
Abstract
The composition of the rhizosphere bacterial communities was compared among different maize cultivars by pyrosequencing. The cultivars were “Ye Dan 4,” “Ben Yu 9,” “Zheng Dan 958,” and “Li Min 33,” popularized in the 1980s, 1990s, 2000s, and 2010s, respectively, in Jilin Province, China. These cultivars harbored different bacterial dominant species. Significant differences were detected in the five dominant phyla, Actinobacteria, Acidobacteria, Bacteroidetes, Chloroflexi, and Planctomycetes, especially between Li Min 33 and the three other cultivars. Li Min 33 had the lowest bacterial α-diversity, which was separated from other cultivars, according to a principal component analysis and the dissimilarity test of ADONIS. The γ-Proteobacteria, and within this, the genus Rhodanobacter, were significantly more abundant around Li Min 33 than around the other maize cultivars. The canonical correlation analysis indicated that the organic matter, soil pH, soil moisture, and leaf area index were important drivers of bacterial diversity. Mantel tests showed that the cultivar was significantly correlated with the microbial community composition. These results may aid in breeding or selecting new generations of plant cultivars that have the potential to support large populations of specific microbiota.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plant breeding programs are generally designed to improve agronomic characteristics, such as yield, nutrient use efficiency, and disease resistance, but generally do not consider the effect of plant-associated soil microflora, which can promote plant growth through phytohormone and vitamin production (Vessey 2003; Ali et al. 2009; Palacios et al. 2014); suppress phytopathogens through competition, antagonism, and hyperparasitism (Weller et al. 2002; Innerebner et al. 2011); and help plants to withstand heat (Castiglioni et al. 2008), salt (Zhang et al. 2008), and other abiotic stresses (Figueiredo et al. 2008). Different plant cultivars or genotypes may have specific phenotypic traits, including root properties (Fan et al. 2001; Czarnota et al. 2003; Bais et al. 2006), which are key factors in shaping microbial assembly and community composition (Grayston and Campbell 1996; Baudoin et al. 2003; Haichar et al. 2008). It would be possible to increase the agronomic potential of crops, while conserving soil and system sustainability, using a breeding strategy directed toward genotypes able to support abundant populations of beneficial rhizobacteria.
The rhizosphere soil of different crop genotypes is inhabited by bacteria, such as Paenibacillus spp. (Araújo da Silva et al. 2003), Pseudomonas fluorescens antagonist of Fusarium wilt (Leeman et al. 1995), Bacillus cereus antagonist of the seed pathogen Pythium torulosum (Smith et al. 1999), and plant growth-promoting Azospirillum (Chamam et al. 2013). Plant genotypes have important roles in the genetic composition of resident fluorescent pseudomonad populations and their biocontrol efficacy on soil-borne diseases (Raaijmakers et al. 2002). The ancient landrace wheat rhizosphere had more abundant pseudomonads than the newer cultivars (Germida and Siciliano 2001). Specific wheat cultivars, such as “Lewjain,” could enhance resident soil populations of the 2,4-diacetylphloroglucinol (DAPG)-producing strains (such as P. fluorescens “LR3-A28”). The two newfound phlD+ genotypes, referred to as genotypes “PfZ” and “PfY”, respectively, inhabited the rhizosphere soil of wheat cultivars Lewjain and “Penawawa” (Mazzola et al. 2004). The wheat cultivar–Pseudomonas interactions may have the potential to efficiently suppress the take-all disease of wheat and the replant disease of apple (root infection or infestation by Pythium spp., Rhizoctonia spp., and Pratylenchus penetrans) (Mazzola and Gu 2000). Different maize cultivars also influence the expression of the DAPG biosynthesis gene phlA in P. fluorescens “CHA0” (Notz et al. 2001). Maize hybrids “Lo964” and “H99” select their own specific DAPG+ diverse populations and facilitate more numerous and, genetically, more diverse, populations of Pseudomonas, which produce the antibiotic DAPG, than those supported by their parental lines under field conditions (Picard et al. 2004; Picard and Bosco 2005, 2006). Meanwhile, the hybrid genotype also has an important effect on the frequency and the diversity of the auxin producers (Chanway et al. 1988; Kucey 1988; Picard et al. 2004). In addition, several studies have reported different responses to pathogen and biocontrol agents among cultivars (Vakili and Bailey 1989; Vakili 1992; King and Parke 1993; Liu et al. 1995; Smith et al. 1997). There also seem to be extensive interactions between cultivar and the plant growth-promoting bacteria involved in nutrient cycling in the agroecosystems (Pathan et al. 2015b). The maize rhizosphere with low N use efficiency (NUE) was dominated by δ-Proteobacteria with β-glucosidase genes, whereas the high NUE maize rhizosphere mainly selected α-, β-, and γ-Proteobacteria with β-glucosidase genes. This difference in the diversity of β-glucosidase encoding genes may be due the difference in root exudates of the two maize lines (Pathan et al. 2015a, b). Rice cultivar-specific differences were found in the activity and diversity of ammonia-oxidizing bacteria in rhizospheres by a multiphasic approach (Briones et al. 2002). Compared with rhizoplane and endosphere compartments, the greatest effect of the rice cultivar on the microbiome was in the rhizosphere where the most operational taxonomic units (OTUs) exhibit a notable difference of α-diversity (Edwards et al. 2015). In field-grown potato, 4% of the OTUs from three cultivars show quantitative cultivar dependence (Weinert et al. 2011). Genetically modified plants with altered root exudates also exhibit cultivar-specific effects in root-associated bacterial, as well as in the composition of fungal communities (Mansouri et al. 2002; Milling et al. 2004). In the transgenic Bt-maize rhizosphere, the microbial communities composition differed from those in their non-transgenic counterpart, including the rare taxa (contributing <0.5% of the genera-assigned sequences) at the genus level (Brusetti et al. 2004; Castaldini et al. 2005; Dohrmann et al. 2013).
Using plant host genetics to understand plant–microbe interactions in the rhizosphere soil has been studied for the last 10 years, but studies were time-consuming, and the findings are often difficult to be compared due to differences in the experimental plan. Some responsive genera might have been overlooked by conventional cloning and sequencing approaches owing to their rareness (Dohrmann et al. 2013). With the advances in pyrosequencing technology, it is possible to have better insights in the microbial community composition and further elucidate how the rhizosphere microflora varies with plant cultivars. In northeast China, different cultivars, touting improved yield, adaptability, or disease-resistance were released and popularized in different decades. Our objectives were to determine whether there were differences in the composition of the rhizospheres’ bacterial community among these maize cultivars in the field. If the differences were detected in relation to the cultivar, then it may be useful to exploit plant germplasms stemming from crop diversification (Den Herder et al. 2010) and to conserve soil and system sustainability by reducing the fertilizer use (Gahoonia and Nielsen 2004) and pesticide inputs (Bradshaw et al. 2003).
Site, sampling, and analyses
Site and sampling
The experimental site was located in Fanjiatun County (44° 45′ N, 125° 01′ E), Jilin Province, northeastern China. This region has a temperate continental monsoonal climate with an average annual temperature of 4–6 °C and annual sunshine hours of approximately 2800 h, with 140 frost-free days. The mean annual precipitation is 567 mm, belonging to the typical rain-fed farming area. The soil is characterized as a thin layer black soil with an average organic matter content of 35.2 g kg−1, total N content of 0.4 g kg−1, available P content of 0.16 g kg−1, available K content of 0.2 g kg−1, and an average pH of 6.12.
The experiment was conducted in 2013. The maize cultivars from different decades were “Ye Dan 4” (“YD4”), “Ben Yu 9” (“BY9”), “Zheng Dan 958” (“ZD958”), and “Li Min 33” (“LM33”) popularized in the 1980s, 1990s, 2000s, and 2010s, respectively, in Jilin Province (Table 1). The maximum planting areas of YD4, BY9, and ZD958 were 1,228,667, 964,667, and 3,450,667 ha, respectively, in China.
The four cultivars were planted at the same time in the same field. Three replications were included and fully randomized in a 4 × 3 Latin rectangle, with each plot having a size of 31.2 m2 in the experiment. The experimental blocks were separated by 1 m walkways. Crop management was the same as adopted by local farmers. All of the treatments were fertilized with urea (N) at 60 kg ha−1, phosphorus pentoxide (P2O5) at 75 kg ha−1, and potassium oxide (K2O) at 90 kg ha−1 as the base fertilizer, with N at 100 kg ha−1 at jointing and 40 kg ha−1 at tasseling, as topdressings.
All of the soil samples were collected at the tasseling stage, July 12 in 2013. One maize plant showing uniform growth was selected and dug out from each plot. The soil loosely adhering to the root systems of the plants was discarded by vigorously shaking. Each root system still held some rhizosphere soil (tightly adhering soil), which was collected (Sanguin et al. 2009; Aira et al. 2010) and a portion was stored at −20 °C for DNA extraction. The remaining soil was air-dried, sieved (<2 mm), and analyzed for pH (1:2 soil to H2O ratio), total organic C, total N, and available P and K, as previously described (Sparks et al. 1996).
Soil moisture and plant characteristic measurements
At the time of soil sampling, the soil moisture at a depth of 10 cm was recorded by time domain reflectometry (Model Diviner-2000, Sentek Pty Ltd., Stepney, Australia). The aboveground plant was dried and weighed and the leaf area was measured. The plant leaf area index (LAI) is the leaf area divided by the ground area.
DNA extraction and amplification of the 16S rDNA variable region
DNA was extracted from the soil samples (1 g wet weight) using the E.Z.N.A. Soil DNA Kit (Omega Bio-tek, Norcross, GA, USA) according to the manufacturer’s instructions.
An aliquot of the extracted DNA from each sample was used as a template for amplification. To sequence the V1-V3 region of the bacterial 16S rDNA amplicon, each DNA was amplified with the 27F and 533R primers containing the B and A sequencing adaptors, respectively. The forward primer (B-27F), 5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAG AGA GTT TGA TCC TGG CTC AG-3′, contained the B adaptor, which is shown in italics and underlined. The reverse primer (A-533R), 5′-CCATCTCATCCCTGCGTGTCTCCGACTCAG NN NNN NNN NNT TAC CGC GGC TGC TGG CAC-3′ (Kumar et al. 2011), contained the A adaptor, which is shown in italics and underlined, and the Ns represent an eight-base sample-specific barcode sequence.
PCRs was performed and replicated three times in a 20-μL mixture containing 4 μL of 5× FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu polymerase, and 10 ng of template DNA. The following thermal program was used for amplification: 95 °C for 2 min, followed by 25 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, followed by an extension at 72 °C for 5 min. The PCR products were pooled and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA). An equal amount of the PCR product from the same sample was combined in a single tube to be run on Roche Genome Sequencer GS FLX Titanium platform at Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China.
Processing the pyrosequencing data
Raw fastq files were demultiplexed, quality-filtered using QIIME (version 1.17) with the following criteria: (i) 300-bp reads were truncated at any site receiving an average quality score <20 over a 50-bp sliding window, discarding the truncated reads that were shorter than 50 bp; (ii) exact barcode matching, two nucleotide mismatches in primer matching, and reads containing ambiguous characters were removed; and (iii) only sequences that had overlaps longer than 10 bp were assembled using their overlapping sequences. Reads that could not be assembled were discarded.
OTUs were clustered with a 97% similarity cutoff using UPARSE (version 7.1 http://drive5.com/uparse/), and chimeric sequences were identified and removed using UCHIME. The taxonomy of each 16S ribosomal RNA (rRNA) gene sequence was analyzed by RDP Classifier (http://rdp.cme.msu.edu/) against the silva (SSU115)16S rRNA database using a confidence threshold of 70% (Amato et al. 2013).
Statistical analysis
A principal component analysis (PCA) was used to determine the overall structural changes at the OTU level in the microbial communities. Bray–Curtis dissimilarity-based distance matrices were used with the ADONIS algorithm to compare the 454 pyrosequencing data of the four cultivars. The Mantel test and canonical correspondence analysis (CCA) were used to evaluate the linkages between microbial compositions and environmental attributes. To select attributes in CCA modeling, we used variation inflation factors (VIFs) to examine whether the variances of the canonical coefficients were inflated by the presence of correlations to other attributes. If an attribute had a VIF value higher than 20, then we deemed it to be affected by other attributes and, consequently, removed it from CCA modeling. All analyses were performed by functions in the Vegan package (v.1.15-1) in R v. 2.8.1 (R C Team 2013).
Data accession numbers
All the 454 sequencing data (.fq files) were uploaded to the National Center for Biotechnology Information sequence reads archive and can be accessed with the BioSample number SAMN06118453 under BioProject PRJNA356669 (http://www.ncbi.nlm.nih.gov/bioproject/PRJNA356669).
Results
A total of 153,048 valid reads and 108,650 OTUs were obtained from the 12 samples through a 454 pyrosequencing analysis. These OTUs were assigned to 44 different phyla. Each of the 12 samples contained between 9581 and 15,198 reads, with OTU richness values ranging from 6700 to 11,262. Rarefaction curves seemed to approach a saturation plateau by increasing the sample size, indicating that the sequencing depth was sufficient to wholly capture the richness (Fig. S1).
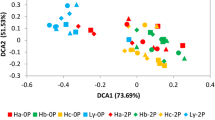
The cultivar LM33 had significantly low Chao1 richness and Shannon diversity indices (P < 0.05) (Fig. 1). The results revealed that the LM33 rhizosphere had the lowest levels of bacterial richness and diversity. The PCA showed that the microbial community in LM33 was well separated from those of the other three cultivars (Fig. 2), which was supported by the dissimilarity test using the ADONIS algorithm (Table S1).

Estimated bacterial α-diversity for 16S rDNA libraries of rhizosphere soil of different cultivars. a, b Significant differences among the four cultivars within one field using Tukey’s test (P < 0.05). YD4, “Ye Dan 4” cultivar; BY9, “Ben Yu 9” cultivar; ZD958, “Zheng Dan 958” cultivar; LM33, “Li Min 33” cultivar

Principle component analysis (PCA) of 454 pyrosequencing data. The values of PCA 1 and 2 are percentages of total variations that can be attributed to the corresponding axis. YD4, “Ye Dan 4” cultivar; BY9, “Ben Yu 9” cultivar; ZD958, “Zheng Dan 958” cultivar; LM33, “Li Min 33” cultivar
Most of the sequences (84%) of the four cultivars belonged to the seven phyla Proteobacteria, Actinobacteria, Acidobacteria, Bacteroidetes, Chloroflexi, Planctomycetes, and Gemmatimonadetes (Fig. S2). Each phylum accounted for 2 to 45% of the classified sequences. Other phyla were less abundant accounting for less than 2%.
The most prevalent phylum Proteobacteria showed significant differences at the class and genus levels between LM33 and other cultivars, although no differences were observed at the phylum level. LM33 had a lower proportion of β-Proteobacteria than BY9 and ZD958 and a lower proportion of δ-Proteobacteria than ZD958. Additionally, LM33 had a significantly higher proportion of γ-Proteobacteria (18.41%) than the other cultivars (less than 7.10%), with Rhodanobacter (12.43%) being the most abundant genus compared with in the other cultivars (less than 2.80%) (P < 0.05) (Fig. S3a).
The four cultivars exhibited significant differences in the percentage of five majority phyla, Actinobacteria, Acidobacteria, Bacteroidetes, Chloroflexi, and Planctomycetes, especially between LM33 and the three other cultivars (P < 0.05) (Table 2). LM33 had significantly lower percentage of the Actinobacteria phylum than YD4, with the genus Microlunatus being less abundant than BY9 (P < 0.05) (Fig. S3b). It also had a significantly lower relative abundance of Acidobacteria than BY9, with less of the Holophagae class than BY9 and ZD958 (P < 0.05) (Fig. S3c). This cultivar had a higher relative abundance of the Bacteroidetes phylum than BY9 and the most abundant Sphingobacteriia class, with more of Mucilaginibacter genus than ZD958 and less of Flexibacter genus than ZD958 (P < 0.05) (Fig. S3d). It had a lower relative abundance of Chloroflexi phylum than ZD958, with Sphaerobacter being the most abundant genus among the cultivars (P < 0.05) (Fig. S3e). Additionally, it had a lower relative abundance of Planctomycetes phylum than BY9, with less of Planctomycetacia class than BY9 (P < 0.05) (Fig. S3f) and none of Phycisphaerae class, which was present in the other cultivars.
To gain more insights into the potential effects of cultivars on rhizosphere bacteria, the most abundant OTUs were analyzed. LM33 had the highest total percentage of the top 10 abundant OTUs. Additionally, it had the highest OTU18710 abundance, which was the second dominant OTU in ZD958 and fourth in BY9, but was not in the top 10 OTUs in YD4. Furthermore, it contained 6 OTUs that were not in the top 10 of the 3 other cultivars. The distributions of the top 10 OTUs indicated that different cultivars had their own dominant species (Fig. 3).

Relative read abundance of the most abundant OTUs within the different cultivar communities. YD4, “Ye Dan 4” cultivar; BY9, “Ben Yu 9” cultivar; ZD958, “Zheng Dan 958” cultivar; LM33, “Li Min 33” cultivar
To compare the relationships among the composition of these bacterial communities in detail, the shared and specific species were determined using a Venn diagram (Fig. 4a). The species shared among the four cultivars represented small percentages of each bacterial community, 24.35% for YD4, 21.25% for BY9, 22.69% for ZD958, and 29.70% for LM33. The percentage of specific species were a little greater than shared species, accounting for 30.33% of YD4, 34.44% of BY9, 34.84% of ZD958, and 31.99% of LM33.

Specific OTU analyses of the different cultivar libraries. a Venn diagram showing the unique and shared OTUs (3% distance level); b specific species existing in each cultivar community. YD4, “Ye Dan 4” cultivar; BY9, “Ben Yu 9” cultivar; ZD958, “Zheng Dan 958” cultivar; LM33, “Li Min 33” cultivar
The compositions of specific species were different (Fig. 4b). Again, the LM33 rhizosphere had a distinctly high percentage of specific Proteobacteria, containing a high percentage of γ-Proteobacteria class and Rhodanobacter genus. It had a greater percentage of specific Actinobacteria, with more of genus Nocardioides and Streptomyces than BY9 and ZD958. Additionally, it had a greater abundance of Bacteroidetes, with more of Sphingobacteriia class and Mucilaginibacter genus than BY9 and ZD958.
The CCA was used to identify soil chemical and vegetation factors, including plant biomass and LAI controlling the soil microbial composition (Table S2). Significant model at the confidence level of P < 0.05 indicated that soil organic C, soil pH, plant LAI, and soil moisture were important environmental factors controlling the microbial community composition because they were significantly correlated with axis 1 (P < 0.05), which represented the major variations among the composition of microbial communities (Fig. 5).

Canonical correspondence analysis (CCA) of 454 pyrosequencing data and soil and plant characteristics. The percentage of variation explained by each axis is shown, and the relationship is significant (P < 0.05). YD4, “Ye Dan 4” cultivar; BY9, “Ben Yu 9” cultivar; ZD958, “Zheng Dan 958” cultivar; LM33, “Li Min 33” cultivar; TN, soil total N; AP, soil available P; SOC, total soil organic C; LAI, plant leaf area index; Biomass, aboveground plant dry weight
Discussion
The microbiota diversity may reflect resource allocation
LM33 was very different from the other cultivars for its low bacteria α-diversity, which may be related to its different crossing model (Table 1) and high plant density-tolerant (PDT) genotypes. Both of its crossing parents could be planted at high population, namely being density-tolerant, while at least one of other three cultivars’ parents is not density-tolerant (Li 2013). LM33 was also higher density-tolerant than other three cultivars. These PDT genotypes accumulated more assimilates to the aboveground than the belowground tissues (Herbert et al. 2001). The root system of PDT maize quickly accumulated dry matter by rapid nutrition uptake at the seedling stage and the accumulated amount remained stable later (Liu et al. 1994; Li 2013). After short vegetative growth (emergence-tassel stage) (Table S3), the PDT genotypes could accumulate more aboveground dry matter and maintained a green leaf area thus allocating more dry matter to the grain during the long grain filling period (grain filling–maturity) (Table S3) (Tollenaar and Daynard 1978; Lee and Tollenaar 2007; Mansfield and Mumm 2014). Thus, LM33 had a high harvest index (HI) (Table S2) that is the ratio of grain weight to total plant weight. High HI reflects the partitioning of more photosynthates to the grain than to the vegetative part such as the root system for rhizosphere bacterial, which means less food sources for bacterial community and consequently low bacterial diversity. Therefore, the bacteria diversity indirectly reflects photosynthate allocation within the plant. For example, a non-diverse microbiota accompanied by a high HI may indicate less overall C exudation because more carbohydrates are diverted to the kernels than to the roots (Peiffer et al. 2013). In the same way, BY9 had the highest bacterial α-diversity (Fig. 1) and the lowest harvest index (HI) (Table S3).
Statistically enriched bacteria in particular cultivars
Proteobacteria are generally considered to be r-selected or weedy fast-growing bacteria, adapted to the plant rhizosphere across diverse plant species. Their populations fluctuate opportunistically because of their response to labile C sources (Fierer et al. 2007; Lauber et al. 2009). However, some subsets of Proteobacteria exhibited statistically different percentages among cultivars.
A significant enrichment of the genus Rhodanobacter was observed in LM33. Rhodanobacter was first proposed by Nalin et al. (1999) as belonging to the Xanthomonadaceae family and the γ-Proteobacteria class. Described species have been isolated under aerobic conditions from surface soils, indicating their ubiquitous presence (Im et al. 2004; De Clercq et al. 2006; An et al. 2009; Bui et al. 2010; Wang et al. 2011). Rhodanobacter denitrificans was reported to carry out complete denitrification in low pH, high nitrate, and uranium polluted soil (Kostka et al. 2012), as well as reducing the N2O emissions from low pH soils (van den Heuvel et al. 2010). The low pH tolerance is a key physiological capability of the Rhodanobacter genus (Weon et al. 2007; An et al. 2009; Bollmann et al. 2010; van den Heuvel et al. 2010). The pH of LM33 rhizosphere was lower than those of three other cultivar rhizosphere soils (Table S3) probably due to the root-mediated pH changes, such as proton extrusion to compensate for unbalanced cation–anion uptake (Hinsinger et al. 2003; Sangabriel-Conde et al. 2014; Pathan et al. 2015a). Root exudates from maize are composed of 65% sugars, 33% organic acids, and 2% amino acids, but the composition varies according to maize lines (Corrales et al. 2007). Some microbial species inhabiting the rhizosphere are likely attracted by species- and genotype-specific molecular signals released from the plant, probably as specific components of the root exudates (Merbach et al. 1999; Haichar et al. 2008). Two maize genotypes differing in two genes responsible for encoding the amount and type of sugar in the endosperm, and probably in the amount and type of sugar exuding to the rhizosphere, strongly promoted rhizosphere microbial community differing in composition and activity (Aira et al. 2010). The same conclusion was recently obtained using Arabidopsis mutants or ecotypes that could alter root phytochemical secretions and then strongly change composition and succession of the fungal and bacterial community in the rhizosphere (Badri et al. 2009; Channer et al. 2009). Such progress highlights the possibility to unravel the plant genotypic traits able to select specific functional strains that contribute to plant growth, disease resistance, and bioremediation (Bell et al. 2014).
Cultivar was an important driver of bacterial communities
Mantel tests showed that only the cultivar type significantly correlated with the bacterial composition (Table S4), confirming that the genotypic factors directly affect microbial community in the rhizosphere soil (Edwards et al. 2015). However, we could not identify the key traits responsible for the significant differences in the composition of rhizosphere bacterial community probably due to the low heritability and the fact that only four cultivars were used. These cultivars exhibited low levels of heritable variance (Table 1) and were being used in modern agricultural management, based on heavy fertilization. Root colonization by rhizobacteria with disease suppressive functions may be an inherited trait, probably related to heterosis (Picard et al. 2004; Picard and Bosco 2006). It has been hypothesized that the composition and diversity of rhizosphere microbiota are influenced by a few major alleles and environmental conditions which are field-specific (Peiffer et al. 2013). Therefore, more effective maize genotypes including landraces are necessary to explore the functional alleles and heritable plant–microbe interactions under natural environmental conditions. Future advances in these areas will ultimately allow rhizosphere microbiota to be incorporated into plant breeding.
References
Aira M, Gómez-Brandón M, Lazcano C, Bååth E, Dominguez J (2010) Plant genotype strongly modifies the structure and growth of maize rhizosphere microbial communities. Soil Biol Biochem 42:2276–2281
Ali B, Sabri AN, Ljung K, Hasnain S (2009) Auxin production by plant associated bacteria: impact on endogenous IAA content and growth of Triticum aestivum L. Appl Microbiol Lett 48:542–547
Amato KR, Yeoman CJ, Kent A, Righini N, Carbonero F, Estrada A, Gaskins HR, Stumpf RM, Yildirim S, Torralba M, Gillis M, Wilson BA, Nelson KE, White BA, Leigh SR (2013) Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J 7:1344–1353
An DS, Lee HG, Lee ST, Im WT (2009) Rhodanobacter ginsenosidimutans sp. nov., isolated from soil of a ginseng field in South Korea. Int J Syst Evol Microbiol 59:691–694
Araújo da Silva KR, Salles JF, Seldin L, van Elsas JD (2003) Application of a novel Paenibacillus-specific PCR-DGGE method and sequence analysis to assess the diversity of Paenibacillus spp. in the maize rhizosphere. J Microbiol Meth 54:213–231
Badri DV, Quintana N, El Kassis EG, Kim HK, Choi YH, Sugiyama A, Verpoorte R, Martinoia E, Manter DK, Vivanco JM (2009) An ABC transporter mutation alters root exudation of phytochemicals that provoke an overhaul of natural soil microbiota. Plant Physiol 151:2006–2017
Bais HP, Weir TL, Perry LG, Gilroy S, Vivanco JM (2006) The role of root exudates in rhizosphere interactions with plants and other organisms. Annu Rev Plant Biol 57:233–266
Baudoin E, Benizri E, Guckert A (2003) Impact of artificial root exudates on the bacterial community structure in bulk soil and maize rhizosphere. Soil Biol Biochem 35:1183–1192
Bell TH, Hassan SE, Lauron-Moreau A, Al-Otaibi F, Hijri M, Yergeau E, St-Arnaud M (2014) Linkage between bacterial and fungal rhizosphere communities in hydrocarbon-contaminated soils is related to plant phylogeny. ISME J 8:331–343
Bollmann A, Palumbo AV, Lewis K, Epstein SS (2010) Isolation and physiology of bacteria from contaminated subsurface sediments. Appl Environ Microbiol 76:7413–7419
Bradshaw JE, Dale MFB, Mackay GR (2003) Use of mid-parent values and progeny tests to increase the efficiency of potato breeding for combined processing quality and disease and pest resistance. Theor Appl Genet 107:36–42
Briones AM, Okabe S, Umemiya Y, Ramsing N, Reichardt W, Okuyama H (2002) Influence of different cultivars on populations of ammonia-oxidizing bacteria in the root environment of rice. Appl Environ Microbiol 68:3067–3075
Brusetti L, Francia P, Bertolini C, Pagliuca A, Borin S, Sorlini C, Abruzzese A, Sacchi G, Viti C, Giovannetti L, Giuntini E, Bazzicalupo M, Daffonchio D (2004) Bacterial communities associated with the rhizosphere of transgenic Bt 176 maize (Zea mays) and its non transgenic counterpart. Plant Soil 266:11–21
Bui TP, Kim YJ, Kim H, Yang DC (2010) Rhodanobacter soli sp. nov., isolated from soil of a ginseng field. Int J Syst Evol Microbiol 60:2935–2939
Castaldini M, Turrini A, Sbrana C, Benedetti A, Marchionni M, Mocali S, Fabiani A, Landi S, Santomassimo F, Pietrangeli B, Nuti MP, Miclaus N, Giovannetti M (2005) Impact of Bt corn on rhizospheric and soil eubacterial communities and on beneficial mycorrhizal symbiosis in experimental microcosms. Appl Environ Microbiol 71:6719–6729
Castiglioni P, Warner D, Bensen RJ, Anstrom DC, Harrison J, Stoecker M, Abad M, Kumar G, Salvador S, D’Ordine R, Navarro S, Back S, Fernandes M, Targolli J, Dasgupta S, Bonin C, Luethy MH, Heard JE (2008) Bacterial RNA chaperones confer abiotic stress tolerance in plants and improved grain yield in maize under water-limited conditions. Plant Physiol 147:446–455
Chamam A, Sanguin H, Bellvert F, Meiffren G, Comte G, Wisniewski-Dyé F, Bertrand C, Prigent-Combaret C (2013) Plant secondary metabolite profiling evidences strain dependent effect in the Azospirillum–Oryza sativa association. Phytochemistry 87:65–77
Chanway CP, Nelson LM, Holl FB (1988) Cultivar specific growth promotion of spring wheat Triticum aestivum L. by coexistent Bacillus species. Can J Microbiol 34:925–929
Corrales I, Amenós M, Poschenrieder C, Barceló J (2007) Phosphorus efficiency and root exudates in two contrasting tropical maize varieties. J Plant Nutr 30:887–900
Czarnota MA, Rimando AM, Weston LA (2003) Evaluation of root exudates of seven sorghum accessions. J Chem Ecol 29:2073–2083
De Clercq D, Van Trappen S, Cleenwerck I, Ceustermans A, Swings J, Coosemans J, Ryckeboer J (2006) Rhodanobacter spathiphylli sp. nov., a gammaproteobacterium isolated from the roots of Spathiphyllum plants grown in a compost-amended potting mix. Int J Syst Evol Microbiol 56:1755–1759
Den Herder G, Van Isterdael G, Beeckman T, De Smet I (2010) The roots of a new green revolution. Trends Plant Sci 15:600–607
Dohrmann AB, Küting M, Jünemann S, Jaenicke S, Schlüter A, Tebbe CC (2013) Importance of rare taxa for bacterial diversity in the rhizosphere of Bt- and conventional maize varieties. ISME J 7:37–49
Edwards J, Johnson C, Santos-Medellín C, Lurie E, Podishetty NK, Bhatnagar S, Eisen JA, Sundaresan V (2015) Structure, variation, and assembly of the root-associated microbiomes of rice. Proc Natl Acad Sci U S A 112:E911–E920
Fan TW, Lane AN, Shenker M, Bartley JP, Crowley D, Higashi RM (2001) Comprehensive chemical profiling of gramineous plant root exudates using high-resolution NMR and MS. Phytochemistry 57:209–221
Fierer N, Bradford MA, Jackson RB (2007) Toward an ecological classification of soil bacteria. Ecology 88:1354–1364
Figueiredo MV, Burity HA, Martínez CR, Chanway CP (2008) Alleviation of drought stress in the common bean (Phaseolus vulgaris L.) by co-inoculation with Paenibacillus polymyxa and Rhizobium tropici. Appl Soil Ecol 40:182–188
Gahoonia TS, Nielsen NE (2004) Root traits as tools for creating phosphorus efficient crop varieties. Plant Soil 260:47–57
Germida JJ, Siciliano SD (2001) Taxonomic diversity of bacteria associated with the roots of modern, recent and ancient wheat cultivars. Biol Fert Soils 33:410–415
Grayston S, Campbell C (1996) Functional biodiversity of microbial communities in the rhizospheres of hybrid larch (Larix eurolepis) and Sitka spruce (Picea sitchensis). Tree Physiol 16:1031–1038
Haichar FE, Marol C, Berge O, Rangel-Castro JI, Prosser JI, Balesdent J, Heulin T, Achouak W (2008) Plant host habitat and root exudates shape soil bacterial community structure. ISME J 2:1221–1230
Herbert Y, Guingo E, Loudet O (2001) The response of root/shoot partitioning and root morphology to light reduction in maize genotypes. Crop Sci 16:162–211
Hinsinger P, Plassard C, Tang C, Jaillard B, Tang CX (2003) Origins of root-mediated pH changes in the rhizosphere and their responses to environmental constraints: a review. Plant Soil 248:43–59
Im WT, Lee ST, Yokota A (2004) Rhodanobacter fulvus sp. nov., a beta-galactosidase-producing gammaproteobacterium. J Gen Appl Microbiol 50:143–147
Innerebner G, Knief C, Vorholt JA (2011) Protection of Arabidopsis thaliana against leaf-pathogenic Pseudomonas syringae by Sphingomonas strains in a controlled model system. Appl Environ Microbiol 77:3202–3210
King EB, Parke JL (1993) Biocontrol of Aphanomyces root rot and Pythium damping-off by Pseudomonas cepacia AMMD on four pea cultivars. Plant Dis 77:1185–1188
Kostka JE, Green SJ, Rishishwar L, Prakash O, Katz LS, Mariño-Ramíreze L, Jordana IK, Munk C, Ivanova N, Mikhailova N, Watson DB, Brown SD, Palumbo AV, Brooks SC (2012) Genome sequences for six Rhodanobacter strains, isolated from soils and the terrestrial subsurface, with variable denitrification capabilities. J Bacteriol 194:4461–4462
Kucey RMN (1988) Plant growth-altering effects of Azospirillum brasilense and Bacillus C-11-25 on two wheat cultivars. J Appl Bacteriol 64:187–196
Kumar PS, Brooker MR, Dowd SE, Camerlengo T (2011) Target region selection is a critical determinant of community fingerprints generated by 16S pyrosequencing. PLoS One 6:e20956
Lauber CL, Hamady M, Knight R, Fierer N (2009) Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75:5111–5120
Lee EA, Tollenaar M (2007) Physiological basis of successful breeding strategies for maize grain yield. Crop Sci 47:S202–S215
Leeman M, van Pelt JA, den Ouden FM, Heinsbroek M, Bakker PAHM, Schippers B (1995) Induction of systemic resistance by Pseudomonas fluorescens in radish cultivars differing in susceptibility to Fusarium wilt, using a novel bioassay. Eur J Plant Pathol 101:655–664
Li J (2013) The identification of density-tolerance and genetic study of maize under different density selection pressure. Ph.D Dissertation. Jinlin Agricultural University
Liu L, Kloepper JW, Tuzun S (1995) Induction of systemic resistance in cucumber by plant growth-promoting rhizobacteria: duration of protection and effect of host resistance on protection and root colonization. Phytopathology 85:1064–1068
Liu P, Lin Q, Sui F, Sun Z (1994) Study on the characteristics of root system in high-yield upright-leaf maize. J Maize Sciences 3:36–39
Mansfield BD, Mumm RH (2014) Survey of plant density tolerance in US maize germplasm. Crop Sci 54:157–173
Mansouri H, Petit A, Oger P, Dessaux Y (2002) Engineered rhizosphere: the trophic bias generated opine-producing plants is independent of the opine, the soil origin, and the plant species. Appl Environ Microbiol 68:2562–2566
Mazzola M, Funnell DL, Raaijmakers JM (2004) Wheat cultivar-specific selection of 2,4-diacetylphloroglucinol-producing fluorescent Pseudomonas species from resident soil populations. Microb Ecol 48:338–348
Mazzola M, Gu Y-H (2000) Phyto-management of microbial community structure to enhance growth of apple in replant soils. Acta Hortic 532:73–78
Merbach W, Mirus E, Knof G, Remus R, Ruppel S, Russow R, Gransee A, Schulze J (1999) Release of carbon and nitrogen compounds by plant roots and their possible ecological importance. J Plant Nutr Soil Sc 162:373–383
Micallef SA, Channer S, Shiaris MP, Colon-Carmona A (2009) Plant age and genotype impact the progression of bacterial community succession in the Arabidopsis rhizosphere. Plant Signal Behav 4:777–780
Milling A, Smalla K, Xaver F, Maidl K, Schloter M, Munch JC (2004) Effects of transgenic potatoes with an altered starch composition on the diversity of soil and rhizosphere bacteria and fungi. Plant Soil 266:23–39
Nalin R, Simonet P, Vogel TM, Normand P (1999) Rhodanobacter lindaniclasticus gen. Nov., sp. nov., a lindane-degrading bacterium. Int J Syst Bacteriol 49:19–23
Notz R, Maurhofer M, Schnider-Keel U, Duffy B, Haas D, Défago G (2001) Biotic factors affecting expression of the 2,4-diacetylphloroglucinol biosynthesis gene phlA in Pseudomonas fluorescens biocontrol strain CHA0 in the rhizosphere. Phytopathology 91:873–881
Palacios OA, Bashan Y, de-Bashan LE (2014) Proven and potential involvement of vitamins in interactions of plants with plant growth-promoting bacteria—an overview. Biol Fert Soils 50:415–432
Pathan SI, Ceccherini MT, Hansen MA, Giagnoni L, Ascher J, Arenella M, Sørensen SJ, Pietramellara G, Nannipieri P, Renella G (2015a) Maize lines with different nitrogen use efficiency select bacterial communities with different β-glucosidase-encoding genes and glucosidase activity in the rhizosphere. Biol Fert Soils 51:995–1004
Pathan SI, Ceccherini MT, Pietramellara G, Puschenreiter M, Giagnoni L, Arenella M, Varanini Z, Nannpieri P, Renella G (2015b) Enzyme activity and microbial community structure in the rhizosphere of two maize lines differing in N use efficiency. Plant Soil 387:413–424
Peiffer JA, Spor A, Koren O, Jin Z, Tringe SG, Dangl JL, Buckler ES, Ley RE (2013) Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci U S A 110:6548–6553
Picard C, Bosco M (2006) Heterozygosis drives maize hybrids to select elite 2,4-diacethylphloroglucinol-producing Pseudomonas strains among resident soil populations. FEMS Microbiol Ecol 58:193–204
Picard C, Bosco M (2005) Maize heterosis affects the structure and dynamics of indigenous rhizospheric auxins producing Pseudomonas populations. FEMS Microbiol Ecol 53:349–357
Picard C, Frascaroli E, Bosco M (2004) Frequency and biodiversity of 2,4-diacetylphloroglucinol-producing rhizobacteria are differentially affected by the genotype of two maize inbred lines and their hybrid. FEMS Microbiol Ecol 49:207–215
Raaijmakers JM, Vlami M, De Souza JT (2002) Antibiotic production by bacterial biocontrol agents. Antonie Van Leeuwenhoek 81:537–547
R Core Team. R (2013) A language and environment for statistical computing R Foundation for Statistical Computing: Vienna, Austria. http://www.R-project.org/
Sangabriel-Conde W, Negrete-Yankelevich S, Maldonado-Mendoza IE, Trejo-Aguilar D (2014) Native maize landraces from Los Tuxtlas, Mexico show varying mycorrhizal dependency for P uptake. Biol Fert soils 50:405–414
Sanguin H, Sarniguet A, Gazengel K, Moёnne-Loccoz Y, Grundmann GL (2009) Rhizosphere bacterial communities associated with disease suppressiveness stages of take-all decline in wheat monoculture. New Phytol 184:694–707
Smith KP, Handelsman J, Goodman RM (1997) Modeling dose-response relationships in biocontrol: partitioning host responses to the pathogen and biocontrol agent. Phytopathology 87:720–729
Smith KP, Handelsman J, Goodman RM (1999) Genetic basis in plants for interactions with disease-suppressive bacteria. Proc Natl Acad Sci U S A 96:4786–4790
Sparks DL, Page AL, Helmke PA, Loeppert RH, Soltanpour PN, Tabatabai MA, Johnston CT, Sumner ME (1996) Methods of soil analysis. Part 3—chemical methods. Soil Science Society of America Inc, Madison, WI
Tollenaar M, Daynard TB (1978) Relationship between assimilate source and reproductive sink in maize grown in a short season environment. Agron J 70:219–223
Vakili NG (1992) Biological seed treatment of corn with mycopathogenic fungi. J Phytopathol 134:313–323
Vakili NG, Bailey TB Jr (1989) Yield response of corn hybrids and inbred lines to phylloplane treatment with mycopathogenic fungi. Crop Sci 29:183–190
van den Heuvel RN, van der Biezen E, Jetten MS, Hefting MM, Kartal B (2010) Denitrification at pH 4 by a soil-derived Rhodanobacter-dominated community. Enviro Microbiol 12:3264–3271
Vessey JK (2003) Plant growth promoting rhizobacteria as biofertilizers. Plant Soil 255:571–586
Wang L, An DS, Kim SG, Jin FX, Lee ST, Im WT (2011) Rhodanobacter panaciterrae sp. nov., a bacterium with ginsenoside-converting activity isolated from soil of a ginseng field. Int J Syst Evol Microbiol 61:3028–3032
Weinert N, Piceno Y, Ding GC, Meincke R, Heuer H, Berg G, Schloter M, Andersen G, Smalla K (2011) PhyloChip hybridization uncovered an enormous bacterial diversity in the rhizosphere of different potato cultivars: many common and few cultivar-dependent taxa. FEMS Microbiol Ecol 75:497–506
Weller DM, Raaijmakers JM, Gardener BBM, Thomashow LS (2002) Microbial populations responsible for specific soil suppressiveness to plant pathogens. Annu Rev Phytopathol 40:309–348
Weon HY, Kim BY, Hong SB, Jeon YA, Kwon SW, Go SJ, Koo BS (2007) Rhodanobacter ginsengisoli sp. nov. and Rhodanobacter terrae sp. nov., isolated from soil cultivated with Korean ginseng. Int J Syst Evol Microbiol 57:2810–2813
Zhang H, Kim M, Sun Y, Dowd SE, Shi H, Paré PW (2008) Soil bacteria confer plant salt tolerance by tissue-specific regulation of the sodium transporter HKT1. Mol Plant 21:737–744
Acknowledgements
This work was supported by the Special Fund for Agro-Scientific Research of the Ministry of Agriculture of China (201103001) and the National Key Research and Development Program of China (2016YFD0300203).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Xinya Wen and Meng Wang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Wen, X., Wang, M., Ti, J. et al. Bacterial community composition in the rhizosphere of maize cultivars widely grown in different decades. Biol Fertil Soils 53, 221–229 (2017). https://doi.org/10.1007/s00374-016-1169-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00374-016-1169-6