
Abstract
The rhizosphere is a critical interface for pollutant remediation in soils. Association between biodegradation of organic pollutants and spatial pattern of degraders along the rhizosphere gradient is, however, still unclear. This study investigated the phenanthrene-degrading bacterial consortia in a Phragmites australis rhizosphere using DNA-stable isotope probing (DNA-SIP). The relative abundance of Sphingomonadales in the 13C-labeled consortia decreased with the distance from roots, suggesting that its contribution in phenanthrene degradation was decreased with the distance from roots. Conversely, the relative abundance of Rhizobiales, Rhodobacterales, Lactobacillales, and Enterobacteriales in 13C-labeled consortia increased with the distance from roots, suggesting that their contributions in phenanthrene degradation were increased with the distance from roots. The linkage numbers of bacterial species in the co-occurrence network increased with the percentages of 13C-labeled reads, suggesting the critical role of syntrophic interactions for phenanthrene degraders. These results suggest the niche partition of phenanthrene degraders, which leaded to the non-linear variation of phenanthrene degradation rates along the rhizosphere gradient. These findings will help us to better understand rhizo degradation of organic pollutants and optimize bioremediation technology by achieving a trade-off among different degraders.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous products of the combustion of carbon-based substances. They pose a significant risk to human health by genotoxicity and ecosystems by affecting natural biodiversity (Wilcke 2007). Increasing human activity leads to a massive use of fossil fuels and generates vast quantities of anthropogenic PAHs. Bioremediation of PAHs has potential advantages over physical and chemical remediation approaches, with lower cost, smaller risk, and fewer disturbances (Peng et al. 2008). Their low bioavailability, however, might limit bioremediation effects (El Amrani et al. 2015). The long history of evolutionary processes allows many microorganisms to adapt metabolically in ecosystems containing complex aromatic molecules produced by plants as cell wall components and soluble secondary products. Many PAH-degrading bacteria have been isolated and their PAH degradation pathways have been fully elucidated (Peng et al. 2008). For instance, they can adapt to low bioavailability of PAHs by active migration and chemotaxis toward PAHs, biofilm formation near PAHs sources, and high-affinity PAH-uptake pathways (Johnsen et al. 2005).
Plants can trigger increases in both the diversity and abundance of PAH degraders in PAH-contaminated soils and rhizosphere processes have been widely reported to stimulate the degradation of PAHs (Ma et al. 2010). One important reason for the stimulation of PAH degradation in the rhizosphere is the release of root exudates, which have the potential to enhance the rate of PAH degradation by providing substrates for soil microorganisms and by increasing the bioavailability of PAHs (Cébron et al. 2011). Plant roots release a myriad of compounds, such as flavonoids and fatty acids, which increase microbial growth by providing carbon resources and enhance PAH degradation by stimulating co-metabolism (Philippot et al. 2013). The organic acids released from roots enhance the bioavailability of PAHs by increasing the desorption of PAHs from soils (Gao et al. 2010). In addition, root growth also enhances PAH degradation by increasing soil aeration (Gerhardt et al. 2009). Some wetland plants transport oxygen from above-ground parts to below-ground parts by aerenchyma to enable them to survive in anoxic environments and to increase the oxygen concentration in the rhizosphere (Shiono et al. 2011). Another reason for the stimulation of PAH degradation in the rhizosphere is the depletion of mineral nutrients due to root absorption (Xu et al. 2015). For example, the addition of phosphorus can decrease the degradation rates of phenanthrene in soils (Johnson and Scow 1999), indicating that PAH degraders are capable of metabolizing PAHs under low-nutrient conditions. The complex interactions between plants and rhizosphere microorganisms play a critical role in the biodegradation of PAHs. Our previous study demonstrated that the stimulation effect of the rhizosphere on PAH degradation was non-linear in the rhizosphere gradient, where the highest degradation rate was found at 2–4 mm from the root surface (Ma et al. 2012). However, the association between the spatial patterns of the PAH-degrading bacteria and the non-linear degradation rates along the rhizosphere gradient is still unclear. It is therefore important to understand the mechanisms involved in forming this spatial pattern for optimizing PAH-bioremediation.
Syntrophic interactions, in which microorganisms exchange metabolic byproducts and share metabolic pathways, also play a key role in hydrocarbon degradation (Gieg et al. 2014). Syntrophy commonly occurs in anaerobic environments but can also occur in higher redox environments (Kleinsteuber et al. 2012). Correlation networks provide a useful tool for predicting species interactions in the environments (Faust and Raes 2012). The syntrophy between microorganisms is a mutualism interaction, which represents a positive correlation in the network. The correlation relationships of PAH degraders in the community have yet to be studied.
Our previous study demonstrated significant changes in microbial community compositions along the rhizosphere gradient (Ma et al. 2015), but the changes in functional microorganisms involved in PAH degradation is still unclear. Stable isotope probing (SIP) provides an opportunity to link the functions of microorganisms with their identities. A number of studies have applied the SIP technique in an attempt to identify bacteria in contaminated soils involved in phenanthrene degradation (Jones et al. 2011; Ma et al. 2006; Martin et al. 2012; Song et al. 2016; Tauler et al. 2016). Betaproteobacteria are prevalent as phenanthrene degraders in a constructed wetland collecting road runoff (Martin et al. 2012). Members of Pseudomonas have been found to play an important role in phenanthrene degradation in Antarctic soils (Ma et al. 2006). The dominant phenanthrene degraders in a gas plant soil were related to Pigmentiphaga, Acidovorax, and Sphingobium (Jones et al. 2011). The members of Sphingomonads were also associated with the utilization of phenanthrene in a creosote polluted soil (Tauler et al. 2016). The putative phenanthrene degraders were classified as belonging to the genus Sphingomona in a forest soil (Song et al. 2016). In this study, we labeled the phenanthrene degraders with stable isotope probing using [U−13C] phenanthrene. The aim of this study was to elucidate the association between the spatial pattern of the dominant phenanthrene degraders and phenanthrene degradation rates along the rhizosphere gradient of Phragmites australis, one of the most widely distributed flowering plants on earth.
Material and methods
SIP incubation in the rhizobox
Uniform-sized P. australis seedlings collected from the yellow river delta in China were transplanted into the root chambers of three rhizoboxes as described previously (He et al. 2005). The soil near root surface separated into five 1-mm-thick layers by nylon mesh (pore size < 25 μm) referred to here as the 1- to 5-mm rhizosphere soil from roots. Distilled water was supplied daily to ensure that soils were covered in water throughout the experiment, which was carried out in a growth chamber at 25 °C with light for 14 h and 15 °C and darkness for 10 h. The P. australis formed stable root meshes after 30 days of growth. The soil was sampled from the 20-cm topsoil of wetland in the yellow river delta. The soil pH was 8.1 and the salt concentration was 0.26%. The total C and N were 2.1% and 0.16%, respectively. The particle size distribution was 14% clay, 61% silt, and 25% sand. The soil was air-dried and passed through a 0.25-mm sieve. [U−13C]-phenanthrene (Sigma, 99%) was dissolved in hexane and mixed with 5 g of dry soil and then with 25 g of dry soil. After solvent volatilization under a fume hood, 270 g of dry soil was added and mixed to obtain 10 mg kg−1 [U−13C]-phenanthrene. Ten grams of spiked soil was put into each rhizosphere layer (100 mm × 100 mm × 1 mm).
Phenanthrene degradation
Five layers of 1-mm-thick rhizosphere soils were collected from each rhizobox at 20 days after the spike addition. Five grams of soil samples were extracted twice with 25 ml dichloromethane (DCM) in an ultrasonic bath, each for 30 min. The phenanthrene concentrations were determined with a gas chromatograph coupled to a mass spectrometer (GC-MS) (Agilent Technologies 6890 equipped with a capillary column in silica glass DB 5-MS, and Agilent technologies 5973 Inert detector on the full scan mode). The temperature program was as follows: from 70 to 130 °C at 15 °C min−1, then from 130 to 210 °C at 5 °C min−1. The carrier gas was helium at 1.4 ml min−1 constant flow.
Soil DNA extraction, CsCl gradient ultracentrifugation, and identification of 13C-labeled DNA
Total soil DNA was extracted from 0.5 g of soil with a FastDNA spin kit for soil (MP biomedicals, Solon, OH, USA) according to the provided protocol. Heavy (the 13C-labeled) DNA was separated from the light (the unlabeled) DNA by density gradient ultracentrifugation using CsCl (Sigma, 99.9%). The gradient mixture consisted of sterile CsCl stock solution with a gradient buffer (0.1 M Tris, 0.1 M KCl, and 1 mM EDTA) and DNA (2 μg) to a final density of 1.725 g ml−1. Density gradient centrifugation was performed in 5.1-ml tubes in a near-vertical rotor (VTi 65.2, Beckman) at 20 °C, 177000g, for 72 h. Centrifuged gradients were fractionated into 12 fractions of 400 μl, from the bottom of the gradient using a pump. DNA was precipitated by adding 400 μl milliQ water, 800 μl isopropanol, and 2 μl polyacryl carrier for 1 h at − 20 °C, and then recovered by centrifugation (14,000g, 30 min). The pellets were washed with 70% (v/v) ethanol and suspended in 50 μl milliQ water.
Real-time PCR quantification
The real-time PCR quantifications were performed following the procedure described previously (Ma et al. 2015). Copy numbers of 16S rRNA genes in the unlabeled and the 13C-labeled DNA were quantified using primer sets 338F/518R. The reactions with TransStartTM Green qPCR SuperMix (TransGen Biotech, Beijing, China) were performed on a Roche LightCycler 2.0 (Roche Diagnostics GmbH, Mannheim, Germany) using the following protocol: 50 °C for 2 min, 95 °C for 15 min, then 40 cycles of denaturing at 94 °C for 15 s and annealing at 56 °C for 30 s. Amplification reactions were carried out in a volume of 20 μl, with 19 μl of PCR mix prepared using the SYBR green PCR Master Mix (Bio-Rad), and 1 μl of the template DNA (unlabeled and 13C-labeled DNA and standard plasmids with 103 to 108 gene copies μl−1).
16S rRNA gene high-throughput sequencing
Bacterial 16S rRNA genes in the unlabeled and the 13C-labeled DNA were amplified by the polymerase chain reaction (PCR) from DNA samples using V3 oligonucleotide primers specific for domain bacteria (Vestergaard et al. 2017). We amplified a standard 180-bp V3 region using primers 338F and 518R and standard amplification conditions (Schöler et al. 2017). The forward primers 338F were modified by the addition of a PGM sequencing adaptor, a “GT” spacer and a unique error correcting Golay barcode. Prior to sequencing, all amplicons were assessed for fragment size distribution and DNA concentration using a Bioanalyser 2100 (Agilent Technologies, USA). The samples were attached to the surface of ion sphere particles (ISPs) using an Ion Xpress Template 100 kit (Life Technologies, USA) according to the manufacturer’s instructions. Manual enrichment of the resulting ISPs resulted in > 95% templated ISPs. These were sequenced on a “316” (100 Mbp) micro-chip using an Ion Torrent Personal Genome Machine (Life Technologies, USA) for 65 cycles (260 flows). After sequencing, the individual sequence reads were filtered within the PGM software to remove low-quality and polyclonal sequences. Sequences matching the PGM 3′ adaptor were also automatically trimmed. All PGM quality-filtered data were exported as FastQ files and subsequently analyzed using the UPARSE pipeline (Edgar 2013). Sequence counts were adjusted based on the 16Stimator (Perisin et al. 2015) and were normalized with the negative binomial model, which minimized bias associated with sequencing coverage and allowed for the comparison of results for all samples (McMurdie and Holmes 2014).
Correlation network inferring
We combined the 13C-labeled and unlabeled OTU abundance matrices normalized with 16S rRNA gene copy numbers. To reduce rare OTUs in the dataset, we removed OTUs with relative abundances less than 0.01% of total sequences and OTUs that were not present in all 15 samples. The correlation network was inferred based on the Spearman correlation matrix. The nodes in this network represented OTUs and the edges that connect these nodes represent correlations between OTUs. The indirect correlation dependencies were distinguished using the network deconvolution method (Feizi et al. 2013). The cutoff of correlation coefficients was determined by the RM Threshold package of R (https://CRAN.R-project.org).
Statistical analysis
All statistical analyses and graphics were done using the R program (http://www.r-project.org). The variation of the phenanthrene degradation rate and the 16S rRNA copy numbers were analyzed by the analysis of variance (ANOVA) and Tukey’s honest significant difference (HSD) test. The proportion of the 13C-labeled 16S rRNA genes of taxa groups was calculated from the 16S rRNA gene copy numbers and the relative abundance of taxa groups in the unlabeled and the 13C-labeled bacterial consortia. The taxonomic diversities of communities and metagenomes were calculated with the vegan package. The phylogenetic diversities of communities were calculated using the picante package. Non-matric multidimensional scaling (NMDS) was used for ordination based on the UniFrac phylogenetic distance matrix for bacterial community structure (OTUs). Analysis of similarity (ANOSIM) was employed for group difference tests between community groups. The abundance of genes of the 13C-labeled bacterial consortia was involved in the phenanthrene degradation pathway was normalized with the 16S rRNA gene copy numbers. All sequence data was deposited in the Public National Center for Biotechnology Information (NCBI) database with the accession number PRJNA293484.
Results
Phenanthrene degradation rates and bacterial degrader abundance along the rhizosphere gradient
The degradation rates increased in the rhizosphere soils from 1 to 3 mm from the root surface but decreased from 3 to 5 mm from the root surface (Fig. 1a). The degradation rate was significantly greater at 3 mm from the root surface (Tukey HSD test, P = 0.05). The bacterial degrader abundance, represented as the copy numbers of the 13C-labeled 16S rRNA genes, was significantly smaller than those of the unlabeled 16S rRNA genes, except in the rhizosphere soils at 1 mm from the root surface and decreased with distance from the root surface (Fig. 1b).

The degradation rates and the 16S rRNA gene copy numbers in the bacterial communities along the rhizosphere gradient. a The degradation rates (mean ± sd) were identified by the difference between initial and residual phenanthrene concentrations in three replicate rhizoboxes. Different letters indicated P < 0.05 in Tukey HSD test following ANOVA. b The copy numbers of bacterial 16S rRNA gene were determined by qPCR. *P < 0.05, **P < 0.01, ***P < 0.001 show the copy number in the unlabeled consortia (C12) versus the 13C-labeled consortia (C13) using Wilcoxon’s tests
Diversity and composition of the phenanthrene-degrading bacterial consortia along the rhizosphere gradient
Bacterial 16S rRNA genes were profiled by high-throughput sequencing, and a total of 453,906 were generated after quality control, including 143,244 reads for the 15 13C-labeled DNA samples and 310,662 for the 15 unlabeled DNA samples. At 97% sequence identity, a total of 3464 OTUs were classified. Diversity of the 13C-labeled bacterial consortia was significantly lower than that of the unlabeled bacterial consortia at 1 to 4 mm from the root surface (Fig. 2a). For the 13C-labeled bacterial consortia, the observed OTU numbers and the phylogenetic diversity (PD) values were not significantly different along the rhizosphere gradient, but the values of the Shannon and Simpson indices increased along the rhizosphere gradient at 1 to 4 mm from the root surface. The observed OTU numbers and PD values of the unlabeled bacterial consortia increased at 3 mm from the root surface, but the values of the Shannon and Simpson indices decreased along the rhizosphere gradient.

The diversity and composition of the 13C-labeled and unlabeled bacterial consortia in rhizosphere gradient. a The alpha diversity in the 13C-labeled (C13) and the unlabeled (C12) bacterial consortia along rhizosphere gradient. b The non-matric multidimensional scaling (NMDS) of the 13C-labeled (C13) and unlabeled (C12) bacterial consortia along rhizosphere gradient based on weighted UniFrac dissimilarity
We used the weighted UniFrac distance matrix to evaluate the similarities between the microbial communities. The phylogenetic composition of the 13C-labeled bacterial consortia were significantly different from the unlabeled bacterial consortia (PERMANOVA, P = 0.02, Fig. 2b). The differences in the community compositions between the corresponding 13C-labeled and unlabeled bacterial consortia decreased with the distance from the root surface, being non-significant at 5 mm from the root surface (PERMANOVA, P = 0.09).
Composition variations of the phenanthrene-degrading bacterial consortia along the rhizosphere gradient
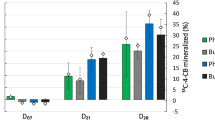
The bacterial composition profiles were different between the 13C-labeled and the unlabeled bacterial consortia (Fig. 3a). The 13C-labeled bacterial consortia were dominated by Rhizobiales (36.7%) and Flavobacteriales (24.7%), followed by Sphingomonadales (13.1%), Enterobacteriales (9.0%), Lactobacillales (8.1%), Bacillales (6.9%), Clostridiales (5.3%), and Rhodobacterales (5.2%). The unlabeled bacterial consortia were, however, dominated by Flavobacteriales (31.8%), followed by Actinomycetales (8.9%), Bacillales (7.8%), Enterobacteriales (6.0%), and Bacteroidales (5.5%). The proportions of phenanthrene degraders, represented as the percentages of the 13C-labeled 16S rRNA genes, were greater than 20% for the orders Rhizobiales, Rhodobacterales, Lactobacillales, Sphingomonadales, and Enterobacteriales (Fig. 3b).

The composition of the 13C-labeled and unlabeled bacterial consortia in rhizosphere gradient. a The relative abundance of dominant orders in the 13C-labeled (C13) and unlabeled (C12) bacterial consortia along rhizosphere gradient. b The proportion of labeled reads for dominant orders along rhizosphere gradient
The spatial patterns along the rhizosphere gradients were also different in terms of the abundance of the dominant compositions of the unlabeled and the 13C-labeled bacterial consortia (Fig. 3a, b). The abundance of the dominant orders of the unlabeled bacterial consortia increased at 2 to 4 mm from the root surface, except for the abundances of Sphingomonadales, Flavobacteriales, and Rhodobacterales which increased at 4 to 5 mm from the root surface (Fig. 4a). Among the orders with putative high phenanthrene-degrading capacities, the abundance of orders Rhizobiales, Rhodobacterales, Lactobacillales, and Enterobacteriales increased but the abundance of the order Sphingomonadales decreased near the root surface with the 13C-labeled bacterial consortia (Fig. 4b). The proportion of the 13C-labeled 16S rRNA genes in the dominant orders also changed along the rhizosphere gradients (Fig. 3b). The proportions of degraders decreased with the distance from the root surface with the orders Rhizobiales and Rhodobacteriales, but increased with the orders Flavobacteriales, Actinomycetales, Desulfuromonadales, Bacteoidales, Sphingomonadales, Anaerolineales, Acenthopleuribacterales, Enterobacteriales, and Coriobacteriales.

The variation of relative abundance of dominant orders: a in the unlabeled bacterial consortia and b in the 13C-labeled bacterial consortia along the rhizosphere gradient and biomarkers c in the 13C-labeled (C13) and unlabeled (C12) bacterial consortia and d in rhizosphere soils from 1 to 5 mm from root surface
Based on the difference of the OTU relative abundance between the 13C-labeled and unlabeled bacterial consortia, we identified the orders Rhizobiales, Rhodobacterales, and Lactobacillales as biomarkers for the 13C-labeled bacterial consortia (Fig. 4c). Based on the difference in the OTU relative abundance of the 13C-labeled bacterial consortia with different distances from root surface, the orders Rhizobiales and Lactobacillales were identified as biomarkers for the 13C-labeled bacterial consortia at 1 and 2 mm from the root surface, respectively, and the orders Rhodobacterales were identified as the biomarkersfor the 13C-labeled bacterial consortia at 3 mm from root surface (Fig. 4d).
Relationship between the proportions of phenanthrene degraders and their associations in the correlation network
To construct a correlation network for the rhizosphere microbiomes, we combined the 13C-labeled and unlabeled bacterial consortia weighted by 16S rRNA gene copy numbers in each consortium. The resulting co-occurrence network consisted of 646 vertices (OTUs) and 3599 edges (significantly direct correlation between vertices) (Fig. 5a). The average path length (average network distance between all pairs of vertices) was 2.96 edges with a diameter (longest distance) of 7.2 edges. The clustering coefficient (how vertices are embedded in the neighborhood and the degree to which they tend to cluster together) was 0.41 and the modularity index was 0.33. Overall, these topological properties suggested that this network was comprised of highly connected OTUs (weighted average degree 10.0) with densely connected groups of vertices and clustered topological structures. The degree of vertices increased with the proportion of the 13C-labeled reads in OTUs (Spearman’s rank correlation R = 0.47, P < 0.001), which suggested a relatively higher association for phenanthrene degraders (Fig. 5b).

The correlation interactions of dominant operating taxonomic units (OTUs) in the soil bacterial communities in rhizosphere gradient. a The connection in the correlation network stands for a strong (Spearman’s ρ > 0.98) correlation. b The relationship between degree of nodes in the network and the proportion of 13C-labeled reads of the corresponding OTUs. The nodes represented OTUs in the community. The size of each node is proportional to the relative abundance. The color indicates the proportion of 13C-labeled reads of the corresponding OTUs
Discussion
Here, we provide evidence that the bacteria mediating the phenanthrene degradation in the P. australis rhizosphere were mainly members of the orders Rhizobiales, Flavobacteriales, Sphingomonadales, Enterobacteriales, Lactobacillales, Bacillales, Clostridiales, and Rhodobacterales. They have not been identified as dominant phenanthrene degraders in previous studies, except Sphingomonadales (Jones et al. 2011; Ma et al. 2006; Martin et al. 2012; Song et al. 2016; Tauler et al. 2016). The distinction of the dominant degraders identified from various studies suggests that the taxonomy of phenanthrene-degrading bacteria varies with different environments. This is supported by the differences in associated bacterial communities during PAH degradation in different soils types (Regonne et al. 2013; Ren et al. 2016).
Given that the taxonomic groups with higher proportions of 13C-labeled reads could have higher phenanthrene-degrading capacities, the orders Rhizobiales, Sphingomonadales, Enterobacteriales, Lactobacillales, and Rhodobacterales would have higher degradation activities than other orders in the 13C-labeled bacteria consortia. Those orders were also identified as biomarkers for the 13C-labeled consortia along the rhizosphere gradient. Many PAH-degrading bacteria have been isolated based on cultivation-dependent methods, mainly belonging to a limited number of genera groups, such as Sphingomonas, Burkholderia, Pseudomonas, and Mycobacterium (Johnsen et al. 2005). The genus Sphingomonas, belonging to the order Sphingomonadales, was one of the dominant phenanthrene degraders in the 13C-labeled consortia of the present study. The proportions of the genera Burkholderia, Pseudomonas, and Mycobacterium were, however, relatively low in the 13C-labeled consortia. Given that only small proportion of bacteria have been cultivated in the laboratory (Ling et al. 2015), one explanation for the low proportion of these genera is that the majority of phenanthrene-degrading bacteria are still uncultured. The orders Rhizobiales, Enterobacteriales, Lactobacillales, and Rhodobacterales have been widely found in phenanthrene-degrading bacterial communities by using cultivation-independent methods (Chakraborty et al. 2012; Singleton et al. 2005; Thomas and Cébron 2016; Zhang et al. 2012). Another explanation is that the syntrophic pathway is also a pivotal mechanism for phenanthrene degradation (El Amrani et al. 2015). Most of the isolated phenanthrene-degrading bacteria can use phenanthrene as the sole source of carbon and energy.
The composition of phenanthrene-degrading bacterial community shifted with the distance from the root surface, suggesting that the influence of the root on phenanthrene-degrading bacteria changed along the rhizosphere gradient. The influence of rhizosphere effects on phenanthrene-degrading consortia has been demonstrated by the shift of phenanthrene degraders from Pseudoxanthomonas and Microbacterium to Pseudomonas and Arthrobacter by root exudate addition in the previous study (Cébron et al. 2011). The short-term influence of rhizosphere effects on the phenanthrene-degrading bacterial communities is in agreement with the rapid response of phenanthrene degraders to shifts in root activity (Thomas and Cébron 2016).
The greater association numbers of the species with higher degrading potential also suggest the critical role of microbial interactions and syntrophy in phenanthrene degradation. Firstly, the degradation of aromatic compounds could trigger the production and the exchange of metabolic factors required for microbial interactions, such as quorum sensing factors (El Amrani et al. 2015). Accordingly, the phenanthrene degraders could play critical roles in bacterial interactions and have more association with other components in the community. Secondly, the bacterial species involved in the syntrophic degrading processes, such as exchange of byproducts, sharing metabolic pathways, and contributing to the regulation of expression of genes involved in catabolism, could be expected to be positively correlated with each other.
The spatial pattern of phenanthrene degradation rates along the rhizosphere gradient in the present study was consistent with previous studies that higher degradation rates of organic pollutants can be observed at 2–4 mm from the root surface of ryegrass (He et al. 2007) and rice (Ma et al. 2012). These studies suggest that non-linear spatial patterns relate with the root exudates, which induce modifications of microbial community compositions and the dominant species within these communities along the rhizosphere gradient (He et al. 2007). The present study shows that the non-linear spatial pattern of degradation along the rhizosphere gradient was synchronous with the spatial pattern of the Shannon and Simpson diversity indices. The higher diversity could facilitate the syntrophic degrading pathway, resulting in higher degradation rates. The PD values were, however, non-significantly different along the rhizosphere gradient. The composition profiles of the 13C-labeled consortia in the present study suggest that the integration between various phenanthrene-degrading bacterial taxa might induce the non-linear degradation patterns along the rhizosphere gradient. Given that the phenanthrene-degraders groups with opposite behavior along the rhizosphere gradient might have different degradation pathways, both groups could be stimulated in the rhizosphere soils within 2–4 mm from the root surface. Furthermore, the higher 13C-labeled reads number near root surface indicated that phenanthrene in the rhizosphere soils at 2–4 mm from the root surface might be transported to the root surface. The dissolved organic materials released from plant roots have been reported to promote the translocation of phenanthrene in the rhizosphere (Magee et al. 1991).
The DNA of non-PAH degraders can be labeled with 13C through utilizing 13CO2 or other 13C-labeled metabolites from PAH degraders. This is one of most important aspects that all SIP experiments need to be taken in account. In the present study, we set the endpoint as the degradation rates reached to more than 75%. The optimal endpoints for phenanthrene DNA-SIP experiments in soil microcosms reported in the previous studies ranged from 12 to 14 days when the phenanthrene degradation rate reached to 65 to 80% (Cébron et al. 2011, Song et al. 2016). Given that the degradation speed was lower under anaerobic condition in the flooding soils than in the soil microcosms without flooding, the endpoint was set at 20-day after spike. Another concern from the reviewer is incomplete separation of heavy and unlabeled DNA, which could happen due to the DNA genomes with high GC contents or insufficient centrifugation time. We tested DNA samples extracted from soils spiked with unlabeled phenanthrene under the same centrifugation condition. For these samples, the copy numbers of 16S rRNA gene in the density gradient fractions at the positions of heavy fractions were only less than 5% of copy numbers in the heavy fractions of the DNA-SIP samples. This indicates that the light and heavy DNA had been efficiently separated under this centrifugation condition. The bias introduced by high GC content of genomes should be less than 5% in all heavy DNA fraction.
In conclusion, our findings elucidate, for the first time, the association between the shifts of phenanthrene-degrading bacteria identified by SIP and the previously reported non-linear spatial pattern of organic pollutant degradation along the rhizosphere gradient. The opposite spatial patterns for the orders Rhizobiales, Enterobacteriales, Lactobacillales, and Rhodobacterales and the order Sphingomonadales suggest the niche partition of phenanthrene degraders. The trade-off between the those phenanthrene-degrading bacteria groups might induce the optimal phenanthrene degradation in rhizosphere. The higher association property of degraders in the correlation network indicates that the microbial interactions and syntrophy might play an important role in phenanthrene degradation in the rhizosphere. Given the gap between taxonomic composition and community functions, our results indicate a need for integrating metagenomics, metatranscriptomics, metabolomics, and metaproteomics to decipher molecular mechanisms of rhizo degradation in the light of developing “omics” approaches.
References
Cébron A, Louvel B, Faure P, France-Lanord C, Chen Y, Murrell JC, Leyval C (2011) Root exudates modify bacterial diversity of phenanthrene degraders in PAH-polluted soil but not phenanthrene degradation rates. Environ Microbiol 13:722–736
Chakraborty R, Borglin SE, Dubinsky EA, Andersen GL, Hazen TC (2012) Microbial response to the MC-252 oil and Corexit 9500 in the Gulf of Mexico. Front Microbiol 3:357
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998
El Amrani A, Dumas A-S, Wick LY, Yergeau E, Berthomé R (2015) “Omics” insights into PAH degradation toward improved green remediation biotechnologies. Environ Sci Technol 49:11281–11291
Faust K, Raes J (2012) Microbial interactions: from networks to models. Nat Rev Microbiol 10:538–550. https://doi.org/10.1038/nrmicro2832
Feizi S, Marbach D, Médard M, Kellis M (2013) Network deconvolution as a general method to distinguish direct dependencies in networks. Nat Biotechnol 31:726–733
Gao Y, Ren L, Ling W, Gong S, Sun B, Zhang Y (2010) Desorption of phenanthrene and pyrene in soils by root exudates. Bioresour Technol 101:1159–1165
Gerhardt KE, Huang X-D, Glick BR, Greenberg BM (2009) Phytoremediation and rhizoremediation of organic soil contaminants: potential and challenges. Plant Sci 176:20–30
Gieg LM, Fowler SJ, Berdugo-Clavijo C (2014) Syntrophic biodegradation of hydrocarbon contaminants. Curr Opin Biotechnol 27:21–29. https://doi.org/10.1016/j.copbio.2013.09.002
He Y, Xu J, Tang C, Wu Y (2005) Facilitation of pentachlorophenol degradation in the rhizosphere of ryegrass (Lolium perenne l.). Soil Biol Biochem 37:2017–2024
He Y, Xu J, Ma Z, Wang H, Wu Y (2007) Profiling of PLFA: implications for nonlinear spatial gradient of PCP degradation in the vicinity of Lolium perenne L. roots. Soil Biol Biochem 39:1121–1129
Johnsen AR, Wick LY, Harms H (2005) Principles of microbial PAH-degradation in soil. Environ Pollut 133:71–84. https://doi.org/10.1016/j.envpol.2004.04.015
Johnson CR, Scow KM (1999) Effect of nitrogen and phosphorus addition on phenanthrene biodegradation in four soils. Biodegradation 10:43–50. https://doi.org/10.1023/A:1008359606545
Jones MD, Crandell DW, Singleton DR, Aitken MD (2011) Stable-isotope probing of the polycyclic aromatic hydrocarbon-degrading bacterial guild in a contaminated soil. Environ Microbiol 13:2623–2632
Kleinsteuber S, Schleinitz KM, Vogt C (2012) Key players and team play: anaerobic microbial communities in hydrocarbon-contaminated aquifers. Appl Microbiol Biotechnol 94:851–873
Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schäberle TF, Hughes DE, Epstein S, Jones M, Lazarides L, Steadman VA, Cohen DR, Felix CR, Fetterman KA, Millett WP, Nitti AG, Zullo AM, Chen C, Lewis K (2015) A new antibiotic kills pathogens without detectable resistance. Nature 517:455–459. https://doi.org/10.1038/nature14098
Ma Y, Wang L, Shao Z (2006) Pseudomonas, the dominant polycyclic aromatic hydrocarbon-degrading bacteria isolated from Antarctic soils and the role of large plasmids in horizontal gene transfer. Environ Microbiol 8:455–465
Ma B, He Y, Chen H, Xu J, Rengel Z (2010) Dissipation of polycyclic aromatic hydrocarbons (PAHs) in the rhizosphere: synthesis through meta-analysis. Environ Pollut 158:855–861
Ma B, Wang J, Xu M, He Y, Wang H, Wu L, Xu J (2012) Evaluation of dissipation gradients of polycyclic aromatic hydrocarbons in rice rhizosphere utilizing a sequential extraction procedure. Environ Pollut 162:413–421
Ma B, Lyu XF, Zha T, Gong J, He Y, Xu JM (2015) Reconstructed metagenomes reveal changes of microbial functional profiling during PAHs degradation along a rice (Oryza sativa) rhizosphere gradient. J Appl Microbiol 118:890–900
Magee BR, Lion LW, Lemley AT (1991) Transport of dissolved organic macromolecules and their effect on the transport of phenanthrene in porous media. Environ Sci Technol 25:323–331
Martin F, Torelli S, Le Paslier D, Barbance A, Martin-Laurent F, Bru D, Geremia R, Blake G, Jouanneau Y (2012) Betaproteobacteria dominance and diversity shifts in the bacterial community of a PAH-contaminated soil exposed to phenanthrene. Environ Pollut 162:345–353
McMurdie PJ, Holmes S (2014) Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol 10:e1003531. https://doi.org/10.1371/journal.pcbi.1003531
Peng R-H, Xiong A-S, Xue Y, Xue Y, Fu XY, Gao F, Zhao W, Tian YS, Yao QH (2008) Microbial biodegradation of polyaromatic hydrocarbons. FEMS Microbiol Rev 32:927–955. https://doi.org/10.1111/j.1574-6976.2008.00127.x
Perisin M, Vetter M, Gilbert JA, Bergelson J (2015) 16Stimator: statistical estimation of ribosomal gene copy numbers from draft genome assemblies. ISME J 10:1020–1024
Philippot L, Raaijmakers JM, Lemanceau P, van der Putten WH (2013) Going back to the roots: the microbial ecology of the rhizosphere. Nat Rev Microbiol 11:789–799. https://doi.org/10.1038/nrmicro3109
Regonne RK, Martin F, Mbawala A, Ngassoum MB, Jouanneau Y (2013) Identification of soil bacteria able to degrade phenanthrene bound to a hydrophobic sorbent in situ. Environ Pollut 180:145–151
Ren G, Teng Y, Ren W, Dai S, Li Z (2016) Pyrene dissipation potential varies with soil type and associated bacterial community changes. Soil Biol Biochem 103:71–85
Schöler A, Jacquiod S, Vestergaard G, Schulz S, Schloter M (2017) Analysis of soil microbial communities based on amplicon sequencing of marker genes. Biol Fert Soil 53:485–489
Shiono K, Ogawa S, Yamazaki S, Isoda H, Fujimura T, Nakazono M, Colmer TD (2011) Contrasting dynamics of radial O2-loss barrier induction and aerenchyma formation in rice roots of two lengths. Ann Bot 107:89–99. https://doi.org/10.1093/aob/mcq221
Singleton DR, Powell SN, Sangaiah R, Gald A, Ball LM, Aitken MD (2005) Stable-isotope probing of bacteria capable of degrading salicylate, naphthalene, or phenanthrene in a bioreactor treating contaminated soil. Appl Environ Microbiol 71:1202–1209
Song M, Jiang L, Zhang D, Luo C, Wang Y, Yu Z, Yin H, Zhang G (2016) Bacteria capable of degrading anthracene, phenanthrene, and fluoranthene as revealed by DNA based stable-isotope probing in a forest soil. J Hazard Mater 308:50–57. https://doi.org/10.1016/j.jhazmat.2016.01.009
Tauler M, Vila J, Nieto JM, Grifoll M (2016) Key high molecular weight PAH-degrading bacteria in a soil consortium enriched using a sand-in-liquid microcosm system. Appl Microbiol Biotechnol 100:3321–3336
Thomas F, Cébron A (2016) Short-term rhizosphere effect on available carbon sources, phenanthrene degradation, and active microbiome in an aged-contaminated industrial soil. Front Microbiol 7:92
Vestergaard G, Schulz S, Schöler A, Schloter M (2017) Making big data smart—how to use metagenomics to understand soil quality. Biol Fert Soil 53:479–484
Wilcke W (2007) Global patterns of polycyclic aromatic hydrocarbons (PAHs) in soil. Geoderma 141:157–166
Xu M, He Z, Zhang Q, Liu J, Guo J, Sun G, Zhou J (2015) Responses of aromatic-degrading microbial communities to elevated nitrate in sediments. Environ Sci Technol 49:12422–12431. https://doi.org/10.1021/acs.est.5b03442
Zhang SY, Wang QF, Xie SG (2012) Molecular characterization of phenanthrene-degrading methanogenic communities in leachate-contaminated aquifer sediment. Int J Environ Sci Technol 9:705–712
Funding
This work was supported by the National Natural Science Foundation of China (41301333), the China Postdoctoral Science Foundation (2017 M621945), and the Fundamental Research Funds for the Central Universities (2018QNA6009).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lv, X., Kankan, Z., Li, H. et al. Niche partition of phenanthrene-degrading bacteria along a Phragmites australis rhizosphere gradient. Biol Fertil Soils 54, 607–616 (2018). https://doi.org/10.1007/s00374-018-1287-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00374-018-1287-4