
Abstract
Introduction
Neural tube defects (NTDs) are a group of common and severe congenital birth defects that occur during early embryonic development due to incomplete closure of the neural tube. The genetic architecture of human NTDs, including spina bifida and hydrocephalus, is highly heterogeneous, with multiple genes/loci and both gene-gene and gene-environment interactions involved. Hence, the variation in outcomes also most likely relates to a combination of the severity of different variants in multiple genes and genetic modifiers affecting the biochemical traits.
Methods
Here, we present a multiple-spouse family with one pedigree lineage where three brothers are affected with NTDs—two lumbar spina bifidas without hydrocephalus and one obstructive hydrocephalus. We sequenced the exomes of three NTD patients and their parents.
Results
The analysis revealed a heterozygous c.844ins68 variant in CBS, which was carried by all affected individuals and inherited from their mother. All affected individuals had a variable set of additional low frequency deleterious variants in PTK7, PLCD4, IL4I1 or RASSF4 as likely causal loci contributing to the disease development.
Conclusion
This report extends the current knowledge of the genetic background of NTDs and proposes that common and low frequency variants in genes involved mostly in one-carbon metabolism or planar cell polarity (PCP) pathways can act in an additive manner to increase the genetic risk of the disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neural tube defects (NTDs) represent common and severe congenital birth defects that result from failure of closure of the neural tube during early embryonic development [1]. The worldwide incidence of NTDs, including myelomeningocele and hydrocephalus, is on average 1–2 per 1000 live births [2]. Despite being the second most common developmental birth defect, the aetiology of NTDs remains unclear due to its combined effects of genetic factors and environmental influences [3]. In humans, the majority of NTDs are sporadic, with occasional familial cases fitting a multifactorial polygenic or oligogenic inheritance pattern [4]. Most NTD cases result from an additive contribution of multiple, mostly common genetic factors, which are each individually insufficient to disrupt neural tube closure [5]. Multiple studies have been carried out to investigate numerous candidate genes in cohorts of patients, referring particularly to those that participate in one-carbon metabolism pathway [1, 4]. In multiplex NTD families, whole-exome sequencing (WES) is an alternative approach to investigate the genetic heritability of both rare and common coding variants [6]. To date, there is one published WES study in sporadic NTD patients [7].
The aim of the current study was to shed a light upon the genetic background of familial NTDs by conducting exome analysis in an Estonian family with three NTD patients.
Materials and methods
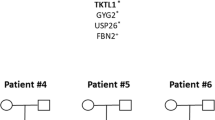
In this study, we analysed one family living in Estonia (Fig. 1). The patients were between 8 and 12 years of age and were clinically evaluated by an experienced paediatric surgeon in the North Estonia Medical Centre and by an experienced medical geneticist to exclude the likelihood of an underlying syndrome. Ethical approval for the study was obtained from the Ethics Review Committee on Human Research of the University of Tartu. All participants signed an informed consent form prior to enrolment in this study. For minors, consent was obtained from their parents. Genomic DNA was extracted from peripheral lymphocytes using standard techniques.

The pedigree of an Estonian family with NTDs
The patients III:4 and III:6 had lumbar spina bifida without hydrocephalus and patient III:5 had obstructive hydrocephalus. The patients’ III:4 and III:6 walking ability was satisfactory, they were independent from a wheelchair or crutches. The patient III:5 was submitted to a shunt operation at the age of 7 months. No other unusual phenotypic manifestations were detected in affected individuals. Relevant clinical data are presented in Table 1 .
In all patients, the folate level was significantly below normal. The homocysteine level was slightly elevated in patients III:5 and III:6. The patient’s height and weight measurements did not exceed ±2 SD boundary.
Exome sequencing
A total of 3 μg of genomic DNA was used to prepare next-generation sequencing libraries according to the SureSelectXT Human All Exon V5 protocol (Agilent Technologies Inc., Santa Clara, CA, USA) and was sequenced on HiSeq2500 platform (Illumina, San Diego, CA, USA) in the Estonian Genome Center resulting in 2 × 150 bp paired-end reads. 86.5% of calls had base sequence quality Q > 30 (mean score 36). Reads were aligned on the human reference genome (GRCh37/hg19) using BWA-MEM version 0.7.7. PCR duplicates were marked using Picard version 1.136. Resulting BAM files were realigned around known indels and base quality scores recalibrated using Genome Analysis Toolkit (GATK) version 3.4–46. Genotype calling was performed on all samples jointly using GATK HaplotypeCaller algorithm. Genotypes were filtered based on the GATK Variant Quality Score Recalibration (VQSR) truth sensitivity values and only PASS sites were considered in the further analysis. Variants were annotated with Variant Effect Predictor version 84. We used the PolyPhen-2, SIFT, MutationTaster and CADD scores to predict the functional effects of mutations.
Results
In the present study, we sequenced and analysed the exomes of an Estonian multiple-spouse family presenting one lineage where three brothers are affected with NTDs (one obstructive hydrocephalus and two lumbar spina bifidas without hydrocephalus). We performed WES in three affected individuals (III:4, III:5 and III:6) and their unaffected parents (II:6 and II:9). The average individual coverage of exome captured was 101.5–128.8× and 77.2% (range 74–82%) of the exomes were covered at least 50-fold. Here we report clinically relevant genetic variants (Table 2) found in the exomes of three NTD patients.
All patients were heterozygous carriers for c.844ins68 variant in CBS which was inherited from their mother (Table 2). Among all other variants (Table S1), both spina bifida patients (III:4 and III:6) had a heterozygous missense variant c.2938G>A in SCRIB, and one homozygous missense variant c.1324C>T in MTRR. Patient III:4 had two heterozygous missense variants (c.665C>T and c.1286A>C) in MTHFR, one heterozygous missense variant c.401A>G in MTHFD1, a heterozygous missense variant c.383C>T in IL4I1 and a heterozygous missense variant (c.236G>A) in PLCD4 which was shared with patient III:5. Patient III:6 had additional heterozygous missense variant c.2259G>C in PTK7 which was shared with patient III:5. Patient III:5 had a heterozygous missense variant c.665C>T in MTHFR, and another heterozygous missense variant c.1265C>T in SCRIB. Interestingly, the patient III:5 had three homozygous missense variants (c.1513G>A, c.1598G>T and c.1792C>T) in TCEB3B. In silico prediction showed that variants in IL4I1, SCRIB, PTK7, MTRR, MTHFR (c.665C>T) and PLCD4 had a deleterious effect. Full list of contributing genetic variants is shown in Table S1.
Discussion
We sequenced the exomes of three affected and two unaffected individuals of one Estonian family to identify the genetic background of three NTD patients with spina bifida or hydrocephalus (an example that both phenotypes may occur in the same family). The heritability of NTDs has been estimated to 60%, which implies a strong genetic component [8]. Candidate gene studies in NTDs have faced difficulties in identifying major causative clinically relevant variants participating in the development of NTDs, suggesting the need for novel approaches. A recent WES study, conducted with 43 sporadic NTD patients of European descent, demonstrated enrichment of loss-of-function de novo variants particularly in SHROOM3, compared to control cohorts [7]. However, none of these loss-of-function variants was found in our study.
A previous study has found that a relatively small number of genes (e.g. Vangl2, Celsr1, Scrb1, Ptk7), fit into well-established biological or molecular functions, cluster in integrated molecular-cellular pathways essential for neural tube closure [9]. Multiple candidate gene studies have shown that variants in one-carbon metabolism pathway and PCP signalling pathway increase the risk of NTDs [1].
Folate and methionine pathway-related genes
The baseline finding in this study was c.844ins68 in CBS which was present in all patients and was inherited from their mother. CBS is essential for the degradation of homocysteine to cysteine by linking the metabolism of sulphur-containing amino acids with more than hundred S-adenosyl methionine-dependent methylation reactions, redox-control, the folate cycle and the metabolism of signalling molecules through the trans-sulphuration pathway and is therefore considered as an important candidate gene for NTDs [10]. The co-occurrence of c.844ins68 with other one-carbon metabolism pathway gene variants, especially the polymorphism c.665C>T in MTHFR, has been claimed to be higher than expected in spina bifida cases [11, 12]. So far, one meta-analysis has demonstrated that c.844ins68 in CBS alone is not a good predictor for NTD risk [13].
In addition to the c.844ins68 in CBS, spina bifida patients (III:4 and III:6) had other contributing variants in MTRR, MTHFR and IL4I1. Although both MTHFR variants (c.665C>T and c.1286A>C) are common, it is previously known that both these polymorphisms lower the enzyme activity and thereby lower the folate level in homozygous state [14]. The MTHFR c.665C>T has been shown to increase the risk of NTDs by 2–4 fold, whereas the c.1286A>C has milder effects [15]. In addition to other NTD risk factors, the MTRR role in one-carbon metabolism pathway is to participate in maintaining the B12-dependent conversion of homocysteine to methionine. Therefore, variants in MTRR could alter the homocysteine levels in NTD patients [16]. There is also evidence suggesting that decreased B12 vitamin and increased total choline or homocysteine in maternal blood are associated with increased NTDs risk [17]. In addition to B12, it is well-known that maternal folate status is also a risk factor in NTD pregnancies and an inverse relationship between blood folate concentration and risk of an affected pregnancy has been shown [1]. In one-carbon metabolism pathway, during the decreased methionine levels, it is important to maintain sustained and balanced methionine usage, which is secured through methionine salvage pathway. One of the key genes in that pathway is IL4I1, which is essential for the methionine recovery during the absence of exogenous methionine [18].
Planar cell polarity genes
The PCP pathway mediates cell polarity by signal transduction through DVL2 which then modulates actin cytoskeleton through the small GTPases RhoA and Rac and the downstream Rho kinase. Hence, the PCP pathway genes are partially responsible for a variety of changes in cell adhesion, polarity and short-range tissue movements participating in the regulation of cytoskeletal changes that are directly involved in the neural tube closure, and therefore are associated with several forms of NTDs [19,20,21]. Candidate gene studies of PCP pathway genes in humans have identified mutations in CELSR1, VANGL1, VANGL2, FZD6, SCRIB1 and DVL2 in some patients with different forms of NTDs [1, 22, 23].
In our study, all patients carried one deleterious variant in the SCRIB gene. SCRIB domains interact with other PCP proteins (including PCP core protein Vangl2) in spina bifida patients [24]. Another PCP gene, PTK7 has been proposed to act as a molecular switch that activates the non-canonical Wnt/PCP pathway and at the same time inhibits the canonical Wnt/β-catenin pathway [25]. Additionally, all patients carried modifier variants in other PCP pathway genes, including FZD1, VANGL2 and DVL2. Interestingly, patient III:5 carried three variant homozygous deleterious variants in TCEB3B which encodes transcription elongation factor B polypeptide 3B (elongin A2). Evidence from interactome mapping has suggested that TCEB3B interacts with DVL2 (dishevelled 2) which participates in Wnt signalling by binding to the cytoplasmic C-terminus of frizzled family members and transducing the Wnt signal to downstream effectors [26].
Lastly, the patients had variants (Table 2) in genes which are expressed in embryonic neural tube. Patients III:4 and III:5 had maternally inherited deleterious variants in the PLCD4, RASSF4 and FARS2 genes which are expressed in neural tube during early embryogenesis [LifeMap® Discovery, http://discovery.lifemapsc.com/in-vivo-development/neural-tube].
To date, it is widely accepted that genetic variants participating in neural tube closure are mainly connected with one-carbon metabolism (important for cell proliferation and/or cell survival) and/or PCP signalling (required for initiation of neural tube closure) pathways. The involvement of variants in multiple genes and pathways denotes a scenario describing variable expression of a single shared genetic variant in the presence of sibling-specific sets of multiple other variants, each having weak individual effect, in modifier genes that underlie the phenotypic variation. Likewise, we assume that combined sets of common and low frequency variants in folate metabolism, sulphur amino acid metabolism and PCP signalling pathway genes found in our patients might be sufficient to disrupt the development and closure of the neural tube.
Conclusions
The multifactorial complexity of NTDs aetiology presents a great challenge for researchers. In this study, we describe genetic variants in major pathways involved in genetic predisposition of developing human NTDs. Although no loss-of-function variants or novel high-impact variants with in biologically meaningful genes were found, the results of current study might add new knowledge of the genetic background of familial cases of NTDs. It is important to find more patients from familial forms of NTDs to assure a causality of reported variants and genes. Moreover, investigators will need to integrate genetic data with information on epigenetic and environmental factors to obtain a more complete understanding of the cause of individual NTDs.
In conclusion, this study provides new evidence that genetic variants in one-carbon metabolism, sulphur amino acid metabolism and planar cell polarity pathways are implicated in familial non-syndromic NTDs. Advances in next-generation sequencing may help researchers to better understand the genetic basis of NTDs in humans.
References
Greene NDE, Copp AJ (2014) Neural tube defects. Annu Rev Neurosci 37:221–242
Kibar Z, Capra V, Gros P (2007) Toward understanding the genetic basis of neural tube defects. Clin Genet 71:295–310
Detrait ER, George TM, Etchevers HC, Gilbert JR, Vekemans M, Speer MC (2005) Human neural tube defects: developmental biology, epidemiology, and genetics. Neurotoxicol Teratol 27(3):515–524
Greene NDE, Stanier P, Copp AJ (2009) Genetics of human neural tube defects. Hum Mol Genet 18(R2):R113–R129
Harris MJ, Juriloff DM (2007) Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth defects res. A Clin Mol Teratol 79(3):187–210
Krupp DR, Soldano KL, Garrett ME, Cope H, Ashley-Koch AE, Gregory SG (2014) Missing genetic risk in neural tube defects: can exome sequencing yield an insight? Birth Defects Res A Clin Mol Teratol 100(8):642–646
Lemay P, Guyot MC, Tremblay É, Dionne-Laporte A, Spiegelman D, Henrion É, Diallo O, De Marco P, Merello E, Massicotte C, Désilets V, Michaud JL, Rouleau GA, Capra V, Kibar Z (2015) Loss-of-function de novo mutations play an important role in severe human neural tube defects. J Med Genet 52(7):493–497
Bassuk AG, Kibar Z (2009) Genetic basis of neural tube defects. Semin Pediatr Neurol 16:101–110
Andrew J Copp, Nicholas DE Greene, (2009) Genetics and development of neural tube defects. The Journal of Pathology:n/a-n/a
Sponholz C, Kramer M, Schöneweck F, Menzel U, Inanloo Rahatloo K, Giamarellos-Bourboulis EJ, Papavassileiou V, Lymberopoulou K, Pavlaki M, Koutelidakis I, Perdios I, Scherag A, Bauer M, Platzer M, Huse K (2016) Polymorphisms of cystathionine beta-synthase gene are associated with susceptibility to sepsis. Eur J Hum Genet 24(7):1041–1048
De Franchis R, Botto LD, Sebastio G, Ricci R, Iolascon A, Capra V, Andria G, Mastroiacovo P (2002) Spina bifida and folate-related genes: a study of gene-gene interactions. Genet Med 4:126–130
Rubini M, Brusati R, Garattini G, Magnani C, Liviero F, Bianchi F, Tarantino E, Massei A, Pollastri S, Carturan S, Amadori A, Bertagnin E, Cavallaro A, Fabiano A, Franchella A, Calzolari E (2005) Cystathionine beta-synthase c.844ins68 gene variant and non-syndromic cleft lip and palate. Am J Med Genet 136A:368–372
Ouyang S, Liu Z, Li Y, Ma F, Wu J (2014) Cystathionine beta-synthase 844ins68 polymorphism is unrelated to susceptibility to neural tube defects. Gene 535(2):119–123
Nazki FH, Sameer AS, Ganaie BA (2014) Folate: metabolism, genes, polymorphisms and the associated diseases. Gene 533(1):11–20
Yaliwal LV, Desai RM (2012) Methylenetetrahydrofolate reductase mutations, a genetic cause for familial recurrent neural tube defects. Indian J Hum Genet 18(1):122–124
Shaw GM, Lu W, Zhu H, Yang W, Briggs FBS, Carmichael SL, Barcellos LF, Lammer EJ, Finnell RH (2009) 118 SNPs of folate-related genes and risks of spina bifida and conotruncal heart defects. BMC Med Genet 10:49
Imbard A, Benoist J-F, Blom HJ (2013) Neural tube defects, folic acid and methylation. Int J Environ Res Public Health 10(9):4352–4389
Witham KL, Minchin RF, Butcher NJ (2016) Role for human arylamine N-acetyltransferase 1 in the methionine salvage pathway. Biochem Pharmacol 125:93–100
Komiya Y, Habas R (2008) Wnt signal transduction pathways. Organ 4(2):68–75
Wu G, Huang X, Hua Y, Mu D (2011) Roles of planar cell polarity pathways in the development of neural tube defects. J Biomed Sci 18:66
Claudia Kappen, Anne M. Molloy, Diana M. Juriloff, Muriel J. Harris, (2012) A consideration of the evidence that genetic defects in planar cell polarity contribute to the etiology of human neural tube defects. Birth Defects Research Part A: Clinical and Molecular Teratology 94 (10):824-840
Allache R, De Marco P, Merello E, Capra V, Kibar Z (2012) Role of the planar cell polarity gene CELSR1 in neural tube defects and caudal agenesis. Birth Defects Res A Clin Mol Teratol 94(3):176–181
Robinson A, Escuin S, Doudney K, Vekemans M, Stevenson RE, Greene NDE, Copp AJ, Stanier P (2012) Mutations in the planar cell polarity genes CELSR1 and SCRIB are associated with the severe neural tube defect craniorachischisis. Hum Mutat 33(2):440–447
Lei Y, Zhu H, Duhon C, Yang W, Ross ME, Shaw GM, Finnell RH (2013) Mutations in planar cell polarity gene SCRIB are associated with spina bifida. PLoS One 8(7):e69262
Wang M, De Marco P, Merello E, Drapeau P, Capra V, Kibar Z (2015) Role of the planar cell polarity gene protein tyrosine kinase 7 in neural tube defects in humans. Birth Defects Res A Clin Mol Teratol. 103(12):1021–1027
Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li S, Albala JS, Lim J, Fraughton C, Llamosas E, Cevik S, Bex C, Lamesch P, Sikorski RS, Vandenhaute J, Zoghbi HY, Smolyar A, Bosak S, Sequerra R, Doucette-Stamm L, Cusick ME, Hill DE, Roth FP, Vidal M (2005) Towards a proteome-scale map of the human protein-protein interaction network. Nature 437:1173–1178
Acknowledgments
We would like to acknowledge High Performance Computing Center of University of Tartu. This study was funded by EU H2020 grants 692145, 676550, 654248, the Estonian Research Council Grant IUT20-60 and European Union through the European Regional Development Fund (Project No. 2014-2020.4.01.15-0012 GENTRANSMED).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval for the study was obtained from the Ethics Review Committee on Human Research of the University of Tartu. All participants signed an informed consent form prior to enrolment in this study. For minors, consent was obtained from their parents.
Conflict of interest
The authors of this article have no conflict of interest to declare.
Electronic supplementary material
Table S1
(DOCX 35 kb)
Rights and permissions
About this article

Cite this article
Pappa, L., Kals, M., Kivistik, P.A. et al. Exome analysis in an Estonian multiplex family with neural tube defects—a case report. Childs Nerv Syst 33, 1575–1581 (2017). https://doi.org/10.1007/s00381-017-3491-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-017-3491-1