwards MS, Wara WM et al (1983) Primary cerebral neuroblastoma. Long-term follow-up review and therapeutic guidelines. J Neurosurg 59:418–423. https://doi.org/10.3171/jns.1983.59.3.0418
Davis PC, Wichman RD, Takei Y, Hoffman JC (1990) Primary cerebral neuroblastoma: CT and MR findings in 12 cases. AJNR Am J Neuroradiol 11:115–120
Lannering B, Rutkowski S, Doz F, Pizer B, Gustafsson G, Navajas A, Massimino M, Reddingius R, Benesch M, Carrie C, Taylor R, Gandola L, Björk-Eriksson T, Giralt J, Oldenburger F, Pietsch T, Figarella-Branger D, Robson K, Forni M, Clifford SC, Warmuth-Metz M, von Hoff K, Faldum A, Mosseri V, Kortmann R (2012) Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial. J Clin Oncol Off J Am Soc Clin Oncol 30:3187–3193. https://doi.org/10.1200/JCO.2011.39.8719
Latchaw RE, L’Heureux PR, Young G, Priest JR (1982) Neuroblastoma presenting as central nervous system disease. AJNR Am J Neuroradiol 3:623–630
McLendon RE, Bentley RC, Parisi JE, Tien RD, Harrison JC, Tarbell NJ, Billitt AL, Gualtieri RJ, Friedman HS (1997) Malignant supratentorial glial-neuronal neoplasms: report of two cases and review of the literature. Arch Pathol Lab Med 121:485–492
Torres LF, Grant N, Harding BN, Scaravilli F (1985) Intracerebral neuroblastoma. Report of a case with neuronal maturation and long survival. Acta Neuropathol (Berl) 68:110–114
Yariş N, Yavuz MN, Reis A, Yavuz AA, Okten A (2004) Primary cerebral neuroblastoma: a case treated with adjuvant chemotherapy and radiotherapy. Turk J Pediatr 46:182–185
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Bianchi, F., Tamburrini, G., Gessi, M. et al. Central nervous system (CNS) neuroblastoma. A case-based update. Childs Nerv Syst 34, 817–823 (2018). https://doi.org/10.1007/s00381-018-3764-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-018-3764-3